

Abstract
Message RNA (mRNA) carries a large number of local secondary structures, with structural stability to participate in the regulations of gene expression. A worthy question is how the local structural stability is maintained under the constraint that multiple selective pressures are imposed on mRNA local regions. Here, we performed the first genome-wide study of natural selection operating on high structural stability regions (HSRs) of mRNAs in Escherichia coli. We found that HSR tends to adjust the folded conformation to reduce the harm of mutations, showing a high level of mutational robustness. Moreover, guanine preference in HSR was observed, supporting the hypothesis that the selective constraint for high structural stability may partly account for the high percentage of G content in Escherichia coli genome. Notably, we found a substantially reduced synonymous substitution rate in HSRs compared with that in their adjacent regions. Surprisingly and interestingly, the non-key sites in HSRs, which have slight effect on structural stability, have synonymous substitution rate equivalent to background regions. To explain this result, we identified compensatory mutations in HSRs based on structural stability, and found that a considerable number of synonymous mutations occur to restore the structural stability decreased heavily by the mutations on key sites. Overall, these results suggest a significant role of local structural stability as a selective force operating on mRNA, which furthers our understanding of the constraints imposed on protein-coding RNAs.
Introduction
RNA molecules tend to adopt a folded conformation through the formation of Watson-Crick base pairing between complementary nucleotides. The resulting so-called RNA secondary structure emerges to be a key player in the regulations of gene expression [1]–[4]. By surveying secondary structures in various genomes, previous studies have revealed that a large number of genomes are being transcribed to produce non-coding RNAs that generally contain a conserved secondary structure [5]–[9]. The structural conformation of the molecule is often necessary for its functions. Precursor microRNAs (pre-miRNAs) are among the largest examples that illustrate the functions of secondary structure in non-coding RNAs. The pre-miRNA contains a ∼70-bp hairpin, which is recognized by the Dicer protein and then the loop region is removed to leave a dsRNA [10], [11]. The secondary structure in pre-miRNA is conserved during evolution [12]–[14], suggesting an important role of structural conformation in miRNA maturation. Interestingly, in recent years, a considerable number of protein-coding RNAs have been reported to contain local secondary structures [15]–[18]. Some translational processes, including translation initiation [19]–[21], co-translational folding of protein [22], [23], are sensitive to the variation of local structural stability. Moreover, a strong association between structural stability and protein abundance was observed in yeast [24]. These results suggest an important role of mRNA structural stability, which might be different from the roles of conserved secondary structures reported by previous studies. Besides its functions, numerous studies have focused on the evolution of RNA secondary structure [14], [25]–[29] and revealed several mechanisms to maintain the secondary structure, including lower substitution rate [30] and compensatory mutations [27].
Mutations that occur in the primary sequence might lead to a disruption of the paired regions, thus changing the structural conformation or structural stability of the molecule and impairing its original function. Various studies focused on the selective constraints in folded RNAs that are mostly located in non-coding regions [14], [30], [31]. They found a lower substitution rate in paired regions in comparison with that in unpaired regions [30]. Moreover, previous studies aimed at attributing the variation of GC content to the selection for high structural stability of RNA [32], [33]. The association has been observed in several types of non-coding RNA, such as miRNA. In miRNA, GC content is positively correlated with the organism's physiological temperature [33], suggesting a possible association between the base-pairing strength of miRNA-targets and the temperature of an organism. Unlike non-coding RNAs, multiple selective constraints, including structural stability [34], [35] and translation efficiency [36], [37], operate on mRNAs. Both two constraints influence the pattern of synonymous mutation. If there is selection on local protein-coding region for high structural stability, codons in such region might be under conflicting selective pressures: the codons promoting RNA folding with high structural stability might be translationally non-optimal. In this case, the locations of synonymous substitutions might be non-random with respect to the translational efficiency and structural stability. Knowledge of this conflict can further our understanding of constraints imposed on protein-coding RNAs. The initial studies of secondary structure, in mammals as well as yeast [38], [39], considered the thermodynamic stability of mRNA mediated by the changes in secondary structure, and revealed that C preference at the four-fold degenerate sites might be partly driven by the selection for RNA stability [38]. However, these studies did not focus on local protein-coding regions that form local secondary structures, exhibiting high structural stability. Numerous lines of evidence indicate that mRNA folding windows are small [40], [41]. Therefore, it is reasonable to investigate the substitution patterns of structural regions based on local folding instead of global folding. Moreover, the analysis of selective constraints on local structural stability of protein-coding region has not been performed in the genome wide scale. Therefore, the aim of this study is to analyze the natural selection related to the local structural stability from a genome wide perspective.
In recent years, there has been a sharp growth in evidence showing widespread secondary structures in protein-coding region. In our previous study, we found that most of these structures exhibit high structural stability, while their structural conformations are non-conserved across different species [16]. We therefore identified the regions with high structural stability in Escherichia coli (HSR, high structural stability regions) using a loose threshold (Figure 1, Table S1-S2) [16], and revealed that number variation of HSR is correlated with gene functions, probably involving the regulations on the rhythm of translation elongation. However, the evolutionary pattern of HSR is still undetermined in that study. In particular, it remains unclear that how the structural stability of HSR is maintained under the constraint that multiple selective pressures are imposed on mRNA local regions. The selective pressure on HSR might be relaxed compared with that on structural RNA (e.g. 5 s rRNA), because there is a higher probability that a second mutation restores the structural stability disrupted by the first mutation. It is also of interesting to investigate the difference in the patterns of compensatory mutations between HSR and structural RNA. Therefore, in current study, we focus on the natural selections on HSRs, and aim at addressing the following questions that might advance our knowledge of selective pressures on mRNA: 1) Does selection for structural stability of HSR favor synonymous codons with high G/C? 2) How does HSR influence the local substitution rate of mRNA? 3) Since it is the structural stability rather than the structural conformation that is conserved, is the pattern of compensatory mutations in HSR different from that in structural RNA?
Figure 1. An example of HSRs in Escherichia coli and Escherichia fergusonii.

A) Z-score is the normalized MFE. The threshold used to define HSR is marked by blue dashes. B) shows the secondary structures of HSRs. Although the HSRs between the two species are conserved (see Materials and Methods for details), the secondary structures are non-conserved.
Materials and Methods
Coding sequence (CDS) and orthologs
Protein coding sequences of Escherichia coli K12 MG1655, Escherichia fergusonii ATCC and Salmonella enterica subsp. enterica serovar Typhi CT18 were downloaded from the National Center for Biotechnology Information FTP server (ftp://ftp.ncbi.nih.gov/genomes/). Sequences with length <200 nucleotides (nt) were excluded. In total, we obtained 4152, 4126 and 4246 coding sequences in Escherichia coli, Escherichia fergusonii and Salmonella enterica, respectively.
Definition of HSR
HSR exhibits high structural stability, while not all HSRs have a conserved secondary structure. We thus identified HSRs on mRNAs only based on the minimum folding free energy (MFE) of local regions. The method was described previously [16]. The main steps are as follows. First, we calculated the normalized MFE, z-score, along CDS. For each CDS, we shuffled synonymous codons among sites with identical amino acids, controlling for amino-acid sequence, codon usage bias, and GC content. This process was repeated 100 times to generate 100 random sequences. We calculated MFE along CDS and the corresponding random sequences using a sliding window with 50 nt (approximately equal to the length of region (40 nt) covered by ribosome during elongation) in length and a step of 10 nt. MFE in each sliding window was calculated by RNAfold [42]. Z-score was calculated by equation (1):
| (1) |
where 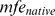 is the MFE of native sequence,
is the MFE of native sequence, 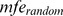 and
and  are the mean and standard deviation of MFE of 100 random sequences, respectively. Second, we used the following criteria to define HSR: 1) if a region contains more than two continuous sliding windows, in which the z-scores were all below the threshold of -0.65 [16], the region was defined as HSR. 2) If the percentage of the overlapping sites of two adjacent HSRs was higher than 50%, the two HSRs were combined.
are the mean and standard deviation of MFE of 100 random sequences, respectively. Second, we used the following criteria to define HSR: 1) if a region contains more than two continuous sliding windows, in which the z-scores were all below the threshold of -0.65 [16], the region was defined as HSR. 2) If the percentage of the overlapping sites of two adjacent HSRs was higher than 50%, the two HSRs were combined.
Mutational robustness of HSR
Mutational robustness of HSR refers to the sensitivity of structural stability to point mutations. We used two measures to estimate the mutational robustness of HSR. The first measure is the mean relative change of MFE over all single point mutations [35]. The second is the number of key sites. Key site indicates those sites, mutations on which result in more than 15% (other thresholds were also considered) increase in MFE. The two measures were obtained by performing the following analyses. First, for each site in HSR, three mutational sequences were generated by replacing original nucleotide with the other three nucleotides. Second, the MFE of the four sequences (one native and three mutational sequences) was calculated. The relative change of MFE was computed by equation (2):
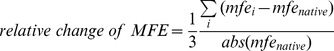 |
(2) |
where 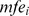 is the MFE of the ith mutational sequence.
is the MFE of the ith mutational sequence. 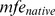 is the MFE of the native sequence.
is the MFE of the native sequence. 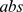 refers to the absolute value. Third, the relative changes at all sites were averaged and the number of key sites was computed.
refers to the absolute value. Third, the relative changes at all sites were averaged and the number of key sites was computed.
As a control, for each HSR, we generated 30 random HSRs (rHSRs) by shuffling all synonymous codons with identical amino acid, maintaining amino acid sequence and codon usage. Moreover, the MFE of rHSR is similar to that of the corresponding HSR (located in MFEHSR±10% MFEHSR). The relative change of MFE and the number of key sites in random sequence are the average values of 30 random sequences.
Calculation of local translation efficiency
We used tRNA adaptation index (tAI) [43] to measure the local translation efficiency. tAI was calculated by equation (3):
| (3) |
where n is the length (codons) of HSR, wi is the relative adaptiveness value of codon i, which was calculated according to the work of dos Reis et al. [43].
Conserved HSRs and flank regions
First, we obtained the orthologous relationship between Escherichia coli and Escherichia fergusonii from the KEGG database [44]. Only one-to-one orthologs were used in the analyses. In total, 3138 orthologs were extracted. Amino acid sequences of orthologs were aligned using MUSCLE [45]. The alignments were subsequently converted into mRNA sequence alignments. Considering that insertions and deletions (indels) strongly affect the positions of HSRs, we discarded the alignments with total indels >10 nt. 2676 alignments were left. Second, we defined the conserved HSRs between the two species. For each HSR in Escherichia coli (HSR-eco), we searched for the homologous HSR near the corresponding region of orthologs in Escherichia fergusonii (HSR-efe). If HSR-efe was found and the percentage of overlapping sites between HSR-eco and HSR-efe was higher than 50%, the overlapping regions of the two HSRs were defined as conserved HSRs (Figure 2A). The other HSRs existing in only one species were defined as specific HSRs (Figure 2B). Two background regions: FAR and BAR (FAR: forward adjacent region, BAR: backward adjacent region) were extracted and used as controls. The three regions have the same length. Considering that the substitution rate and GC content in the first 200 nt are significantly different from that in other regions (Figure S1), we discarded the data set FAR-HSR-BAR if FAR is located in the first 200 nt of CDS.
Figure 2. Definitions of conserved and specific HSRs.
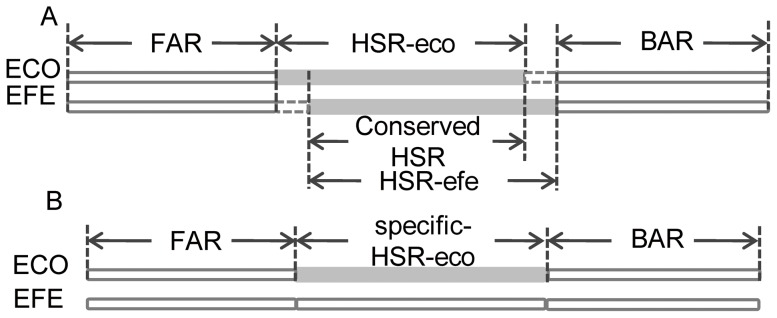
Conserved HSR is the overlapping region of the homologous HSRs between Escherichia coli and Escherichia fergusonii (HSR-eco and HSR-efe). ECO: Escherichia coli; EFE: Escherichia fergusonii; FAR (forward adjacent region) and BAR (backward adjacent region) refer to the two background regions, which have the same length to the corresponding HSR.
Estimation of substitution rate
For each orthologs, all sub-alignments covered by conserved HSRs were concatenated. The concatenated alignments with length <100 nt were excluded. In total, 1217 alignments were remained. The concatenated alignments of FAR and BAR were obtained using the same method. The estimation for synonymous substitution rate (dS) was performed using the CODEML program of the PAML package [46] with runmode -2. Alignments with dS >3 were excluded.
Identification of compensatory mutations
We only considered single point mutations to investigate the pattern of compensatory mutations occurring in HSRs. First, we reconstructed the ancestral sequences of Escherichia coli and Escherichia fergusonii using the maximum likelihood method [47]. Alignments with idels >10 nt were not included. The indels in ancestral sequences were inferred by parsimony method using Salmonella enterica as outgroup. Again, HSRs in ancestral sequences and the conserved HSRs between ancestral and extant sequences, termed ancestral-extant HSRs, were defined using the same method as described above. Ancestral HSRs containing indels were discarded. In total, 11583 pairs of ancestral-extant HSRs were obtained.
Second, we defined the first mutations in ancestral-extant HSRs. For this purpose, we generated the pre-first-mutated HSR by introducing one mutation to ancestral HSR based on the synonymous substitutions in ancestral-extant HSRs (Figure 3A-3B). The relative change of MFE between pre-first-mutated and ancestral HSRs was calculated by equation (4):
| (4) |
where 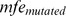 is the MFE of mutated HSR.
is the MFE of mutated HSR. 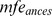 refers to the MFE of ancestral HSR.
refers to the MFE of ancestral HSR. 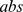 refers to the absolute value. Mutations with relative change >10% were defined as the first mutations, and the corresponding mutated HSRs were termed first-mutated HSRs.
refers to the absolute value. Mutations with relative change >10% were defined as the first mutations, and the corresponding mutated HSRs were termed first-mutated HSRs.
Figure 3. Overview of the method to identify the compensatory mutations.

A-B: we generated the pre-first-mutated HSR by introducing one mutation to ancestral HSR based on the synonymous mutations in ancestral-extant HSRs. The mutated site in pre-first-mutated HSR is marked in red. First mutations were defined as the mutations that increase the MFE of ancestral-HSR, the corresponding pre-first-mutated was termed first-mutated HSR. C-D: second-mutated HSR was generated by introducing one mutation to the first-mutated HSR. First mutation is marked in blue. The mutated site in second-mutated HSR is marked in red.
Third, we identified the second mutations occurring in the first-mutated HSRs using the same method (Figure 3C-3D). The second mutation refers to the mutation that decreases the MFE of the first-mutated HSR (i.e. the relative change of MFE between the first-mutated and second-mutated HSR <0, Figure 3). Moreover, the second mutations, which decrease MFE of ancestral HSR without the occurrence of the first mutation (in this case, the second-mutated HSR can be treated as the pre-first mutated HSR), were excluded. The left second mutations were defined as the compensatory mutations corresponding to the first mutation (Figure 3).
As a control, for each pair of ancestral-extant HSRs, we simulated random extant HSR by randomly generating the synonymous mutations on ancestral HSR based on the number of synonymous substitutions in ancestral-extant HSRs and the genomic frequency of codons in extant species. This process was repeated 30 times to generate 30 pairs of ancestral-random HSRs. The compensatory mutations in ancestral-random HSRs were defined using the same method as described above.
Results
High mutational robustness of HSR
Mutational robustness is the ability of genotypes to display high tolerance against mutations, which is considered a fundamental feature of biological systems, from single molecules to gene regulatory networks [48], [49]. To maintain its functions, HSR is assumed to evolve to keep a high level of mutational robustness. Table 1 summaries the two measures to estimate the mutational robustness. In total, we identified 6352 conserved HSRs between Escherichia coli and Escherichia fergusonii (Table 1, Table S3), which are located in 2256 genes (84.3% of all orthologs). In addition, 4202 and 4306 specific HSRs were detected in Escherichia coli and Escherichia fergusonii, respectively. We found that both conserved and specific HSRs have lower mean relative changes of MFE over all point mutations on HSRs (paired t-test, all p-values <10−16, Table 1), compared with the corresponding random HSRs with the same features. Considering that the difference in MFE between the native and random HSR (the MFE of random HSR is similar, not equal, to that of the native HSR) might affect the relative change of MFE, we also calculated the mean absolute change. The difference remains significant (paired t-test, all p-values <10−16, Table 1). The results suggest that HSRs have high tolerance to point mutations. Moreover, we computed the number of key sites, which strongly decrease the structural stability of HSR. Again, we found a lower number of key sites in both conserved and specific HSRs in comparison with that in random HSRs (paired t-test, all p-values <10−16, Table 1, Figure S2), indicating that there is a tendency to adjust the folded conformation of HSR to reduce the number of the sites, which have significant effect on structural stability. Overall, these results indicate that HSRs evolve to maintain high mutational robustness.
Table 1. Mutational robustness of HSRs.
| HSR classification | Number of HSRs | Absolute change of MFE (95% CI b) | Relative change of MFE (95% CI) | Number of key sites (95% CI) |
| Conserved HSR | 6352 | 0.423 (0.409, 0.437) | 0.0148 (0.0143, 0.0154) | 3.337 (3.273, 3.460) |
| Conserved rHSR a | 6352 | 0.501 (0.493, 0.509) | 0.0188 (0.0185, 0.0191) | 3.940 (3.875, 4.006) |
| HSR-eco | 4202 | 0.367 (0.351, 0.384) | 0.0137 (0.0130, 0.0144) | 3.501 (3.391, 3.612) |
| rHSR-eco | 4202 | 0.439 (0.430, 0.449) | 0.0187 (0.0184, 0.0190) | 4.169 (4.094, 4.243) |
| HSR-efe | 4362 | 0.368 (0.352, 0.385) | 0.0138 (0.0131, 0.0146) | 3.778 (3.664, 3.893) |
| rHSR-efe | 4362 | 0.435 (0.425, 0.444) | 0.0176 (0.0172, 0.0180) | 4.357 (4.284, 4.430) |
rHSR: random HSR, which was generated by shuffling synonymous codons among sites with identical amino acids, while maintaining amino acid sequence, codon usage bias, and GC content. In addition, the MFE of rHSR is similar (located in MFEHSR±10%) to that of native HSR. HSR-eco (HSR-efe): the HSRs only exist in Escherichia coli (Escherichia fergusonii).
CI: confidence interval.
Guanine preference at the third sites
Although high percentage of GC content promotes RNA folding with high structural stability because GC pairs are more stable than AT pairs, it is not obvious that HSRs maintain high structural stability by increasing G/C content during evolution. There are multiple selective pressures, including selections for RNA folding and translation efficiency, appearing to affect the patterns of synonymous mutation. Therefore, increasing GC content does not always benefit translational regulation. In particular, codon order in HSR instead of base composition might be adjusted to meet the requirements for structural stability and translation efficiency.
Here, we asked whether GC content in HSR is under selection for high structural stability. We analyzed GC content at the four-fold degenerate sites in the three regions: FAR, HSR and BAR. We found that HSR exhibits significantly higher G content than the other two regions (paired t-test, all p-values <10−16, Table 2). There is also a slightly increasing C content in HSRs (paired t-test, p-values <0.01). These results indicate a selection for increasing G and C (especially for G) in HSRs. Meanwhile, by checking local translation efficiency measured by tAI, we found a trend towards increasing translation efficiency in HSRs (paired t-test, p-values <10−5, Table 2). Therefore, the possibility remains that the selection for translation efficiency might be responsible for the high G/C content observed in HSRs. To resolve this issue, we simulated random mutations on HSRs by replacing the nucleotides at the four-fold degenerate sites with other nucleotides. For each HSR, half of the four-fold degenerate sites were randomly selected and mutated to generate four types of substituted sequences: G-Seq (G rich sequence, replacing nucleotides with G, the same to others), C-Seq, T-Seq, and A-Seq. To exclude the effect of substituted positions, for each type of sequence, we generated 50 substituted sequences, and calculated the mean MFE difference between native and substituted sequences. We found that G-seqs have a lower mean MFE than native sequences (paired t-test, p-value <10−16), whereas other three types of substituted sequences show significantly increased MFE (paired t-test, all p-values <10−16, Figure 4A), suggesting that increasing A/T/C (especially for A/T) or decreasing G in HSR will decrease the structural stability of HSR. Interestingly, we found an opposite pattern when comparing local translation efficiency between substituted and native sequences. Only C-seqs have a higher mean value of tAI, which is approximately equal to that of native sequences (0.257 in C-seqs vs. 0.255 in native sequences). The other three types of substitutions significantly decrease the translation efficiency in HSRs (paired t-test, all p-values <10−16, Figure 4B). In addition, considering that HSRs are G preference, G-seqs have a lower number of substitutions than the other types of sequences, which might influence the significance inferred from G-seqs, we thus performed a similar analysis using the HSRs with G content <0.25. Similar results were obtained (Figure S3). Overall, these results suggest that the increased G content in HSRs results from the selection for maintaining high structural stability.
Table 2. Comparison of base composition among three regions.
| Species | Regions | MFE (kcal/mol) | Base Composition | tAI | |||
| A | T | G | C | ||||
| ECO | FAR | −22.25 (7.55) | 0.131 (0.072 a) | 0.231 (0.092) | 0.355 (0.105) | 0.284 (0.093) | 0.250 (0.033) |
| HSR | −25.21 (7.73) | 0.107 (0.068) | 0.195 (0.088) | 0.412 (0.106) | 0.285 (0.088) | 0.255 (0.033) | |
| BAR | −22.23 (7.47) | 0.136 (0.075) | 0.229 (0.096) | 0.359 (0.108) | 0.277 (0.089) | 0.249 (0.034) | |
| EFE | FAR | −21.57 (7.13) | 0.151 (0.076) | 0.252 (0.095) | 0.333 (0.104) | 0.264 (0.088) | 0.282 (0.039) |
| HSR | −24.73 (7.68) | 0.130 (0.074) | 0.213 (0.088) | 0.385 (0.108) | 0.271 (0.084) | 0.286 (0.039) | |
| BAR | −20.94 (7.08) | 0.155 (0.081) | 0.249 (0.092) | 0.332 (0.104) | 0.263 (0.085) | 0.280 (0.039) | |
The standard deviations are shown in parentheses.
Figure 4. Effect of base composition on MFE and tAI.

Four types of sequences refer to the substituted HSRs replacing nucleotides at the four-fold degenerate sites with A, T, G, or C, respectively. The mean MFE (A) and mean tAI (B) of native HSRs are indicated by dashes.
Substitution rate variation
Mutational robustness is correlated with structural functionality and complexity. The results in previous sections showed that the number of key sites in HSR is lower than that in the corresponding random HSR. In addition, we compared base compositions of key sites and non-key sites, and found that key sites are G preference. About 70.2% key sites are G, while the percentage in non-key sites is about 24.7% (Figure 5). Note that HSRs with high percentage of G are sensitive to mutation, making it difficult to maintain high mutational robustness. In this case, one might expect lower dS in HSRs to reduce the harm of mutations. Indeed, we found a reduced dS in HSRs compared with that in the other two regions (Wilcoxon test, all p-values <0.05, Figure 6). Moreover, we estimated the dS of the codons that contain key sites. Again, the dS is significantly lower than that of the other codons in HSR (Wilcoxon test, p-value <10−5, Figure 6). Considering that the codons with key sites are G preference, which might affect the inferred substitution pattern, we randomly extracted the codons in other regions (non-HSRs), which are identical to the codons with key sites in HSRs. We estimated the dS of these “random” codons, and found a higher dS compared with that of the codons with key sites (Wilcoxon test, p-value <10−5). Overall, the results that HSRs have lower synonymous substitution rate, especially for the codons with key sites, suggest a selective constraint imposed on HSRs to reduce the harm of mutations.
Figure 5. Comparison of base composition between key and non-key sites.

Key sites indicate those sites, mutations on which result in >15% increase in MFE.
Figure 6. Comparison of synonymous substitution rates among three regions.

FAR: forward adjacent region; BAR: backward adjacent region. Random indicates the random codons, which are identical to the codons of key sites while are located in other regions. The 95% confidence intervals of synonymous substitution rates of key and non-key sites were estimated by bootstrap methods.
Compensatory mutations in HSRs
Distinct positions in structural RNA may not evolve independently because of shared structural or functional constraints. Considering the RNA with functional secondary structure, mutations on some sites (key sites) might disrupt the secondary structure, thus disabling its original function. Besides the strategy involving the reduction of the substitution rate on key sites (as suggested in previous section), another strategy is used to decrease the harm of the mutations on key sites. That is, a second (so called “compensatory”) mutation occurs on the specific site to restore the original conformation [27], [50], [51]. Previous studies on conserved secondary structure have revealed that the compensatory mutations should have a fitness equivalent to the wild type, resulting in an increasing of the substitution rate among these specific sites compared with that of key sites [52].
By analogy with the conserved secondary structure, compensatory mutations might be observed in HSRs, although it is the structural stability instead of the structure conformation that is maintained during evolution. Indeed, we found that the dS of non-key sites is higher slightly than that of background regions (0.339 in non-key sites vs. 0.283 in FAR and 0.295 in BAR, Figure 6), suggesting a relaxed selective constraint on non-key sites in HSRs compared with that on key sites. Consequently, we asked whether the compensatory mutations occurring on non-key sites account for this result. We first compared the effect of observed synonymous mutations on MFE between the ancestral-extant and ancestral-random HSRs. We found that the mean relative change of MFE caused by synonymous mutations in ancestral-extant HSRs is significantly lower than that in ancestral-random HSRs (0.028 vs. 0.060, paired t-test, p-value <10−16), indicating that the locations and patterns of synonymous mutations are non-random with respect to structural stability. This result also indicates a possibility that compensatory mutations occur to decrease the overall relative changes of MFE caused by mutations.
We then identified the compensatory mutations for each synonymous mutation that strongly increases the MFE of HSR. In two species, 2294 synonymous mutations (9.60% of all synonymous mutations) were extracted and treated as the first mutations (Table 3). In 41.70% of the first mutations in Escherichia coli, compensatory mutations were observed. The percentage is higher (t-test, p-value <10−16, Table 3) than that in ancestral-random HSRs. A similar pattern was observed in Escherichia fergusonii (Table 3). Note that only single point mutations were used and the conjugated effect of two or more than two point mutations was not considered. The true percentage of the compensatory mutations could be substantially higher than the observed value.
Table 3. Summary of compensatory mutations.
| Type | Species | Number of first mutations | Number of compensatory mutations | Percentage c (%) | Paired/Unpaired d |
| Ia | ECO b | 1115 | 691 | 41.70 | 0.578 |
| Random | 1693.3 (34.6) | 637.0 (28.8) | 28.59 (1.08) | 0.657 (0.045) | |
| EFE | 1719 | 842 | 33.92 | 0.520 | |
| Random | 1668.8 (24.2) | 639.7 (31.7) | 28.77 (1.19) | 0.614 (0.050) | |
| II | ECO | 931 | 337 | 9.47 | – |
| Random | 607.0 (25.3) | 823.5 (30.8) | 67.6 (1.96) | – | |
| EFE | 778 | 364 | 9.35 | – | |
| Random | 695.8 (15.9) | 950.2 (39.4) | 67.9 (1.66) | – |
Type refers to the two types of first mutations. I indicates the first mutations, which increase the MFE of ancestral HSR; II indicates the first mutations, which decrease the MFE of ancestral HSR (see text for details).
ECO (EFE) refers to the pair of ancestral-extant HSRs in Escherichia coli (Escherichia fergusonii); Random refers to the pair of ancestral-random HSRs, which were obtained by randomly generating the synonymous mutations based on the number of synonymous substitutions in corresponding ancestral-extant HSRs and the genomic frequency of codons.
Percentage refers to the percentage of the first mutations, which decrease the MFE of ancestral HSR while compensatory mutations exist to partly restore this disruption. Two or more compensatory mutations were observed in a few first mutations. Thus, the percentage is lower than the ratio: Number of compensatory mutations/Number of first mutations.
Paired means that the first mutated base is paired with the second mutated base, while unpaired means that the two mutated bases are unpaired.
We classified all compensatory mutations into two types: paired and unpaired. Paired means that the first mutated base is paired with the second mutated base, while the unpaired means that the two mutated bases are unpaired. We calculated the ratio paired/unpaired and surprisingly found that the ratio is lower than the expected value in the two species (t-test, all p-values <10−7, Table 3), which is different from the pattern inferred from the conserved secondary structure (the ratio is higher than the expected value) [27]. This result is consistent with the idea that structural stability instead of structural conformation in HSR is conserved during evolution [16], [35].
In addition, we redefined the first mutations as the synonymous mutations that result in >10% decrease in MFE of ancestral HSR. We performed a similar analysis as above based on the new first-mutated HSRs to test whether there are compensatory mutations that restore the MFE decreased by the first mutation. Interestingly, compensatory mutations were found only in 9.47% of the first-mutated HSRs in Escherichia coli, which is significantly lower (t-test, p-value <10−16, Table 3) than that in random mutations. A similar pattern was found in Escherichia fergusonii (Table 3). The results suggest that only a few mutations that increase structural stability of HSR are subject to negative selection. Overall, the findings suggest that a compensatory mechanism exists to maintain the high structural stability of HSRs.
Discussion
mRNA is a key component of a complex regulatory network. It accommodates numerous regulatory signals delineated along the protein coding regions in an intricate overlapping manner [53]. A worthy issue is that how these signals evolve to meet the requirements of the regulations on different levels of translation, and how the evolutionary patterns of these regulatory elements affect the observed evolutionary pathway of genome. In this study, we focused on one of the most important regulatory elements, mRNA secondary structure, and investigated their evolutionary patterns. HSR is a special region on mRNA containing a local secondary structure. A considerable proportion of mRNAs are covered by HSRs (about 30% on average). Therefore, base compositions and substitution rate of mRNA might be remarkably affected by HSRs.
In current study, we found that HSRs have high mutational robustness compared with random HSRs. It suggests that the folded conformation of HSR is adjusted to reduce the harm of mutation. We subsequently asked how the mutational robustness is maintained. Since base composition has strong effect on structural stability of HSR, we thus focused on the GC content of HSR. The results showed that HSRs are G preference, supporting the hypothesis that the selective constraint for high structural stability might partly account for high percentage of G in Escherichia coli genome. Note that the observed G contents in the other two background regions are higher than the other three bases. This suggests that there are other selective pressures imposed on mRNA, resulting in the variation of G content among sites, as suggested by the previous studies [4], [34], [54].
Our result is different from the claim in the work of Chamary et al. [38], which showed that mRNA stability partly drives C preference in Mus musculus. To explain the difference, we analyzed the relationship between the local stability and base composition of the 70 coding sequences used in their study. Again, we found that G content at the four-fold degenerate sites in HSRs is higher than that in background regions (0.276 in HSR vs. 0.238 in FAR, Wilcoxon test, p-value = 0.050, and 0.227 in BAR, p-value = 0.011, ). Although there is a universal excess of C over G, the difference in C contents among FAR, HSR and BAR is not significant (Wilcoxon test, all p-values values >0.1, Figure S4). In addition, we compared the mean MFE of the four types of substituted sequences, and found that the pattern is similar with that in Escherichia coli (Figure S5). Note that global mRNA was folded in the work of Chamary et al. [38], which might be involved in the global regulation of translation, such as RNA decay. We focused on local structural stability, which regulates a series of co-translational processes. Therefore, the difference between the two studies might result from the different methods dealing with RNA folding.
Although high percentage of G promotes HSR folding with high stability, increasing G will decrease the mutational robustness of HSR. An efficient strategy is to keep low substitution rate in HSR, especially for the key sites. As expected, a lower dS in HSR was observed. This result is consistent with the findings in a recent work [55], which revealed a significant correlation between mRNA structural stability and synonymous rate, as well as structural stability and non-synonymous substitution rate [55]. Moreover, note that horizontal gene transfer (HGT) occurs frequently in Escherichia coli [56]. The results might be affected by HGT events. Therefore, we discarded the predicted HGT genes, obtained from the works of Garcia-Vallvé et al. [57], and re-estimated substitution rate and GC content in HSRs. Similar results were obtained (Figure S6).
The dS in non-key sites is approximately equal to that in background regions. We proposed that compensatory mutations, occurring in about 40% first-mutated HSRs, partly account for this result (Table 3). Moreover, we calculated the number of compensatory mutations, and found that two or more compensatory mutations were detected in more than 30% first-mutated HSRs (Figure 7). This result suggests a different type of compensatory evolution compared with that occurring in the structural RNAs, in which the compensatory mutation is site-specific, and co-evolution would be observed during evolution. In HSRs, however, the substitution patterns of a large number of sites are affected by key sites, making it difficult to detect an obvious co-evolution. In addition, we only considered single point mutations to detect the compensatory mutations. In fact, it is more likely that multiple mutations coordinate the folded conformation to restore the disrupted MFE. The conjugated effect of multiple mutations are worth pursuing at a deeper level.
Figure 7. Distribution of the number of compensatory mutations in HSR.

In more than 30% of the first-mutated HSR with compensatory mutations, two or more compensatory mutations were detected.
Supporting Information
GC content and sequence identity along mRNA. In the first 30 codons of mRNA, GC content (A) at the three positions of codon is significantly lower than that in other regions. The sequence identity (B) in the first 50 codons is different from the latter regions.
(TIF)
Number of key sites under different thresholds. The number of key sites in native HSRs is significantly lower (paired t-test, all p-values <10−16) than that in random HSRs when threshold <0.2. In both native and random HSRs, the numbers of key sites are close to 0 when threshold >0.2.
(TIF)
Comparison of MFE and tAI among four types of substituted sequences. Four types of sequences refer to the substituted HSRs replacing nucleotides at the four-fold degenerate sites with A, T, G, or C, respectively. The mean MFE (A) and tAI (B) of native HSRs is indicated by dashes, respectively. The data was based on the HSRs with G<0.25.
(TIF)
Comparison of base composition in three regions, showing G preference in HSRs. The data were obtained based on 70 mRNAs in Mus musculus.
(TIF)
Comparison of MFE and tAI among four types of substituted sequences. The data were calculated based on 70 mRNAs in Mus musculus.
(TIF)
Comparisons of synonymous substitution rates and base compositions in the three regions. The data were obtained by excluding the genes detecting the horizontal gene transfer event.
(TIF)
Positions of HSRs in Escherichia coli .
(XLS)
Positions of HSRs in Escherichia fergusonii.
(XLS)
Positions of conserved HSRs between Escherichia coli and Escherichia fergusonii .
(XLS)
Acknowledgments
The authors are grateful to the two anonymous reviewers for their excellent suggestions.
Funding Statement
This work was supported by the PhD Programs Foundation of the Ministry of Education of China (20100204110026) and project from National Science Foundation of China (31125007). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Yu CH, Noteborn MH, Pleij CWA, Olsthoorn RCL (2011) Stem–loop structures can effectively substitute for an RNA pseudoknot in −1 ribosomal frameshifting. Nucleic Acids Res 39: 8952–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Novikova IV, Hennelly SP, Sanbonmatsu KY (2012) Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res 40: 5034–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mao Y, Wang W, Cheng N, Li Q, Tao S (2013) Universally increased mRNA stability downstream of the translation initiation site in eukaryotes and prokaryotes. Gene 517: 230–235. [DOI] [PubMed] [Google Scholar]
- 4. Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY (2011) Understanding the transcriptome through RNA structure. Nat Rev Genet 12: 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, et al. (2006) Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol 2: e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eddy SR (2001) Non-coding RNA genes and the modern RNA world. Nat Rev Genet 2: 919–929. [DOI] [PubMed] [Google Scholar]
- 7. Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, et al. (2012) Genome-wide determination of RNA stability reveals hundreds of short-lived non-coding transcripts in mammals. Genome Res 22: 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y, et al. (2012) Global analysis of RNA secondary structure in two metazoans. Cell Rep 1: 69–82. [DOI] [PubMed] [Google Scholar]
- 9. Washietl S, Hofacker IL, Lukasser M, Hüttenhofer A, Stadler PF (2005) Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol 23: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 10. Pasquinelli AE (2012) MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13: 271–282. [DOI] [PubMed] [Google Scholar]
- 11. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, et al. (2005) TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436: 740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee MT, Kim J (2008) Self containment, a property of modular RNA structures, distinguishes microRNAs. PLoS Comput Biol 4: e1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonnet E, Wuyts J, Rouzé P, Van de Peer Y (2004) Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 20: 2911–2917. [DOI] [PubMed] [Google Scholar]
- 14. Mimouni NK, Lyngsø RB, Griffiths-Jones S, Hein J (2009) An analysis of structural influences on selection in RNA genes. Mol Biol Evol 26: 209–216. [DOI] [PubMed] [Google Scholar]
- 15. Katz L, Burge CB (2003) Widespread selection for local RNA secondary structure in coding regions of bacterial genes. Genome Res 13: 2042–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mao Y, Li Q, Wang W, Liang P, Tao S (2013) Number variation of high stability regions is correlated with gene functions. Genome Biol Evol 5: 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moss WN, Priore SF, Turner DH (2011) Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 17: 991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, et al. (2010) Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gu W, Zhou T, Wilke CO (2010) A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol 6: e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tuller T, Waldman YY, Kupiec M, Ruppin E (2010) Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A 107: 3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kudla G, Murray AW, Tollervey D, Plotkin JB (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, et al. (2009) Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proctor JR, Meyer IM (2013) COFOLD: an RNA secondary structure prediction method that takes co-transcriptional folding into account. Nucleic Acids Res: gkt174v171–gkt174. [DOI] [PMC free article] [PubMed]
- 24. Zur H, Tuller T (2012) Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep 13: 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pereira F, Soares P, Carneiro J, Pereira L, Richards MB, et al. (2008) Evidence for variable selective pressures at a large secondary structure of the human mitochondrial DNA control region. Mol Biol Evol 25: 2759–2770. [DOI] [PubMed] [Google Scholar]
- 26. Price N, Cartwright RA, Sabath N, Graur D, Azevedo RB (2011) Neutral evolution of robustness in Drosophila microRNA precursors. Mol Biol Evol 28: 2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng N, Mao Y, Shi Y, Tao S (2012) Coevolution in RNA molecules driven by selective constraints: evidence from 5S rRNA. PloS one 7: e44376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caetano-Anollés G (2002) Evolved RNA secondary structure and the rooting of the universal tree of life. J Mol Evol 54: 333–345. [DOI] [PubMed] [Google Scholar]
- 29. Lind PA, Andersson DI (2013) Fitness costs of synonymous mutations in the rpsT gene can be compensated by restoring mRNA base pairing. PloS one 8: e63373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piskol R, Stephan W (2011) Selective constraints in conserved folded RNAs of drosophilid and hominid genomes. Mol Biol Evol 28: 1519–1529. [DOI] [PubMed] [Google Scholar]
- 31. Dutheil JY, Jossinet F, Westhof E (2010) Base pairing constraints drive structural epistasis in ribosomal RNA sequences. Mol Biol Evol 27: 1868–1876. [DOI] [PubMed] [Google Scholar]
- 32. Rocha EP, Feil EJ (2010) Mutational patterns cannot explain genome composition: are there any neutral sites in the genomes of bacteria? PLoS Genet 6: e1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carmel I, Shomron N, Heifetz Y (2012) Does base-pairing strength play a role in microRNA repression? Rna 18: 1947–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu W, Wang X, Zhai C, Xie X, Zhou T (2012) Selection on synonymous sites for increased accessibility around miRNA binding sites in plants. Mol Biol Evol 29: 3037–3044. [DOI] [PubMed] [Google Scholar]
- 35. Tuller T, Veksler-Lublinsky I, Gazit N, Kupiec M, Ruppin E, et al. (2011) Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol 12: R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gingold H, Dahan O, Pilpel Y (2012) Dynamic changes in translational efficiency are deduced from codon usage of the transcriptome. Nucleic Acids Res 40: 10053–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah P, Gilchrist MA (2011) Explaining complex codon usage patterns with selection for translational efficiency, mutation bias, and genetic drift. Proc Natl Acad Sci U S A 108: 10231–10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chamary J, Hurst LD (2005) Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol 6: R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stoletzki N (2008) Conflicting selection pressures on synonymous codon use in yeast suggest selection on mRNA secondary structures. BMC Evol Biol 8: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schroeder R, Barta A, Semrad K (2004) Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol 5: 908–919. [DOI] [PubMed] [Google Scholar]
- 41. Pan T, Sosnick T (2006) RNA folding during transcription. Annu Rev Biophys Biomol Struct 35: 161–175. [DOI] [PubMed] [Google Scholar]
- 42. Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL (2008) The vienna RNA websuite. Nucleic Acids Res 36: W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480–D484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- 47. Blanchette M, Diallo AB, Green ED, Miller W, Haussler D (2008) Computational reconstruction of ancestral DNA sequences. Methods Mol Biol 422: 171. [DOI] [PubMed] [Google Scholar]
- 48. Visser J, Hermisson J, Wagner GP, Meyers LA, Bagheri-Chaichian H, et al. (2003) Perspective: evolution and detection of genetic robustness. Evolution 57: 1959–1972. [DOI] [PubMed] [Google Scholar]
- 49. Kitano H (2004) Biological robustness. Nat Rev Genet 5: 826–837. [DOI] [PubMed] [Google Scholar]
- 50. Levin BR, Perrot V, Walker N (2000) Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria. Genetics 154: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hancock JM, Tautz D, Dover GA (1988) Evolution of the secondary structures and compensatory mutations of the ribosomal RNAs of Drosophila melanogaster. Mol Biol Evol 5: 393–414. [DOI] [PubMed] [Google Scholar]
- 52. Knies JL, Dang KK, Vision TJ, Hoffman NG, Swanstrom R, et al. (2008) Compensatory evolution in RNA secondary structures increases substitution rate variation among sites. Mol Biol Evol 25: 1778–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shabalina SA, Spiridonov NA, Kashina A (2013) Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res 41: 2073–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Botzman M, Margalit H (2011) Variation in global codon usage bias among prokaryotic organisms is associated with their lifestyles. Genome Biol 12: R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park C, Chen X, Yang J-R, Zhang J (2013) Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A 110: E678–E686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koonin EV, Makarova KS, Aravind L (2001) Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiology 55: 709–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia-Vallvé S, Romeu A, Palau J (2000) Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res 10: 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GC content and sequence identity along mRNA. In the first 30 codons of mRNA, GC content (A) at the three positions of codon is significantly lower than that in other regions. The sequence identity (B) in the first 50 codons is different from the latter regions.
(TIF)
Number of key sites under different thresholds. The number of key sites in native HSRs is significantly lower (paired t-test, all p-values <10−16) than that in random HSRs when threshold <0.2. In both native and random HSRs, the numbers of key sites are close to 0 when threshold >0.2.
(TIF)
Comparison of MFE and tAI among four types of substituted sequences. Four types of sequences refer to the substituted HSRs replacing nucleotides at the four-fold degenerate sites with A, T, G, or C, respectively. The mean MFE (A) and tAI (B) of native HSRs is indicated by dashes, respectively. The data was based on the HSRs with G<0.25.
(TIF)
Comparison of base composition in three regions, showing G preference in HSRs. The data were obtained based on 70 mRNAs in Mus musculus.
(TIF)
Comparison of MFE and tAI among four types of substituted sequences. The data were calculated based on 70 mRNAs in Mus musculus.
(TIF)
Comparisons of synonymous substitution rates and base compositions in the three regions. The data were obtained by excluding the genes detecting the horizontal gene transfer event.
(TIF)
Positions of HSRs in Escherichia coli .
(XLS)
Positions of HSRs in Escherichia fergusonii.
(XLS)
Positions of conserved HSRs between Escherichia coli and Escherichia fergusonii .
(XLS)