

Abstract
Tax1-binding protein 1 (Tax1bp1) negatively regulates NF-κB by editing the ubiquitylation of target molecules by its catalytic partner A20. Genetically engineered TAX1BP1-deficient (KO) mice develop age-dependent inflammatory constitutions in multiple organs manifested as valvulitis or dermatitis and succumb to premature death. Laser capture dissection and gene expression microarray analysis on the mitral valves of TAX1BP1-KO mice (8 and 16 week old) revealed 588 gene transcription alterations from the wild type. SAA3 (serum amyloid A3), CHI3L1, HP, IL1B and SPP1/OPN were induced 1,180-, 361-, 187-, 122- and 101-fold respectively. WIF1 (Wnt inhibitory factor 1) exhibited 11-fold reduction. Intense Saa3 staining and significant I-κBα reduction were reconfirmed and massive infiltration of inflammatory lymphocytes and edema formation were seen in the area. Antibiotics-induced ‘germ free’ status or the additional MyD88 deficiency significantly ameliorated TAX1BP1-KO mice's inflammatory lesions. These pathological conditions, as we named ‘pseudo-infective endocarditis’ were boosted by the commensal microbiota who are usually harmless by their nature. This experimental outcome raises a novel mechanistic linkage between endothelial inflammation caused by the ubiquitin remodeling immune regulators and fatal cardiac dysfunction.
Introduction
The transcription factor NF-κB is essential for the regulation of the innate and adaptive immune responses. NF-κB is activated in response to a wide variety of stimuli, such as inflammation, DNA damage, or nociception [1], [2], and is involved in embryogenesis and multiple tissue development [3]. The NF-κB family comprises five proteins including RelA (p65), RelB, c-Rel, NF-κB1, and NF-κB2, and their transcriptional activities are tightly controlled to ensure their transient signaling in response to specific stimuli. The NF-κB signaling cascade is usually triggered by sensor molecules, such as toll-like receptor (TLR) family proteins. These proteins can identify the presence of a wide range of microorganisms and then transmit that information through phosphorylation relays to downstream kinases, which eventually culminate at the I-κB kinase (IKK). IKK activates NF-κB via phosphorylation of inhibitory I-κB proteins (primarily I-κBα), which leads to its ubiquitylation and degradation by the 26S proteasome complex and allows NF-κB to enter the nucleus. I-κB is induced by NF-κB to function in a negative feedback loop that terminates NF-κB signaling. Aberrant activation of NF-κB has been linked to several pathological features such as allergic responses, autoimmune diseases, septic shock, and carcinogenesis in a variety of organs [4].
In addition to I-κB, deubiquitinase A20 (also referred to as TNFα-induced protein 3 or TNFAIP3) targets important signaling intermediates upstream of I-κB to terminate NF-κB activation [5], [6]. A20 cleaves Lys63 (K63)-linked polyubiquitin chains on overlapping substrates, such as E3 ubiquitin ligase TRAF6 and adaptor molecule RIP1, with the help of the substrate-specific adaptor Tax1-binding protein 1 (Tax1bp1 [7], [8]). Tax1bp1 intrinsically regulates NF-κB by recruiting A20 to the target molecules to remove their polyubiquitin chains, which play important roles in their assembly into the IKK complex [8], [9]. Deficiencies in A20 or Tax1bp1 lead to uncontrolled and spontaneous systemic inflammation in mice as a result of unchecked NF-κB signaling [8], [10].
Tax1bp1 was originally identified as a host cell factor that binds to the encoded protein of human T-lymphotropic virus type 1 (HTLV-1), known as Tax1 [7]. Tax1 is a potent activator of NF-κB and a major pathogenic factor in HTLV-1 associated diseases (HAD), such as HTLV-1 associated myelopathy (HAM) or HTLV-1 uveitis (HU [11]), and adult T-cell leukemia (ATL [12]). Tax1 interrupts the ability of Tax1bp1 to connect to and recruit A20 to target molecules and thus evokes persistent NF-κB activation [13], [14]. Tax1 also activates NF-κB by binding to the NF-κB essential modulator (NEMO), a regulatory subunit of IKK [15]. The aberrant activation of NF-κB in HADs can therefore be attributed to Tax1, which leads to Tax1bp1 dysfunction, over-activation of IKK, or both. Epidemiological studies provide support for a close link between HTLV-1 infection and HAD or other inflammatory diseases such as Sjögren's syndrome [16], vascular dementia [17], and atherosclerosis [18]. Moreover, recent accumulating evidence strongly suggests that several mutations in the A20 locus are primarily responsible for the development of Crohn's disease, rheumatoid arthritis, systemic lupus erythematosus, psoriasis and type 1 diabetes [19].
For research purposes, we established TAX1BP1-deficient (-KO) mice, which display exacerbation of inflammation (characterized as valvulitis and dermatitis) in an age-dependent manner in addition to functional inadequacies manifested in growth retardation and premature death [8]. To elucidate the molecular mechanisms underlying the manifestation of inflammatory symptoms and their link to premature or possible cardiac abnormalities induced by TAX1BP1-deficiency, we performed a series of pathological evaluations using TAX1BP1-KO mice: (1) laser capture microdissection (LCM)- and gene expression microarray-based profiling of the mitral valves, which was reevaluated using real-time polymerase chain reaction (RT-PCR); (2) multiplex cytokine and chemokine quantitation in sera on systemic inflammatory constitution; (3) histochemical and electron microscopic analyses of multiple pathogenic foci; and (4) antibiotic treatments and cross experimentation with MyD88-deficient mice [20] to examine the role of commensal microbiota in the pathogenesis of TAX1BP1-KO mice.
From our experimental data, we conclude that systemic inflammation and cardiac structural abnormalities in TAX1BP1-KO mice originated from commensal microbiota, which are usually harmless in nature. Furthermore, these results indicate a potential risk to asymptomatic HTLV-1 carriers, which should be addressed by further clinical research.
Materials and Methods
Animals
TAX1BP1-KO mice having replaced their exon 17 region with CMV-driven NEO gene in reverse orientation [8] and their wild-type (WT) littermates as controls were analyzed throughout the experiment. These strains are maintained as F9 or advanced generations of C57BL/6CrSlc or the original 129/+ Ter/SvJcl. MyD88 deficient mice are kind gifts from professor Hitoshi Nakashima from Fukuoka University [21]. Homozygous TAX1BP1-KO mice were crossbred with homozygous MyD88-KO background to generate MyD88/TAX1BP1-KO mutants. Each of the targeted loci was evaluated by PCR. These mice were bred and maintained under specific pathogen-free (SPF) conditions at the animal facility of Oita University Faculty of Medicine. All the mice related manipulations were performed with protocols approved by the animal ethics committee at the Oita University (Justified numbers, daily care, treatment and euthanasia procedures).
Laser capture microdissection
Three mitral valves from 8 or 16 week old (-wk) maleTAX1BP1-KO and their WT littermates were collected by Arcturus XT laser capture microdissection system according to a manufacture's directions.
RNA Isolation and gene expression microarray analysis
Total RNAs were purified from the mitral valves using RNeasy mini kit (Qiagen). RNA quantity and purity were evaluated using a NanoDrop 2000 (NanoDrop Technologies). All RNA samples were labeled, linearly amplified by Low Input Quick Amp Labeling Kit and RNA Spike-In Kit then analyzed with Whole Mouse Genome Microarray Kit (Agilent). Signal intensities were quantitated with laser confocal scanner and analyzed with Feature Extraction software (Version 10.7.3.1, Agilent) and R statistical package (Version 2.15.1). Probe set data were median-normalized per chip. Empirical Bayesian method controlling for false discovery rate (FDR: <3% and logFC >1.0 [22]) for comparison of differentially expressed between TAX1BP1-KO mice and their WT. Principal Component Analysis (PCA) for the systematic trend examination, heatmaps by R Software and volcano plot analysis were applied to identify the single mRNA differentially expressed in TAX1BP1-KO mice (log2-fold expression change on the x-axis and t test p values on the y-axis, negative log). Each dot represents a single probe. The complete gene expression dataset can be viewed in the Gene Expression Omnibus (GEO) repository accession number GSE43932 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43932).
Quantitative real time-polymerase chain reaction (RT-PCR)
Taqman quantitative RT-PCR was performed to validate a subset of genes. Random hexamer-primed cDNA templates were synthesized from purified (RNAs ReverTra Ace®, TOYOBO). The output of RT-PCR reactions were quantitated with LightCycler® R 480 System (Roche). Primer sequences were listed in Table 1. Each reaction was run in triplicate with endogenous control GAPDH on the same reaction plate.
Table 1. Primer sequences.
| SAA3 | acagcctctctggcatcg | atgctcggggaactatgat | #26 |
| TAX1BP1 | ataaaaatgtgtaatagtcacgagcag | cactccaaagattgggttgg | #56 |
| EFCAB2 | tgtccgtcgtggctatgac | cctgcttcaccaccttcttg | #80 |
| GAPDH | tcgaccatgaatcgaataataca | tgcagctctccttcagtcg | #89 |
Multiple cytokine & chemokine quantitation
The 3-, 8-, 16- and 32-wk male TAX1BP1-KO and their WT littermates were anesthetized and an aliquot of serum (12.5 μl) from heart blood were collected (n = 5/groups). Quantitation of 23 cytokines and chemokines was performed by a multiplex ELISA system (Bio-Plex, BioRad) and analyzed by the Bio-Plex Manager Software 6.1 (Bio-Rad) with a five-parameter curve-fitting algorithm for standard curve calculations.
Immunohistochemistry
A standard avidin-biotin-peroxidase technique or hematoxylin and eosin (HE) staining were employed for Saa3 and I-κBα staining or morphological observation of heart, liver and skin tissues of 8- or 16-wk male TAX1BP1-KO and their WT littermates (n = 5/groups). Rabbit polyclonal anti-Saa3 antibody (ab59736, abcum), rabbit monoclonal anti-I-κBα antibody (ab32518, abcum) or control antibody for visualization of antigens with EnVision + System-HRP Labelled Polymer Anti-Rabbit (Dako). DAB + Liquid (Dako) for positive staining and Mayer's hematoxylin solution for counterstaing. Images were captured with BZ-9000 (KEYENCE). Mice whole eye sections were examined with anti-T6BP antibody (ab22049, abcam). Anti-IgG (H+L), rabbit, goat-poly, DyLight 649 (KPL) was used as secondary antibodies.
Electron microscopy
For transmission electron microscopy (TEM), mitral valve, atrioventricular node, sinoatrial node and papillary muscles of the left ventricle of 8-, 16-, 60-wk male TAX1BP1-KO and their WT littermates (n = 3/groups) were fixed with 2.5% glutaraldehyde/2% paraformaldehyde in a 0.1 M cacodylate buffer (pH7.4) for 3 hr or longer at 4°C. After a washing in the cacodylate buffer, specimens were postfixed in 2% osmium tetroxide in cacodylate buffer for 2 hr, washed with cacodylate buffer, dehydrated with ethanol and embedded in epoxy resin. Thin section specimens (80–90 nm) were then stained with uranyl acetate and lead cystate and examined with TEM H-7650 (at 80 kV, HITACHI).
Western blotting
Tissues from liver, heart, spleen, muscle, lung, skin, stomach and brain from WT BL6 were lysed with Co-IP buffer [23] and equal amounts of protein solutions (20 μg/lane) were separated by SDS-PAGE and transferred to immobilion membranes (Millipore) and incubated with primary antibodies, T6BP Antibody (sc-15274, Santa Cruz) or anti-Tubulin antibody (ab6160, abcum) and secondary antibodies, donkey anti-goat IgG-HRP (sc-2033, Santa Cruz) or ZyMAX™ Goat anti-Rat IgG(H+L) HRP conjugate (81–9520, invitrogen) and visualized with ECL Western Blotting Detection System (GE Healthcare Lifesciences) and high-performance chemiluminescence film.
Evaluation of physiological responses to LPS-stimulation
200 μg of Salmonella typhimurium lipopolysaccharide (LPS, Sigma) in 100 μl sterile pyrogen-free saline were injected into the footpads of TAX1BP1-KO or WT littermates (n = 4/groups). Tissue lysates were prepared from eyeball and the expression of Tax1bp1, I-κBα (anti-I-κBα rabbit mAb, #4812, Cell Signaling Technology) and Tubulin were evaluated by western blotting. Total RNAs were prepared from eyeballs of TAX1BP1-KO or WT littermates (n = 4/groups). Taqman quantitative RT-PCR was performed as described above (See Table 2).
Table 2.
| IL6 | gctaccaaactgga tataatcagga | ccaggtagctatgg tactccagaa | #6 |
| CXCL1 | agactccagccacacactccaa | tgacagcgcagctcattg | #83 |
| GAPDH | tcgaccatgaatcgaataataca | tgcagctctccttcagtcg | #89 |
Sera from peripheral blood samples were collected 0, 6 and 12 hr after LPS injection and quantitated with Bio-Plex Pro™ Mouse Cytokine 23-plex kit.
Enzyme-linked immunosorbant assay (ELISA)
The amounts of Saa3 and Cxcl13 Sera from 16-wk mice (n = 5/group) were measured with MOUSE SAA-3 ELISA KIT (Millipore) and Mouse CXCL13/BLC/BCA-1 Quantikine ELISA Kit (R&D Systems).
Telemetric electrocardiogram (ECG)
Sixteen week old male TAX1BP1-KO or WT littermates with or without antibiotic treatment (n = 5/group) were monitored with telemetric electrocardiogram. Telemetric transmitter was implanted into the back of mice under aseptic conditions and the muscle layers and the skin were closed with resorbable sutures. Data were acquired at least 72 hour after the implantation with a receiver placed under the cage and a full-disclosure 72 hour recordings were analyzed off-line and the P-Q intervals were evaluated.
Antibiotic treatment
TAX1BP1-KO or WT littermate male mice were first raised with the normal diets and water for 4 weeks, and then, antibiotic group (n = 5/groups) received ampicillin (1 g/L; Wako), vancomycin hydrochloride (500 mg/L; Wako), neomycin trisulfate salt hydrate (1 g/L; Sigma-Aldrich), and metronidazole (1 g/L; Wako) in drinking water for 12 weeks [24]. The non-antibiotic controls were equally raised and maintained except for antibiotics treatment. Both groups of mice were maintained in flexible film isolators under a strict 12-hour light cycle and fed an autoclaved chow diet and tap water ad libitum. Germ free status was verified regularly by ensuring negative cultures from mouse feces in three media types: nutrient agar (Nissui), pourmedia sheep blood agar M70 (Eiken), and Sabouraud agar (Nissui). Microbial colonies were counted after incubation at 37°C for 48 hour (aerobes) or 72 hour (anaerobes). Both groups of mice were anesthetized and sacrificed at the end of 16 weeks experimental period. Daily fluid consumption, body weight, liver function (ALT, AST), renal function (BUN), nutritional status (TG, GLU, TP) and spleen weight (After 10% formalin fixation) were examined. Caecum surface area was measured with Image J (NIH). In general, there were no particular adverse effects on mice through antibiotic treatment.
Statistical analysis
All numerical data are expressed as means ± SD. Statistical significance was assessed by Student's two-tailed t-test. In the case of ELISA, Statistical analyses were performed by one-way analysis of variance and Steel-Dwass test. Data were considered significant when P<0.05.
Results
LCM- and gene expression microarray array-based profiling of the mitral valves in TAX1BP1-KO mice and reevaluation by RT-PCR and immunostaining
We have previously observed that the mRNA expression level for several inflammatory cytokines, including IL-1β and TNFα, increases in the cardiac and skin tissues of TAX1BP1-KO mice; more importantly, these mice showed mitral valvulitis and premature death compared to their wild-type (WT) littermates. However, the underlying mechanisms involved in these processes remain unknown [8].
To date, information on variations in the levels of gene expression in regions of the heart (more specifically, the mitral valves) showing inflammation in TAX1BP1-KO mice is still lacking. This pathologic event is thought to be linked to premature death, which might be brought on by cardiac failure. In the current study, we employed LCM- and gene expression microarray-based techniques to obtain detailed information on the levels of gene expression in organs showing pathological changes. Total RNA was extracted from three independent tissue samples obtained from the mitral valves of 8- or 16- week-old (-wk) male (WT and TAX1BP1-KO) mice by using LCM, which was followed by total RNA extraction. Then, global mRNA expression profiles were analyzed by an Agilent gene expression microarray.
Principle component analysis, using two principle components, was conducted and the results were represented by a scatterplot (Fig. 1A). The data showed that the results for all samples from TAX1BP1-KO mice clearly deviated from those for control mice, indicating detectable differences in the gene transcription patterns of the two genetic backgrounds. A gene list was compiled on the basis of normalization and statistical analysis (P<0.03, logFC >1.0). Using these criteria, alterations in 588 gene expression profiles were identified. Unsupervised hierarchical clustering analysis (Cluster 3.0; Stanford University) of the 588 genes resulted in the separation of all TAX1BP1-KO from their paired WT controls. In total, 428 probes were upregulated and 160 were downregulated for a total of 24,000 genes (Fig. 1B). We then applied volcano plot analysis to identify the differences in mitral valve mRNA expression in TAX1BP1-KO mice and the controls. The plot showed a log2-fold change in mRNA expression between the two groups on the x-axis and the negative log of the t-test p-values on the y-axis. Each gene was represented by a single dot. Using the plot, we identified 588 probes that showed a more than 2-fold differential expression of mRNA when compared to the controls (p<0.03, Fig. 1C).
Figure 1. Elevated inflammatory profiles in the multiple organs of TAX1BP1-KO mice.

Mitral valve tissues from either 8 or 16(-wk) TAX1BP1-KO mice or their wild-type littermates were collected by Arcturus XT LCM system and total RNAs were prepared by RNeasy mini kit (Qiagen). Each cDNA pool was generated from the individual RNA sample and gene expression profiles were evaluated using Whole Mouse Genome Microarray Kit (Agilent). A) Principal component analysis (PCA) by conditions was performed on R statistical package (Version 2.15.1) and represented as a scatterplots of whole gene expression profiles of 8- or 16-wk TAX1BP1-KO mice (8wKO #1- #3 or 16wKO #1-#3, surrounded by red circles) and their WT littermates (8wWT #1- #3 or 16wWT #1-#3, blue circles). The PCA plot showed that samples clustered based on their genetic backgrounds. Data represent n = 12. Component % variance; PC1 = 34.95%, PC2 = 19.48%. B) Heat map representation of differentially expressed genes in the mitral valves from either 8- or 16-wk TAX1BP1-KO mice or their WT littermates. 588 genes were differentially expressed in TAX1BP1-KO vs. WT littermates (P<0.03). Each column represents the expression profile of either the TAX1BP1-KO mice or WT littermates. Red and green colors indicate high and low expression levels, respectively, relative to the mean (see color bar). C) Volcano plot analysis of microarray revealed that 588 probes were significantly expressed more than 2-fold vs control. Red and green areas indicate significant increasing and decreasing changes in gene expression (p<0.03).
Tables 3 and 4 list the gene symbols, gene descriptions, fold changes, and p-value for all genes upregulated by more than 20-fold or downregulated by more than 5-fold. Most of the upregulated genes were primarily involved in inflammation. The gene showing the highest level of induction, SAA3, (i.e., 1,180 fold induction) along with SAA1 (i.e., 61 fold, 10th induction) are well-known inflammatory markers in patients with autoimmune disease, chronic infection and cancer [25]. SAA3 is also hyperinduced at the site of injury [26], inflammation [27] in mice experimental models. Additionally, genes related to immune modulation, including pathogen recognition, inflammation, chemotaxis [28]–[30], or tissue adhesion, degeneration and rearrangement [31], [32] were induced in the mitral valves of TAX1BP1-KO mice. The characteristics of the downregulated genes also suggested the link between inflammation and tissue degeneration (Table S1); for example, such as WIF1, a Wnt signaling suppressor; UCMA, a gene associated with cartilage development [33]–[35]. EFCAB2 is a functional partner of the voltage-gated Ca2+ channel [36]. TSC22D3 (also known as GILZ: a Glucocorticoid Induced Leucine Zipper) is an IL-10-inducible immune suppressor [37].
Table 3. Gene symbol, gene description, fold change and p-value for all genes up-regulated by >20-fold in TAX1BP1-KO mice.
| SYMBOL | DESCRIPTION | Fold activation | adj.P. Val |
| SAA3 | Serum amyloid A 3 | 1179.5 | 0.006 |
| CHI3L1 | Chitinase 3-like 1 | 361.0 | 0.006 |
| HP | Haptoglobin | 187.2 | 0.007 |
| IL1B | Interleukin 1 beta | 121.9 | 0.007 |
| SPP1/OPN | Secreted phosphoprotein 1/Osteopontin | 100.7 | 0.006 |
| CCL2/MCP1 | Chemokine (C-C motif) ligand 2/Monocyte chemotactic protein-1 | 81.7 | 0.021 |
| CLEC7A/DECTIN1 | C-type lectin domain family 7, member a/Dectin-1 | 81.0 | 0.005 |
| SERPINA3G | Serine (or cysteine) peptidase inhibitor, clade A, member 3G | 73.0 | 0.006 |
| LCN2 | Lipocalin 2 | 65.3 | 0.024 |
| SAA1 | Serum amyloid A 1 | 61.1 | 0.024 |
| CXCL13/BLC | Chemokine (C-X-C motif) ligand 13/B lymphocyte chemo-attractant | 52.9 | 0.0001 |
| SLPI | Secretory leukocyte peptidase inhibitor | 39.8 | 0.009 |
| CLEC4D/DECTIN2 | C-type lectin domain family 4, member d | 39.6 | 0.006 |
| TIMP1 | Tissue inhibitor of metalloproteinase 1 | 37.4 | 0.024 |
| CCL17/TARC | Chemokine (C-C motif) ligand 17/Thymus and activation regulated chemokine | 35.2 | 0.020 |
| CCL7 | Chemokine (C-C motif) ligand 7 | 33.8 | 0.025 |
| LGALS3/GALECTIN3 | Lectin, galactose binding, soluble 3/Galectin-3 | 33.5 | 0.008 |
| SIRPB1A | Signal-regulatory protein beta 1A | 33.3 | 0.006 |
| CHL1 | Cell adhesion molecule with homology to L1CAM | 32.4 | 0.027 |
| CCL8 | Chemokine (C-C motif) ligand 8 | 31.4 | 0.006 |
| BCL2A1B | B-cell leukemia/lymphoma 2 related protein A1b | 27.0 | 0.006 |
| MEFV | Mediterranean fever | 26.7 | 0.006 |
| PLAC8 | Placenta-specific 8 | 21.7 | 0.008 |
| ZMYND15 | Zinc finger, MYND-type containing 15 | 20.6 | 0.007 |
| ITGAX | Integrin alpha X | 20.0 | 0.006 |
Statistical significance (p<0.03) was calculated using the Empirical Bayesian method controlling for false discovery rate (FDR) <3% and logFC >1.0 on R statistical package (Version 2.15.1). Fold change represents a comparison between mean normalized signal intensity for control (n = 6) versus TAX1BP1-KO mice (n = 6).
Table 4. Gene symbol, gene description, fold change and p-value for all genes down-regulated by >5 fold in TAX1BP1-KO mice.
| SYMBOL | DESCRIPTION | Fold suppression | adj.P. Val |
| TAX1BP1 | Tax1 (human T-cell leukemia virus type I) binding protein 1 | 56.7 | 0.0000001 |
| WIF1 | Wnt inhibitory factor 1 | 11.1 | 0.0205 |
| UCMA | Upper zone of growth plate and cartilage matrix associated | 9.1 | 0.0173 |
| EFCAB2 | EF-hand calcium binding domain 2 | 8.0 | 0.0001 |
| FAM107A/DRR1 | Family with sequence similarity 107, member A/down-regulated in renal cell carcinoma 1 | 7.6 | 0.0219 |
| TSC22D3 | TSC22 domain family, member 3 | 7.3 | 0.0197 |
| TAX1BP1 | Tax1 (human T-cell leukemia virus type I) binding protein 1 | 7.2 | 0.0004 |
| MAP3K6/ASK2 | Mitogen-activated protein kinase kinase kinase 6 | 7.1 | 0.0212 |
| 6030422H21RIK | RIKEN cDNA 6030422H21 gene | 6.8 | 0.0124 |
| TSC22D3 | TSC22 domain family, member 3 | 5.9 | 0.0240 |
| PENK | Preproenkephalin | 5.7 | 0.0119 |
| CNTFR | Ciliary neurotrophic factor receptor | 5.3 | 0.0104 |
| COL11A2 | Collagen, type XI, alpha 2 | 5.3 | 0.0069 |
| RXFP3 | Relaxin family peptide receptor 3 | 5.2 | 0.0197 |
| NRXN1 | Neurexin I | 5.1 | 0.0110 |
| CYTL1 | Cytokine-like 1 | 5.0 | 0.0099 |
Statistical significance (p<0.03) was calculated using the Empirical Bayesian method controlling for false discovery rate (FDR) <3% and logFC >1.0 on R statistical package (Version 2.15.1). Fold change represents a comparison between mean normalized signal intensity for control (n = 6) versus TAX1BP1-KO mice (n = 6).
We further confirmed the microarray results for SAA3 and EFCAB2 by using RT-PCR (Fig. 2AB and Figure S1) and for Saa3 (induction) or I-κBα (reduction) by using immunostaining for mitral valve samples from 16-wk TAX1BP1-KO mice (Fig. 2C to F). In addition to these microenvironmental changes, broad-spectrum inflammatory effects, such as lymphocyte accumulation, apoptotic Councilman body formation, and Kupffer cell hyper proliferation in the hepatocyte (Fig. 3A), and thickening of the inflamed skin (Fig. 3C), were observed in 16-wk TAX1BP1-KO mice. Multiplex ELISA quantitation of the sera for homozygous or heterozygous TAX1BP1-KO and their WT littermates showed age-dependent development of systemic inflammation (Table S1). The levels of Il-6 and Cxcl1 were elevated more than 10- fold in the homozygous TAX1BP1-KO mice.
Figure 2. Validation of genes and proteins identified their expression alteration in the mitral valves of TAX1BP1-KO mice.

RT-PCR validation of genes identified their expression alteration in the mitral valves of TAX1BP1-KO mice, A) SAA3 B) EFCAB2 respectively. Gray bar: TAX1BP1-KO, black bar: WT. Mitral valve specimens were prepared from 16-wk TAX1BP1-KO mice or their WT littermates and stained by anti-Saa3 antibody (C and D) or anti-I-κBα antibody respectively (E and F).
Figure 3. Inflammatory properties in the multiple organs of TAX1BP1-KO mice.

The morphologic and functional alterations of the environments of liver (A and B) and skin (C and D) were also examined with HE-staining. Red and white triangles indicate accumulated lymphocytes and Councilman bodies respectively.
Massive infiltration of inflammatory lymphocytes in the mitral valves of TAX1BP1-KO mice
To obtain more detailed images of critical sites of inflammation, tissues obtained from the mitral valves of TAX1BP1-KO mice and their WT littermates at varying time points were examined with electron microscopy (Fig. 4). Surprisingly, the mitral valves TAX1BP1-KO mice showed extensive infiltration of lymphocytes, macrophages and neutrophils and tissue degeneration at only 8 weeks of age (Fig. 4A and 4A'), whereas the mitral valves of the WT littermates exhibited healthy collagen layers (Fig. 4B and 4B'). Extensive disruption of collagen layers and edema were observed at 60 weeks of age for TAX1BP1-KO mice (Fig. 4C, 4D and 4C', 4D').
Figure 4. Massive infiltration of inflammatory cells causes severe tissue lesion in the mitral valves of TAX1BP1-KO mice.

Electron microscopy examinations on the mitral valves of 8-, 16- and 60-wk TAX1BP1-KO mice (A: 8wKO, and C: 60wKO) and their WT littermates (B: 8wWT and D: 60wWT). See Figure S1 for details. Each panel was duplicated with colorized areas in specific cell types and abbreviated descriptions (Fig. 4A' to 4D'). Abbreviations, CL: Collagen layer; EC: Endothelial cell; ED: Edema; FB: Fibroblast; FC: Fibrocyte; GD: Granule deposition; MΦ: Macrophage; NP: Neutrophil; PC: Plasma cell; TC: T cell.
Enhanced inflammatory responses in TAX1BP1-KO mice after the LPS-stimulation
In addition to the chronic inflammation, the acute-phase inflammatory response of TAX1BP1-KO mice was also examined. Salmonella typhimurium lipopolysaccharide (LPS) was injected into the footpads of TAX1BP1-KO mice and their WT littermates. Then, the mice were monitored, and the effects were recorded. We examined the kinetics of mRNA expression in those same eye tissues (Fig. 5A, B: tissue specific) and the translational products in the sera (Fig. 5C, D: systemic) of IL-6 and CXCL1 were monitored. Both data clearly indicate that a deficiency in TAX1BP1 causes significantly enhanced inflammation in responses to LPS in TAX1BP1-KO mice.
Figure 5. Enhanced expression of inflammatory genes after the LPS-stimulation to TAX1BP1-KO mice.

200 μg of Salmonella typhimurium lipopolysaccharide (LPS) in 100 μl sterile pyrogen-free saline were injected into a right footpads of TAX1BP1-KO or WT littermate mice. At the time 2, 6, 12, 24 and 48 hour post-injection (PT), each group of mouse were euthanized and tissues including serum, lymphocytes and eyes were collected. A) LPS-triggered induction of Tax1bp1 in eye tissue was monitored. Ten μg of cell lysates from WT BL6 mice at 2, 6, 12, 24 and 48 hour PT of LPS were probed with anti-Tax1bp1, -I-κBα and -Tubulin antibodies. B, C) Total RNAs of eye tissues from at 6, 12 and 48 hour PT of LPS to TAX1BP1-KO or WT littermates and their untreated controls were prepared and the expressions of IL-6 and CXCL1 were quantitated with RT-PCR. D, E) Sera from at 6, 12 and 48 hour PT of LPS to TAX1BP1-KO or WT littermates and their untreated controls were collected and the amount of Il-6 and Cxcl1 were quantitated with multiplex ELISA system (BioRad). Gray bar: TAX1BP1-KO, black bar: WT littermate.
Amelioration of the inflammatory symptoms and the cardiac conduction defect of TAX1BP1-KO mice by antibiotic treatment and simultaneous MyD88 deficiency
Microbial infections spontaneously cause severe endothelial inflammatory diseases such as rheumatic fever and Kawasaki disease [38]. At the subcellular level, modulation of the threshold of immune cell activation, differentiation, and immune cell activity in response to non-self or self antigens in TAX1BP1-KO mice (Fig. 1 and Tables 3 and 4) might evoke autoimmune profiles and heart dysfunction. To test this hypothesis, we examined the link between the commensal microbiota and mitral valvulitis and endocarditis in TAX1BP1-KO mice. When the mice were 4 weeks old, antibiotics were orally administered to all subjects over a 12-week period. The telemetric electrocardiogram profiles then sacrificed for the pathologic examination. Inflammatory hypertrophy (Fig. 6A) and extensive Saa3 staining (Fig. 6E) of the mitral valves in TAX1BP1-KO mice were abolished with antibiotic treatments (Fig. 6C and G); no changes were observed in their WT littermates (Fig. 6B, D, F and H). Extended PQ-intervals observed by telemetric electrocardiogram in TAX1BP1-KO mice (Fig. 6I, middle panel) were alleviated with the administration of antibiotics (Fig. 6I, bottom panel). The statistical significance of the differences in the PQ-intervals was tested (Fig. 6J). The antibiotic regimen also reduced the secretion of Saa3 and Cxcl13 in the sera of TAX1BP1-KO mice (Fig. 7A, B), and splenic hypertrophy of TAX1BP1-KO mice was almost nonexistent (Fig. 8A). Typical cecum thickening due to antibiotic treatment was also confirmed (Fig. 8B), and fecal microbes were completely disappeared under these conditions (data not shown). If the eradication of microbiota is the main reason for the amelioration of the symptoms in TAX1BP1-KO mice, we hypothesized that the disruption of the innate immune cascade could bring about similar results. We crossbred TAX1BP1-KO mice with MyD88-KO mice [20] and examined the morphological features or immunostaining profiles of marker proteins in the mitral valves of 16-week-old TAX1BP1-KO and MyD88/TAX1BP1-KO mice. MyD88/TAX1BP1-double knockout canceled hyperplasia (Fig. 9A, B), Saa3 induction (Fig. 9C, D) and I-κBα degradation (Fig. 9E, F). Comparisons of ELISA values for TAX1BP1-KO and MyD88/TAX1BP1-KO mice also indicated amelioration of the inflammatory response in MyD88/TAX1BP1-KO mice (Fig. 9G, H).
Figure 6. Amelioration of inflammatory valvulitis and conduction disturbance after the antibiotics treatment on TAX1BP1-KO mice.

TAX1BP1-KO or WT littermate mice (male) were first raised with the normal diets and water for 4 weeks, and then, antibiotic treatment group (C, D, G and H, n = 5/group) provided ampicillin (1 g/L; Wako), vancomycin Hydrochloride (500 mg/L; Wako), neomycin trisulfate salt hydrate (1 g/L; Sigma-Aldrich), and metronidazole (1 g/L; Wako) in drinking water for 12 weeks based on a protocal of the commensal depletion (Rakoff-Nahoum S., Cell 2004). The non-antibiotics controls (A, B, E and F, n = 5/group) were equally raised and maintained except for antibiotics treatment. Each group of mice were anesthetized and sacrificed at the end of 16 weeks experimental period and histochemical representatives of each group were displayed with HE-staining (A to D) or anti-Saa3 immuno-staining (IS, E to H). I). Heart rhythms of 16-week-old TAX1BP1-KO treated with antibiotics over 12 weeks (male, n = 5/group) were monitored with telemetric electrocardiogram (12-lead ECG). J) The average values of PQ-intervals were compared with those of untreated TAX1BP1-KO mice and their WT littermates.
Figure 7. Reduction of the Saa3 and Cxcl1 expression in the sera of TAX1BP1-KO mice after the antibiotics treatment.
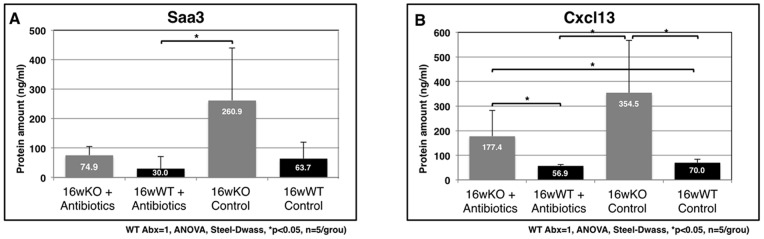
ELISA quantitation of Saa3 (A) or Cxcl13 (B) of the sera on four groups were performed. Gray bar: TAX1BP1-KO mice, black bar: WT littermates (n = 5/group).
Figure 8. Splenic hypertrophy of TAX1BP1-KO mice and its cancellation by antibiotics treatment.

Examinations on the spleen volume (A) and the area of cecum (B) were performed. The average values of spleen volumes (C) and cecum areas (D) were displayed (n = 5/group).
Figure 9. Cancelation of valvulitis in the MyD88/TAX1BP1 double-KO mice.

The HE-staining (A, B) and immunostaining of Saa3 (C, D) and I-κBα (E, F) were compared between TAX1BP1-KO and MyD88/TAX1BP1-KO mice. ELISA quantification of Saa3 (I) and Cxcl13 on the sera of both genetic background.
Discussion
Chronic infection with a retrovirus can have a significant impact on the host immune system. In the case of HTLV-1 infection, the pathological features of the disease are influenced by multiple factors. While HIV causes immune deficiency in the host, HTLV-1 causes a wide range of inflammatory symptoms (HAM and HU) and, in some cases, immunosuppressive ATL, a malignant growth of regulatory T-lymphocytes [39], [40]. Furthermore, HAD patients frequently display impaired immune response such as an ineffective interferon response in HAM patients [41] and frequent development of dermatitis in ATL patients [42].
Multiple inflammatory symptoms, including cardiac valvulitis, dermatitis, and a hypersensitive response to endotoxins and inflammatory cytokines, were noted in our preclinical model involving TAX1BP1-KO mice. More importantly, TAX1BP1-KO mice died prematurely because of unknown mechanisms [8]. In this study, we discovered the hyper-induction of multiple inflammation-related genes including SAA3, CHI3L1, HP, IL1B, SPP1/OPN, and the significant reduction of TSC22D3/GILZ in the mitral valves and microenvironment deterioration in a progressive age-dependent manner for TAX1BP1-KO mice [43]–[47], the significant reduction of EFCAB2 expression was highly implicated in functional defects of the heart [36].
HTLV-1-transgenic mice develop autoimmune symptom closely related to those observed for rheumatoid arthritis [48] or Sjögren's syndrome [49]. A rat model, infected with the HTLV-1 producing cell line, is known to develop HAM-like myelopathies in seronegative carrier rats [50]. A Tax1-transgenic mouse model, which specifically expresses Tax1 in T-lymphocytes, illustrates the development of aggressive ATL-like lymphoma with continuous invasion of lymphomatous cells into multiple organs such as the skin, liver and spleen [51], [52]. Subcutaneous inoculation of HTLV-1 transformed cells into NOG mice also results in ATL-like symptoms [53]. These transgenic/transplant models show symptoms similar to those found in human clinical cases. Furthermore, HTLV-1-driven inflammatory symptoms tend to occur in patients with HAD under normal host immune response conditions, while ATL-like symptoms develop under immunosuppressive conditions [54].
TAX1BP1-KO mice displayed invasive growth of lymphocytes into multiple organs (Fig. 3) and splenic hypertrophy (Fig. 8). We previously observed that transplantation of TAX1BP1-KO bone marrow to γ-irradiated normal mice resulted in the same inflammatory responses [8]. These results imply that TAX1BP1-KO model may be correlated with inflammatory HAD. The novelty of this system is identification of possible risk factors associated with vascular disease in HTLV-1 carriers [17], [18]. Preliminary electrocardiogram experiments using TAX1BP1-KO mice showed an abnormal prolongation of PQ intervals and/or atrioventricular conduction defects (Fig. 6I, J), which might cause fatal cardiac failure. Since the PQ interval and atrioventricular conduction highly depend on the functioning of voltage-dependent L-type Ca2+ channels, L-type Ca2+ channel function may deteriorate in the heart of TAX1BP1-KO mice. Of note, EFCAB2, a functional partner in the voltage-gated Ca2+ channel, was significantly downregulated in the cardiac tissue of TAX1BP1-KO mice (Table 4). Further studies are required to elucidate these defects caused in TAX1BP1-KO mice.
Intensive antibiotic treatment [24] for TAX1BP1-KO mice significantly ameliorated inflammatory symptoms (Fig. 6). TAX1BP1-KO mice crossbred with MyD88-KO mice showed similar results. Since the intrinsic role of Tax1bp1 is to inhibit unnecessarily activated innate immunity responses [8], a functional deficiency of Tax1bp1 through HTLV-1 infection can lead to similar symptoms in humans; that is, commensal microbiota can cause pseudo-Infective endocarditis symptoms [55]. The extent of the deficiency, however, is much more moderate than that of typical infective endocarditis (IE) [56].
A large population-based epidemiological study revealed that the prevalence of heart valve disease in the entire population of the United States is 2.5% [53]. IE is thought to result from the following sequence of events: (1) the formation of nonbacterial thrombotic endocarditis on the surface of a cardiac valve or elsewhere that endothelial damage occurs; (2) bacteremia; and (3) the adherence of the bacteria in the bloodstream to nonbacterial thrombotic endocarditis and proliferation of bacteria within a vegetation [57]. Viridans group streptococci are a part of normal skin, oral, respiratory, and gastrointestinal tract flora, and are responsible for ≥50% of community-acquired native valve IE cases [58]. Another review reported that 20% of IE cases originated from culture-negative or Enterococci [59]. Each of these epidemiological surveys clearly indicates the importance of prevention and control measures with regard to microbial infection and vegetation. However, it is still not known why IE is developed in limited population and it is not clear whether there are any differences in the frequencies of allelic polymorphisms in the immune response genes for IE patients?
In summary, HTLV-1 induces diverse forms of inflammatory disorders [60], [61], which may originate from the functional dysregulation of Tax1bp1. Single-nucleotide polymorphisms (SNPs) in A20 or RNF11, catalytic partners of Tax1bp1, has have linked to many inflammatory diseases [19], [62], [63]. However, in the case of TAX1BP1 SNPs, only one study has linked them to the head and neck cancer [64]. The genetic variations in TAX1BP1 and its partners would provide novel insights on the pathogenic machinery of HADs.
Supporting Information
Validation of genes identified their expression alteration in the mitral valves of TAX1BP1-KO mice. RT-PCR validation of genes identified their expression alteration in the mitral valves of TAX1BP1-KO mice, A) CCL2 B) CHI3L1 respectively. Gray bar: TAX1BP1-KO, black bar: WT. Mitral valve specimens were prepared as described in Fig. 2A. Primers and probes were as indicated.
(PDF)
Age-dependent induction of pro-inflammatory proteins in the sera of TAX1BP1 -KO mice. Sera from four different weeks of age (3, 8, 16 and 32) of TAX1BP1 homozygous knockout (Homo-KO), heterozygous knockout (Hetero-KO) or their WT littermates were collected and examined with multiplex ELISA quantitation kit (Bio-Plex Pro™ Mouse Cytokine 23-plex Assay, BioRad). Each value is an average of four different samples.
(PDF)
Acknowledgments
This paper is dedicated for the memories of the late Dr. Kuan-Teh Jeang who passed away on January 27th, 2013.
Funding Statement
This study is supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology; Okinawa Science and Technology Promotion Center (OSTPC); Miyazaki Prefectural Industrial Support Foundation. E.I. is a research fellow of the OSTPC and was a recipient of the Hita Tenryosui Research Scholarship from Hita Tenryosui Co. Ltd. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed]
- 2. McCool KW, Miyamoto S (2012) DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol Rev 246: 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayden MS, Ghosh S (2012) NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26: 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DiDonato JA, Mercurio F, Karin M (2012) NF-kappaB and the link between inflammation and cancer. Immunol Rev 246: 379–400. [DOI] [PubMed] [Google Scholar]
- 5. Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, et al. (2010) Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol 80: 2009–2020. [DOI] [PubMed] [Google Scholar]
- 6. Shembade N, Harhaj EW (2012) Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol 9: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Valck D, Jin DY, Heyninck K, Van de Craen M, Contreras R, et al. (1999) The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 18: 4182–4190. [DOI] [PubMed] [Google Scholar]
- 8. Iha H, Peloponese JM, Verstrepen L, Zapart G, Ikeda F, et al. (2008) Inflammatory cardiac valvulitis in TAX1BP1-deficient mice through selective NF-kappaB activation. EMBO J 27: 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shembade N, Harhaj NS, Liebl DJ, Harhaj EW (2007) Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J 26: 3910–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ling L, Goeddel DV (2000) T6BP, a TRAF6-interacting protein involved in IL-1 signaling. Proc Natl Acad Sci U S A 97: 9567–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peloponese JM, Yeung ML, Jeang KT (2006) Modulation of nuclear factor-kappaB by human T cell leukemia virus type 1 Tax protein: implications for oncogenesis and inflammation. Immunol Res 34: 1–12. [PubMed] [Google Scholar]
- 12. Giam CZ, Jeang KT (2007) HTLV-1 Tax and adult T-cell leukemia. Front Biosci 12: 1496–1507. [DOI] [PubMed] [Google Scholar]
- 13. Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, et al. (2008) The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol 9: 254–262. [DOI] [PubMed] [Google Scholar]
- 14. Verstrepen L, Verhelst K, Carpentier I, Beyaert R (2011) TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem Sci 36: 347–354. [DOI] [PubMed] [Google Scholar]
- 15. Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, et al. (1998) Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93: 1231–1240. [DOI] [PubMed] [Google Scholar]
- 16. Hida A, Imaizumi M, Sera N, Akahoshi M, Soda M, et al. (2010) Association of human T lymphotropic virus type I with Sjogren syndrome. Ann Rheum Dis 69: 2056–2057. [DOI] [PubMed] [Google Scholar]
- 17. Kira J, Hamada T, Kawano Y, Okayama M, Yamasaki K (1997) An association of human T-cell lymphotropic virus type I infection with vascular dementia. Acta Neurol Scand 96: 305–309. [DOI] [PubMed] [Google Scholar]
- 18. Hayashi J, Furusyo N, Sawayama Y, Murata M (2008) [Chronic infection is one of the etiologies for digestive diseases and atherosclerosis]. Fukuoka Igaku Zasshi 99: 67–73. [PubMed] [Google Scholar]
- 19. Vereecke L, Beyaert R, van Loo G (2009) The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30: 383–391. [DOI] [PubMed] [Google Scholar]
- 20. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11: 115–122. [DOI] [PubMed] [Google Scholar]
- 21. Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, et al. (2007) Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum 56: 1618–1628. [DOI] [PubMed] [Google Scholar]
- 22. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279–284. [DOI] [PubMed] [Google Scholar]
- 23. Iha H, Kibler KV, Yedavalli VR, Peloponese JM, Haller K, et al. (2003) Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: implications for stimulus-specific interruption of oncogenic signaling. Oncogene 22: 8912–8923. [DOI] [PubMed] [Google Scholar]
- 24. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241. [DOI] [PubMed] [Google Scholar]
- 25. Obici L, Merlini G (2012) Amyloidosis in autoinflammatory syndromes. Autoimmun Rev 12: 14–17. [DOI] [PubMed] [Google Scholar]
- 26. Jang SY, Shin YK, Lee HY, Park JY, Suh DJ, et al. (2012) Local production of serum amyloid a is implicated in the induction of macrophage chemoattractants in Schwann cells during wallerian degeneration of peripheral nerves. Glia 60: 1619–1628. [DOI] [PubMed] [Google Scholar]
- 27. Poynter ME (2012) Airway epithelial regulation of allergic sensitization in asthma. Pulm Pharmacol Ther 25: 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geijtenbeek TB, Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9: 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H (2011) Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7: 33–42. [DOI] [PubMed] [Google Scholar]
- 30. Goldszmid RS, Trinchieri G (2012) The price of immunity. Nat Immunol 13: 932–938. [DOI] [PubMed] [Google Scholar]
- 31. Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, et al. (2011) Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 73: 479–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das R, Philip S, Mahabeleshwar GH, Bulbule A, Kundu GC (2005) Osteopontin: it's role in regulation of cell motility and nuclear factor kappa B-mediated urokinase type plasminogen activator expression. IUBMB Life 57: 441–447. [DOI] [PubMed] [Google Scholar]
- 33. Gudjonsson JE, Johnston A, Stoll SW, Riblett MB, Xing X, et al. (2010) Evidence for altered Wnt signaling in psoriatic skin. J Invest Dermatol 130: 1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chien AJ, Conrad WH, Moon RT (2009) A Wnt survival guide: from flies to human disease. J Invest Dermatol 129: 1614–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Surmann-Schmitt C, Dietz U, Kireva T, Adam N, Park J, et al. (2008) Ucma, a novel secreted cartilage-specific protein with implications in osteogenesis. J Biol Chem 283: 7082–7093. [DOI] [PubMed] [Google Scholar]
- 36. Heineke J, Auger-Messier M, Correll RN, Xu J, Benard MJ, et al. (2010) CIB1 is a regulator of pathological cardiac hypertrophy. Nat Med 16: 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, et al. (2003) Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 101: 729–738. [DOI] [PubMed] [Google Scholar]
- 38. Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, et al. (2008) Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. J Am Dent Assoc 139 Suppl: 3S–24S [DOI] [PubMed] [Google Scholar]
- 39. Sandler NG, Douek DC (2012) Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol 10: 655–666. [DOI] [PubMed] [Google Scholar]
- 40. Tattermusch S, Bangham CR (2012) HTLV-1 infection: what determines the risk of inflammatory disease? Trends Microbiol 20: 494–500. [DOI] [PubMed] [Google Scholar]
- 41. Martin F, Taylor GP (2011) Prospects for the management of human T-cell lymphotropic virus type 1-associated myelopathy. AIDS Rev 13: 161–170. [PubMed] [Google Scholar]
- 42. Yagi H, Takigawa M, Hashizume H (2003) Cutaneous type of adult T cell leukemia/lymphoma: a new entity among cutaneous lymphomas. J Dermatol 30: 641–643. [DOI] [PubMed] [Google Scholar]
- 43. Uhlar CM, Whitehead AS (1999) Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265: 501–523. [DOI] [PubMed] [Google Scholar]
- 44. Elias JA, Homer RJ, Hamid Q, Lee CG (2005) Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol 116: 497–500. [DOI] [PubMed] [Google Scholar]
- 45. Quaye IK (2008) Haptoglobin, inflammation and disease. Trans R Soc Trop Med Hyg 102: 735–742. [DOI] [PubMed] [Google Scholar]
- 46. Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: 519–550. [DOI] [PubMed] [Google Scholar]
- 47. Uede T (2011) Osteopontin, intrinsic tissue regulator of intractable inflammatory diseases. Pathol Int 61: 265–280. [DOI] [PubMed] [Google Scholar]
- 48. Iwakura Y, Tosu M, Yoshida E, Takiguchi M, Sato K, et al. (1991) Induction of inflammatory arthropathy resembling rheumatoid arthritis in mice transgenic for HTLV-I. Science 253: 1026–1028. [DOI] [PubMed] [Google Scholar]
- 49. Green JE, Hinrichs SH, Vogel J, Jay G (1989) Exocrinopathy resembling Sjogren's syndrome in HTLV-1 tax transgenic mice. Nature 341: 72–74. [DOI] [PubMed] [Google Scholar]
- 50. Ishiguro N, Abe M, Seto K, Sakurai H, Ikeda H, et al. (1992) A rat model of human T lymphocyte virus type I (HTLV-I) infection. 1. Humoral antibody response, provirus integration, and HTLV-I-associated myelopathy/tropical spastic paraparesis-like myelopathy in seronegative HTLV-I carrier rats. J Exp Med 176: 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hasegawa H, Sawa H, Lewis MJ, Orba Y, Sheehy N, et al. (2006) Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med. 12: 466–472. [DOI] [PubMed] [Google Scholar]
- 52. Ohsugi T, Kumasaka T, Okada S, Urano T (2007) The Tax protein of HTLV-1 promotes oncogenesis in not only immature T cells but also mature T cells. Nat Med 13: 527–528. [DOI] [PubMed] [Google Scholar]
- 53. Dewan MZ, Terashima K, Taruishi M, Hasegawa H, Ito M, et al. (2003) Rapid tumor formation of human T-cell leukemia virus type 1-infected cell lines in novel NOD-SCID/gammac(null) mice: suppression by an inhibitor against NF-kappaB. J Virol 77: 5286–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chervonsky AV (2010) Influence of microbial environment on autoimmunity. Nat Immunol 11: 28–35. [DOI] [PubMed] [Google Scholar]
- 55. Gould FK, Denning DW, Elliott TS, Foweraker J, Perry JD, et al. (2012) Guidelines for the diagnosis and antibiotic treatment of endocarditis in adults: a report of the Working Party of the British Society for Antimicrobial Chemotherapy. J Antimicrob Chemother 67: 269–289. [DOI] [PubMed] [Google Scholar]
- 56. Seckeler MD, Hoke TR (2011) The worldwide epidemiology of acute rheumatic fever and rheumatic heart disease. Clin Epidemiol 3: 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chu VH, Woods CW, Miro JM, Hoen B, Cabell CH, et al. (2008) Emergence of coagulase-negative staphylococci as a cause of native valve endocarditis. Clin Infect Dis 46: 232–242. [DOI] [PubMed] [Google Scholar]
- 58. Moreillon P, Que YA (2004) Infective endocarditis. Lancet 363: 139–149. [DOI] [PubMed] [Google Scholar]
- 59. Oliere S, Douville R, Sze A, Belgnaoui SM, Hiscott J (2011) Modulation of innate immune responses during human T-cell leukemia virus (HTLV-1) pathogenesis. Cytokine Growth Factor Rev 22: 197–210. [DOI] [PubMed] [Google Scholar]
- 60. Roufosse FE, Goldman M, Cogan E (2007) Hypereosinophilic syndromes. Orphanet J Rare Dis 2: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, et al. (2008) Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet 40: 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, et al. (2011) A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 43: 908–912. [DOI] [PubMed] [Google Scholar]
- 63. Sartelet A, Druet T, Michaux C, Fasquelle C, Geron S, et al. (2012) A splice site variant in the bovine RNF11 gene compromises growth and regulation of the inflammatory response. PLoS Genet 8: e1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruiz MT, Balachi JF, Fernandes RA, Galbiatti AL, Maniglia JV, et al. (2010) Analysis of the TAX1BP1 gene in head and neck cancer patients. Braz J Otorhinolaryngol 76: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of genes identified their expression alteration in the mitral valves of TAX1BP1-KO mice. RT-PCR validation of genes identified their expression alteration in the mitral valves of TAX1BP1-KO mice, A) CCL2 B) CHI3L1 respectively. Gray bar: TAX1BP1-KO, black bar: WT. Mitral valve specimens were prepared as described in Fig. 2A. Primers and probes were as indicated.
(PDF)
Age-dependent induction of pro-inflammatory proteins in the sera of TAX1BP1 -KO mice. Sera from four different weeks of age (3, 8, 16 and 32) of TAX1BP1 homozygous knockout (Homo-KO), heterozygous knockout (Hetero-KO) or their WT littermates were collected and examined with multiplex ELISA quantitation kit (Bio-Plex Pro™ Mouse Cytokine 23-plex Assay, BioRad). Each value is an average of four different samples.
(PDF)