

Abstract
Nonresolving inflammation contributes to tissue damage and organ dysfunction in a wide array of pathologies, including cardiovascular disease. At the interface between inflammation and inflammation-resolution is the macrophage. Macrophage engulfment of apoptotic cells (efferocytosis) during immune cell turnover triggers activation of intra- and inter-cellular immunosuppressive signaling networks. In diseases of aging and obesity, accumulating evidence indicates that efferocytosis is impaired; the underlying mechanisms of which are unclear. In the current issue of Circulation Research, Driscoll et al., reveal that deficiency of the metalloproteinase ADAM17 prevents shedding of the apoptotic cell receptor CD36 from macrophages to enhance efferocytosis and inflammation resolution during peritonitis. These findings implicate proteolysis of apoptotic cell receptors as one explanation for defective efferocytosis-directed inflammation resolution during disease. Future studies are warranted to test the significance of these findings during cardiovascular syndromes and in other cases of nonresolving inflammation.
Keywords: Efferocytosis, ADAM17, CD36
Efferocytosis-directed inflammation resolution
Removal of dying cells and pathogens by macrophages (MΦs) is coupled to activation of downstream intracellular signaling pathways that mobilize either pro- or anti-inflammatory signaling networks. In the case of efferocytic1 engulfment of apoptotic cells (ACs), MΦs both suppress 2, 3 pro-inflammatory signaling and activate pro-resolving 4 cascades to uphold tolerance to self-antigen 5. For example, injection of ACs in vivo represses the immune response to cell-associated antigens 6. This is in contrast to MΦ engulfment of IgG-opsonized pathogens7, 8, 9 by Fcγ receptors, which triggers release of pro-inflammatory cytokines 2, 10. Thus, immune cells have evolved distinct mechanisms to differentially recognize and orchestrate inflammation, secondary to engulfment.
In the setting of nonresolving inflammation11, 12, mis-calibration of the immune response (for example, over-exuberant inflammation during sepsis), or failure to clear initiating insults and activate resolving factors, in turn promotes an inflammatory milieu that is counterproductively harmful. In diseases such as cystic fibrosis, COPD, asthma, lupus, and atherosclerosis, prolonged inflammation is also associated with an accumulation of ACs 1, 13-15. AC clearance in the absence of disease is typically efficient. Even in tissues with high rates of cell turnover, only a few ACs can be detected under homeostatic conditions 16. Whether defective efferocytosis is a cause, consequence, or both within these underlying pathologies is not clear and likely context dependent. Nevertheless, inability to activate efferocytosis-directed anti-inflammatory pathways is a candidate contributing factor in the failure to promote healing. Consistent with this notion, studies in experimental gene-targeted rodents support the hypothesis that efferocytosis causally regulates disease progression, as described below.
Defective efferocytosis at the level of apoptotic cell (AC) receptors
Suppressors of efferocytosis can act at multiple stages, including phagocyte recruitment, AC binding to phagocytes, and engulfment. For reasons that are not entirely clear, in some disease states, such as atherosclerosis 17, efferocytosis becomes defective despite abundant MΦ presence, suggesting cell-intrinsic perturbations13. In vitro, inflammatory stimuli such as endotoxin and TNFα directly inhibit efferocytosis, at least in part through generation of superoxides 18. In this scenario, inflammation initiates a reinforcing feedback loop of defective efferocytosis, promoting further inflammation via non-cleared apoptotic cells that become secondary necrotic. Little is known about mechanisms of defective efferocytosis in vivo, however, studies targeting phagocyte AC receptors support the idea that defective efferocytosis can promote inflammation and aggravate underlying disease. These cell-surface AC receptors directly, or through soluble bridging molecules, recognize AC ligands that include phosphatidylserine and oxidized membrane epitopes 19. As an example, deficiency of the AC receptor Mer tyrosine kinase receptor (MERTK) delays AC clearance 20 and increases TNFα levels during endotoxemia. In chronic diseases, MERTK is required to clear dying cells during advanced atherosclerosis 21, 22 and deficiency of MERTK accelerates lupus-like autoimmunity 14.
Besides genetic proofs of principle, natural mechanisms of defective efferocytosis in vivo have eluded discovery. There are however molecular clues that implicate phagocyte AC receptors. Cultures from diseased patients reveal elevated levels of shed, or non-cell associated/soluble AC receptors. Typically, transmembrane AC receptors signal independently or in cooperation through membrane domains. This mobilizes intracellular cytoskeletal signaling to generate force required for AC internalization 23. On the other hand, signal transduction capacity of AC receptors can be uncoupled from AC-binding domains 24. Thus, AC receptor shedding not only depletes phagocytic capacity of the host cell, but also generates a potential competitive inhibitor that can block efferocytosis on neighboring efferocytes. Notable examples of soluble AC receptors during disease include CD36, lectin-like oxidized LDL-receptor-1 (LOX-1), low-density lipoprotein receptor-related protein-1 (LRP-1), and as described above, MERTK. Soluble LRP-1 is increased in patients with acute respiratory distress syndrome 25. Circulating soluble LOX-1 is elevated in patients with acute coronary syndrome 26. Evidence for soluble MerTK is found in patients with lupus, rheumatoid arthritis, cardiovascular disease, and endtoxemia 24, 27-29. Finally, plasma CD36 elevation is associated with diabetes30. These illustrations highlight AC receptor shedding as an important phenotype of diverse inflammatory diseases. In the case of LOX-1, MERTK, and now CD36, the metalloproteinase ADAM17 (a disintegrin and metalloprotease) has been identified as an agent of AC receptor shedding.
Linking ADAM17 to defective efferocytosis and inflammation resolution
Proteases of the ADAM family juxtapose near neighboring membrane-bound proteins and trigger shedding/release of cell surface ectodomans into the extracelluar milieu 31. First described for its role in shedding of pro-TNFα 32, ADAM17 is therefore intimately linked with the regulation of inflammation. However, a direct requirement for ADAMs at the level of efferocytosis and efferocytosis-directed inflammation resolution had previously not been documented. In this context, and independent of effects on cytokine release, Driscoll et. al. 33 hypothesize an additional layer of ADAM17-mediated inflammatory control, specifically through control of efferocytosis efficiency. To test their hypothesis, the investigators measured AC clearance and markers of inflammation in a mouse model of ADAM17 deficiency. To focus their studies on MΦs, ADAM17 deficient cells were transplanted into irradiated mice to generate chimeras deficient for leukocyte ADAM17 in bone marrow. After eliciting inflammatory cells, ADAM17-deficient mice, relative to control, exhibited similar levels and kinetics of neutrophil turnover and monocyte entry in the peritoneum during acute phase peritonitis. Interestingly, after injecting ACs at later stages of inflammation, MΦs from ADAM17-deficient mice displayed a significant enhancement in uptake of exogenous ACs. This led to a marked reduction of MΦ numbers, consistent with resolution of inflammation. Although an alternative explanation for reductions in MΦ content is due to altered interactions with acute-phase neutrophils, it is important to note that ACs were injected after neutrophil egress and that reductions in MΦs were specific to injection of ACs. Evidence for enhanced efferocytosis was also discovered in mixed chimeras containing an equal ratio of control and ADAM17-null MΦs, supporting the notion that increased efferocytosis was MΦ-intrinsic rather than secondary to an altered extracellular milieu. Consistent with an overall anti-inflammatory phenotype, ADAM17 deficient MΦs expressed reduced levels of the pro-inflammatory marker iNOS and increased levels of resolution-associated arginase I 34, 35.
Linking ADAM17 effects on efferocytosis to cleavage of CD36
MΦs express multiple substrates of ADAM17, including some implicated in recognition or uptake of ACs 36-38. Therefore, to focus their mechanistic questions, a screening approach was utilized. AC surrogates with known receptor specificities were loaded onto ADAM17-deficient phagocytes in vitro. Exploiting this approach, ADAM17-null MΦs bound more phosphatidylserine (PS)-containing liposomes, but not liposomes composed of phosphatidylcholine, suggesting that ADAM17 acts to inhibit recognition of PS on ACs. Recognizing that PS-receptors can further be distinguished by their ability to recognize modified atherogenic lipoproteins 39, Driscoll et al. zeroed-in on scavenger receptors after acetylated LDL prevented enhancements in liposome binding. Finally, CD36 was directly implicated after addition of blocking antibodies: CD36-blockade significantly decreased efferocytosis in ADAM17-deficient MΦs relative to control and during peritonitis.
Evidence for ADAM17-mediated shedding of CD36 was found in ADAM17-deficient MΦs after measuring increased surface levels of MΦ CD36 during peritonitis, independent of changes in messenger RNA. In addition, loss of ADAM17 decreased the ratio of soluble protein (by ELISA) from clarified supernatants in vitro, relative to cell-associated CD36. Relative to other AC receptors targeted by ADAM17, such as MERTK and LOX-1, which are type I and type II single-pass transmembrane (TM) proteins respectively, CD36 is unique. CD36 encodes a dual-pass TM, therefore requiring at least two separate cleavage events to liberate its soluble ectodomain. Immunoblot analysis provided initial evidence for proteolytic fragments of CD36 and mass spectrometry identified cleavage sites of CD36 at regions predicted to be proximal to the membrane, as has previously been documented with other ADAM17 substrates 40. However, future experiments are necessary to resolve other candidate cleavage sites that may have been masked by shared trypsin cleavage sites during tryptic mass-spec analysis. Also, considerations for the involvement of other proteases must be made. In fact, this is a likely scenario as significant levels of soluble CD36 were still found in the absence of ADAM17 in vitro. In vivo, studies are also required to measure the extent and biochemical nature of ADAM17-dependent soluble-CD36 levels in serum. Nevertheless, together these experiments provide strong evidence that ADAM17 promotes shedding of CD36, likely by direct proteolytic cleavage, thereby dampening efferocytosis of ACs and leading to delayed inflammation resolution (Figure 1).
Figure 1. Proteolysis of CD36 by ADAM17 delays inflammation resolution by inhibiting efferocytosis.
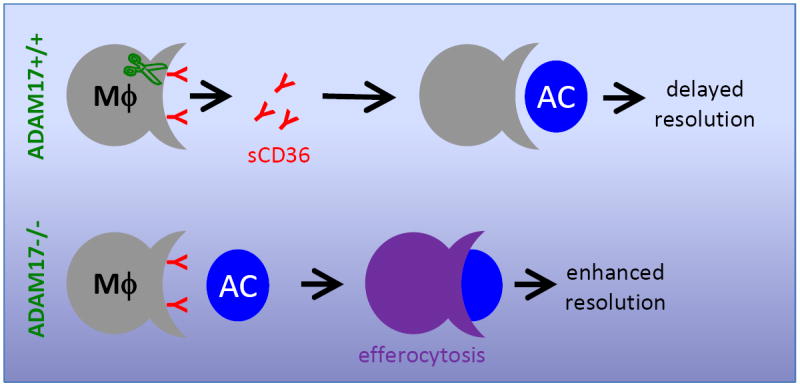
ADAM17 (green) cleaves membrane bound CD36 on macrophages (MΦs), leading to shedding of soluble CD36 (sCD36) into the extracellular milieu. Loss of CD36 reduces efferocytosis, i.e., macrophage engulfment of apoptotic cells (ACs), and in turn delays inflammation resolution. Absence of ADAM17 (ADAM17-/-) in contrast promotes efferocytosis and an anti-inflammatory response in MΦs, thereby enhancing inflammation resolution.
Other future considerations
Interestingly, effects of ADAM17 on efferocytosis were independent of other AC receptors that had primarily been studied as in vitro targets of ADAM17 24, 29. One explanation is that unlike these other reports, CD36 cleavage by Driscoll et al. chiefly occurred independent of inflammatory triggers such lipopolysaccharide and TNFα. Therefore cleavage of alternative AC receptors may play a more significant role in other pathogenic or disease contexts. Also, the relative importance of specific ADAM17 targets may vary in homeostasis versus resolving inflammation versus non-resolving inflammation. In this context, it is tempting to speculate that ADAM17-directed cleavage of AC receptors may selectively target receptors to uniquely regulate efferocytosis and inflammation resolution in disparate inflammatory states. Such specificity could be controlled both spatially and temporally. For example, substrate-specific targeting could be regulated at the level of substrate-site affinity or conformational or proximal accessibility. Temporally, the investigators point out that CD36 expression was highest on MΦs at later stages of inflammation, consistent with involvement during inflammation resolution. This begs additional questions in an evolutionary context: What purpose does ADAM17 serve in blocking inflammation-resolution during efferocytosis, or is such a response maladaptive? One answer may be that cleavage represents a path to quickly reverse inflammation resolution if additional or persistent stimuli are detected. In a similar vein, suppression of efferocytosis could mobilize alternative mechanisms of phagocytic uptake that are more tailored, for example, to the uptake, metabolism, and/or cross-presentation of pathogens. In this setting, uptake of AC self-antigen may also be disadvantageous if “confused” with uptake of non-self, with implications on loss of tolerance and autoimmunity. At the level of CD36, this discussion must also consider the numerous other CD36 functions, independent of AC uptake, including phagocytosis of bacteria, uptake of modified lipoproteins during atherosclerosis, requirements for fatty acid import, and even effects of angiogensis 41-44. Thus, cleavage of CD36 has broader implications, just as the activities of ADAM17 and its multiple substrates.
Finally, singular or combinatorial strategies to resolve inflammation must not only consider issues of specificity, but also the potential for compensatory feedback mechanisms that may further limit efficacy. In addition, therapies must not overly impair innate requirements to ward off infection and repair tissue injury. Thus, a more comprehensive understanding of the mechanisms described by Driscoll et al., beyond efferoctyosis-directed inflammation and at the level of infection and wound healing, will assist in development of future strategies. This is important in the setting of chronic diseases such as aging and obesity, which typically require long-term therapeutic administration. Also to be considered are diseases of acute inflammation, particularly in cases such as inflammation after myocardial infarction, wherein advanced age and therefore limited evolutionary pressures may set the stage for maladaptive responses, with the potential for therapeutic optimization. Thus, the present study by Driscoll et. al. is just the “tip of the iceberg” and provides an intriguing mechanistic starting point to test implications of protease-directed receptor cleavage during efferocytosis and inflammation in multiple settings of disease.
Supplementary Material
Acknowledgments
Sources of Funding: ET is supported by NHLBI 1K99/R00-HL09702.
Footnotes
Disclosures: None
References
- 1.Vandivier RW, Henson PM, Douglas IS. Burying the dead: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 2.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving tgf-beta, pge2, and paf. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maderna P, Godson C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochimica et biophysica acta. 2003;1639:141–151. doi: 10.1016/j.bbadis.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Serhan CN, Savill J. Resolution of inflammation: The beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 5.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med. 2002;196:1091–1097. doi: 10.1084/jem.20021215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown RH, Walters DM, Greenberg RS, Mitzner W. A method of endotracheal intubation and pulmonary functional assessment for repeated studies in mice. J Appl Physiol. 1999;87:2362–2365. doi: 10.1152/jappl.1999.87.6.2362. [DOI] [PubMed] [Google Scholar]
- 8.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 9.Metschnikoff E. Lecture on phagocytosis and immunity. Br Med J. 1891;1:213–217. doi: 10.1136/bmj.1.1570.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto K, Johnston RB., Jr Dissociation of phagocytosis from stimulation of the oxidative metabolic burst in macrophages. J Exp Med. 1984;159:405–416. doi: 10.1084/jem.159.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 12.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 14.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5:e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 1994;372:100–103. doi: 10.1038/372100a0. [DOI] [PubMed] [Google Scholar]
- 17.Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73:470–480. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 18.McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, Bratton DL, Kang JL, Henson P. Tnf-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase a2 and oxidant-dependent mechanisms. J Immunol. 2007;178:8117–8126. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]
- 19.Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: Eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–656. doi: 10.1016/j.tcb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by mer. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 21.Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, Offenstadt G, Leseche G, Cohen PL, Tedgui A, Mallat Z. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1429–1431. doi: 10.1161/ATVBAHA.108.169078. [DOI] [PubMed] [Google Scholar]
- 22.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenberg S, Burridge K, Silverstein SC. Colocalization of f-actin and talin during fc receptor-mediated phagocytosis in mouse macrophages. J Exp Med. 1990;172:1853–1856. doi: 10.1084/jem.172.6.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109:1026–1033. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wygrecka M, Wilhelm J, Jablonska E, Zakrzewicz D, Preissner KT, Seeger W, Guenther A, Markart P. Shedding of low-density lipoprotein receptor-related protein-1 in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2011;184:438–448. doi: 10.1164/rccm.201009-1422OC. [DOI] [PubMed] [Google Scholar]
- 26.Kume N, Mitsuoka H, Hayashida K, Tanaka M, Kominami G, Kita T. Soluble lectin-like oxidized ldl receptor-1 (slox-1) as a sensitive and specific biomarker for acute coronary syndrome--comparison with other biomarkers. J Cardiol. 2010;56:159–165. doi: 10.1016/j.jjcc.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Wu J, Ekman C, Jonsen A, Sturfelt G, Bengtsson AA, Gottsater A, Lindblad B, Lindqvist E, Saxne T, Dahlback B. Increased plasma levels of the soluble mer tyrosine kinase receptor in systemic lupus erythematosus relate to disease activity and nephritis. Arthritis Res Ther. 2011;13:R62. doi: 10.1186/ar3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garbin U, Baggio E, Stranieri C, Pasini A, Manfro S, Mozzini C, Vallerio P, Lipari G, Merigo F, Guidi G, Cominacini L, Fratta Pasini A. Expansion of necrotic core and shedding of mertk receptor in human carotid plaques: A role for oxidized polyunsaturated fatty acids? Cardiovasc Res. 2013;97:125–133. doi: 10.1093/cvr/cvs301. [DOI] [PubMed] [Google Scholar]
- 29.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the mer tyrosine kinase receptor is mediated by adam17 through a pathway involving reactive oxygen species, protein kinase {delta}, and p38 map kinase. J Biol Chem. 2011 doi: 10.1074/jbc.M111.263020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handberg A, Norberg M, Stenlund H, Hallmans G, Attermann J, Eriksson JW. Soluble cd36 (scd36) clusters with markers of insulin resistance, and high scd36 is associated with increased type 2 diabetes risk. J Clin Endocrinol Metab. 2010;95:1939–1946. doi: 10.1210/jc.2009-2002. [DOI] [PubMed] [Google Scholar]
- 31.Blobel CP. Metalloprotease-disintegrins: Links to cell adhesion and cleavage of tnf alpha and notch. Cell. 1997;90:589–592. doi: 10.1016/s0092-8674(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 32.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 33.Driscoll WS, V T, Tang J, Wilson CL, Raines EW. Macrophage adam17 deficiency augments cd36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circulation Research. 2013 doi: 10.1161/CIRCRESAHA.112.300683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willment JA, Lin HH, Reid DM, Taylor PR, Williams DL, Wong SY, Gordon S, Brown GD. Dectin-1 expression and function are enhanced on alternatively activated and gm-csf-treated macrophages and are negatively regulated by il-10, dexamethasone, and lipopolysaccharide. J Immunol. 2003;171:4569–4573. doi: 10.4049/jimmunol.171.9.4569. [DOI] [PubMed] [Google Scholar]
- 35.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Middelhoven PJ, Ager A, Roos D, Verhoeven AJ. Involvement of a metalloprotease in the shedding of human neutrophil fc gammariiib. FEBS Lett. 1997;414:14–18. doi: 10.1016/s0014-5793(97)00959-9. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Brazzell J, Herrera A, Walcheck B. Adam17 deficiency by mature neutrophils has differential effects on l-selectin shedding. Blood. 2006;108:2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Etzerodt A, Maniecki MB, Moller K, Moller HJ, Moestrup SK. Tumor necrosis factor alpha-converting enzyme (tace/adam17) mediates ectodomain shedding of the scavenger receptor cd163. J Leukoc Biol. 2010;88:1201–1205. doi: 10.1189/jlb.0410235. [DOI] [PubMed] [Google Scholar]
- 39.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overall CM, Blobel CP. In search of partners: Linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 41.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A cd36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 42.Savill J, Hogg N, Ren Y, Haslett C. Thrombospondin cooperates with cd36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest. 1992;90:1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren Y, Silverstein RL, Allen J, Savill J. Cd36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med. 1995;181:1857–1862. doi: 10.1084/jem.181.5.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverstein RL, Febbraio M. Cd36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.