

Abstract
The insulin minisatellite (INS VNTR) associates with susceptibility to a variety of diseases. We have developed a high-resolution system for analyzing variant repeat distributions applicable to all known minisatellite alleles, irrespective of size, which allows lineages of related alleles to be identified. This system has previously revealed extremely low structural diversity in the minisatellite among northern Europeans from the United Kingdom, with all alleles belonging to one of only three highly diverged lineages called “I,” “IIIA,” and “IIIB.” To explore the origins of this remarkably limited lineage diversity, we have characterized an additional 780 alleles from three non-African and three African populations. In total, 22 highly diverged lineages were identified, with structural intermediates absent from extant populations, suggesting a bottleneck within the ancestry of all humans. The difference between levels of diversity in Africans and non-Africans is unusually large, with all 22 lineages identified in Africa compared with only three lineages seen not only in the United Kingdom but also in the other non-African populations. We also find evidence for overrepresentation of lineage I chromosomes in non-Africans. These data are consistent with a common out-of-Africa origin and an unusually tight bottleneck within the ancestry of all non-African populations, possibly combined with differential and positive selection for lineage I alleles in non-Africans. The important implications of these data for future disease-association studies are discussed.
Introduction
The insulin minisatellite (INS VNTR [MIM 125852]), located 596 bp upstream of the human insulin gene translation-initiation site, has attracted widespread interest because of its associations with a range of phenotypes, including susceptibility to type 1 diabetes (Bennett and Todd 1996), type 2 diabetes (Huxtable et al. 2000), polycystic ovary syndrome (Waterworth et al. 1997), variation in birth size (Dunger et al. 1998), and obesity (Le Stunff et al. 2001). We have developed a system of minisatellite variant-repeat (MVR) mapping by use of PCR (Jeffreys et al. 1991) to analyze allele structure at the INS VNTR (Stead and Jeffreys 2000; Stead et al. 2000). By detecting six different variant-repeat types and fully analyzing all known alleles irrespective of size, this system generates far more detailed information on allele relationships than could be achieved by analyzing minisatellite length or variation in flanking haplotype. Elsewhere, we have demonstrated a remarkably low level of minisatellite diversity within a type 1 diabetes–associated cohort from the United Kingdom, with all alleles readily assigned by structure to one of just three highly diverged lineages (Stead and Jeffreys 2000; Stead et al. 2000). To investigate the origins and worldwide prevalence of this unusual pattern, we now apply MVR-PCR to the analysis of six additional populations from geographically diverse regions of Africa, Europe, and Asia.
Other studies analyzing genetic variation in different populations have generally revealed greater diversity within Africans than among non-Africans, consistent with an African origin of anatomically modern humans (Cann et al. 1987; Vigilant et al. 1991; Armour et al. 1996; Relethford and Jorde 1999; Seielstad et al. 1999; Jorde et al. 2000; Alonso and Armour 2001; Hollox et al. 2001; Templeton 2002). Furthermore, despite elevated African diversity, all humans show reduced genetic diversity compared with other species, suggesting a bottleneck within the ancestry of all humans (Li and Sadler 1991; Crouau-Roy et al. 1996; Kaessmann et al. 1999). Most recent studies of autosomal genetic variation have examined SNPs arranged into haplotypes (Clark et al. 1998; Fullerton et al. 2000; Alonso and Armour 2001; Hollox et al. 2001; Mateu et al. 2001; Gilad et al. 2002; Nakajima et al. 2002). Although differences between haplotypes allow the inference of phylogenetic relationships between chromosomes and the detection of historical recombination events, the low mutation rate of SNPs limits the temporal resolution of these analyses. Microsatellites have also been used to investigate population diversity, although their higher mutation rates—combined with an approximately stepwise mode of mutation—results in substantial homoplasy, reducing their usefulness for analysis of deeply rooted phylogenies (Nauta and Weissing 1996; Feldman et al. 1997).
At the INS VNTR, major structural differences between alleles reflect ancient divergence between chromosomes, whereas small differences indicate a more recent common ancestor between alleles. Variant-repeat analysis is therefore simultaneously informative on different timescales of population history. Other minisatellites have also been used to investigate population histories. For example, MVR mapping at minisatellite MS205 showed that diversity in non-Africans represents a subset of the diversity within Africa (Armour et al. 1996). However, these studies could not analyze large alleles and detected only a relatively small number of variant-repeat types, limiting their resolution. In the present study, we aimed to provide the most informative description of worldwide minisatellite variability to date, by analyzing six different variant-repeat types within all alleles of the INS VNTR, irrespective of allele size. These data may also have important implications for disease-association studies. Most previous studies on the INS VNTR analyzed cohorts of European descent in which high linkage disequilibrium near the minisatellite (Doria et al. 1996)—combined with low minisatellite lineage diversity (Stead and Jeffreys 2000)—make it difficult to distinguish between etiological variants and associated variants. Analysis of populations with different allele lineages and associated patterns of linkage disequilibrium could therefore facilitate the identification of true etiological variants in the INS region. The present study is therefore a necessary prelude to any future disease-association studies of the insulin region in non-European populations.
Material and Methods
DNA Source
Genomic DNA was extracted from blood or sperm and was analyzed in individuals, sampled at random with respect to disease status, from six populations: United Kingdom (102 individuals), Kazakhstan (40), Japan (59), Ivory Coast (78), Zimbabwe (69), and Kenya (42). The use of human samples was approved by the Leicestershire Local Research Ethics Committee.
Nomenclature
Historically, alleles at the minisatellite have been divided into three groups, on the basis of size: class I (small), II (intermediate), and III (large) (Bell et al. 1981). Here, we reserve the term “class” exclusively for alleles grouped by size, irrespective of allele structure defined by MVR-PCR. Alleles grouped on the basis of similar minisatellite structures are termed “lineages.” These lineages are named with single capital letters, except for lineages I, IIIA, and IIIB, which were previously named according to minisatellite sizes (Stead and Jeffreys 2000). Sublineages are indicated by Roman numerals (e.g., IIIAii is the second sublineage of lineage IIIA), except for lineage I sublineages IC, ID+, ID−, and IE, which were named for consistency with our previous studies (Stead and Jeffreys 2000). The names of specific alleles reflect lineages, size (expressed in repeat number), and a further discriminator; for example, J42.2 is the second different allele of 42 repeats identified in lineage J.
Alleles were assigned, manually and in a hierarchical fashion, to lineages and sublineages. Alleles from different lineages could not be aligned to each other along the majority of their lengths. Within some lineages, subgroups of alleles shared greater similarity to each other than to other alleles within the lineage, owing to large insertions or deletions or to common differences occurring at more than one independent variant position. We refer to these groups as sublineages. Dot matrix analyses of alleles within a lineage and between lineages confirmed the division of alleles into lineage groups. The identity of sublineages was confirmed by multidimensional scaling, as described elsewhere (Stead and Jeffreys 2000). Although it was not possible to fully define a priori the criteria for lineage and sublineage definition, all assignments of alleles to lineage groups were performed by individuals who were blinded with respect to the population of origin, to prevent any bias in the conclusions regarding the differences in genetic variation between populations.
Amplification and Separation of Alleles
Alleles of the INS VNTR were amplified by PCR from 10 ng of genomic DNA, by use of the universal primer INS-1296 and a lineage-specific primer—INS- 23+ (which exclusively amplifies lineage I) or INS-23− (which amplifies all other lineages)—in 10-μl reactions, using the buffer described elsewhere (Jeffreys et al. 1990), supplemented with 12 mM Tris base, 1 μg/ml carrier herring sperm DNA, 0.4 μM of each primer, 0.07 U/μl Taq polymerase (Advanced Biotechnologies), and 0.007 U/μl Pfu polymerase (Stratagene). To determine allele sizes, samples were amplified at 96°C for 40 s, 61°C for 30 s, and 70°C for 5 min for 22 cycles on an MJ Tetrad thermal cycler (MJ Research). Samples were electrophoresed through a 40-cm 1% Seakem LE (FMC Bioproducts) agarose gel in 1× Tris-borate–EDTA buffer (89 mM Tris-borate, pH 8.3, and 2 mM EDTA) at 3 V/cm for 20 h and were detected using Southern blot hybridization, with a 32P-labeled probe generated by PCR amplification of a class I allele. Sized alleles were separated prior to MVR-PCR analysis by band excision after amplification and electrophoresis of products, as described above, with samples amplified for 32 cycles. PCR products were detected by staining with ethidium bromide and were visualized using a Dark Reader (Clare Chemical Research) to prevent UV damage. Alleles >3.5 kb could not be detected on ethidium bromide–stained gels, and their positions were instead estimated using a 1-kb ladder size marker (Gibco BRL) prior to band excision. Alleles <2 kb were released from the gel by adding 50 μl of dilution buffer (5 mM Tris-HCl, pH 7.5, and 5 μg/ml carrier herring sperm DNA) and freezing-thawing-vortexing three times. Larger alleles were gel purified using the QIAquick gel extraction kit (Qiagen), according to the manufacturer's instructions.
MVR-PCR Mapping of Separated Alleles
In an early study, we established a system of MVR-PCR analysis that detects six different variant repeats at the INS VNTR (Stead and Jeffreys 2000). The names of the six variants and their sequences are as follows (nucleotides differing from the A-type repeat are underlined): A, GTGGGGACAGGGGT; B, CCTGGGGACAGGGGT; C, CTGGGGACAGGGGT; E, GTGGGGATAGGGGT; F, CCCGGGGACAGGGGT; and H, GTGGGCACAGGGGT. Individual alleles of the INS VNTR were MVR mapped by PCR amplification with a universal primer flanking the minisatellite (INS-1296) and a primer that specifically detects one of the above variant repeats (MVR primers INS-MA, INS-MB, INS-MC, INS-ME, INS-MF, and INS-MH). Amplification thus generates PCR products whose sizes reflect the positions of a specific variant repeat within the minisatellite. To reduce progressive loss of larger amplicons by internal priming during PCR, MVR primers carry a 20-nt 5′ extension (TAG). During the first round of PCR amplification, this extension becomes incorporated on the end of each amplicon. A third primer with sequence identical to that of this 20-nt extension is included at higher concentration than the MVR primer. Subsequent PCR cycles will therefore predominantly amplify products from the end of each amplicon, using the TAG primer—as opposed to within the amplicon, using the MVR primer. MVR-PCR was performed in 7-μl reactions with ∼0.1 pg of purified allele DNA in the buffer described above, with 0.035 U/μl Taq polymerase and 0.0035 U/μl Pfu polymerase plus one MVR primer at a concentration of 10 nM, together with 0.25 μM INS-1296 and TAG primers. PCR amplifications were performed at 96°C for 40 s, 65°C for 30 s, and 70°C for 2 min for eight cycles, followed by 96°C for 40 s, 58°C for 30 s, and 70°C for 2 min for 12 cycles. Products amplified using each of the six MVR primers were electrophoresed in adjacent lanes through a 40-cm 1.5% LE agarose gel at 3 V/cm for 18 h and were detected by Southern blot hybridization.
This MVR system allows alleles as long as 80 repeats to be fully analyzed. Larger alleles were mapped by creating a range of deletion amplicons, in a way similar to that of our previous analyses of class III alleles (Stead and Jeffreys 2000). In brief, standard MVR analysis produces amplicons between the flanking universal primer and the MVR primer which binds to a variant-repeat unit within the minisatellite. Agarose gel electrophoresis of MVR-PCR products, followed by band excision of a specific MVR product, thus results in purification of a deletion amplicon spanning a region between the flanking universal primer and a specific repeat unit. If the universal primer is located 5′ of the minisatellite (the standard “forward” MVR system), the repeat-specific primer is designed with a 5′ extension that matches the 3′ primer used to amplify alleles (INS-23−). Similarly, if the universal primer is located 3′ of the minisatellite (“reverse” MVR), the repeat-specific primer is designed with a 5′ extension that matches the 5′ primer used to amplify alleles (INS-1296). In this way, deletion amplicons are generated that cover either the 5′ or 3′ end of the repeat array, all of which share identical sequences at each terminus, matching INS-1296 at the 5′ end and INS-23− at the 3′ end. For very large alleles, this process is repeated to produce additional deletion amplicons from previously fragmented alleles. Each deletion amplicon is subsequently mapped using the same system of forward MVR-PCR, and full allele codes are assembled from overlapping codes from various deletion amplicons. In this way, structures of alleles >600 repeats could be determined from as many as 25 overlapping codes with the same level of accuracy as for short alleles. The forward and reverse MVR primers used to generate these deletion amplicons were chosen to detect variant repeats that were present at low frequency within an allele but widely dispersed along the repeat array. The choice of MVR primer was therefore dependent on allele lineage. Full details of the procedure are given elsewhere (Stead and Jeffreys 2000). Deletion amplicons were gel purified, as described above. To ensure that a single band had been excised, products were reamplified, and gel purification was repeated. Since MVR-PCR is very sensitive to the concentration of input DNA, all alleles and deletion amplicons were diluted by between 10-fold and 10,000-fold, depending on concentration of purified product, and a test MVR was performed; the DNA concentration in the full MVR analysis was then adjusted according to signal strength and quality (Stead and Jeffreys 2000).
Supplementary information, including all primer sequences, plus full MVR-PCR codes of the 1,985 alleles typed in this and other studies (Stead and Jeffreys 2000; Stead et al. 2000; authors' unpublished data), are available at the authors' Web site.
In Silico Analysis
Fst and analysis of molecular variance (AMOVA) calculations (Excoffier et al. 1992) were performed using Arlequin v. 2.000 (Schneider et al. 2000). Gene diversity estimates were calculated for each population, using Nei's gene-diversity statistic, which generates an estimate of heterozygosity
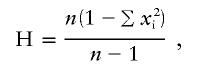 |
where n is the number of gene copies and xi is the frequency of the ith allele or lineage (Nei 1987). Gene diversity was estimated separately for alleles and for lineages. We define an allele as a unique pattern of variant-repeat distribution at the INS VNTR, and we define a lineage as a group of alleles which share similar, though not necessarily identical, structures. Mutation rates within lineages of the INS VNTR were estimated using Ewens's distribution (Ewens 1972), in which the expected number of different alleles defined by MVR structure, na, in a sample of n alleles of a given lineage is given by
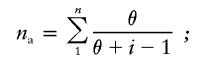 |
θ=4Nefμ, where the effective population size Ne is taken to be 10,000 (Jorde et al. 1998), f is the proportion of all alleles that belong to the lineage being tested, and μ is the mutation rate. In Africans, mutation rates were estimated only for those lineages (15 of 22) that had sufficient numbers of alleles plus instances of two or more identical alleles.
Computer Simulations
Non-African populations contain only 3 of the 22 lineages found among Africans but, nevertheless, retain three of the four sublineages of lineage I seen in Africa. Computer simulations were performed to determine the probability of selecting three of these sublineages while obtaining an overall reduction in lineage diversity from 22 lineages to the I, IIIA, and IIIB lineages present in extant non-African populations. This simulation was thus designed simply to determine the output of a population bottleneck and is not informative as to its size or duration. Chromosomes were sampled at random, with replacement from a pool with the same lineage composition as in the combined African populations in the present study. Sampling was terminated when the number of different lineages represented exceeded three. Three million simulations were performed. Of the simulations that yielded only lineages I, IIIA, and IIIB (0.17% of simulations) only 3% contained at least three different sublineages of lineage I.
Results
Comparisons between Populations
We have produced complete maps of the distribution of six different variant repeats within 780 alleles of the INS VNTR from populations from the United Kingdom (204 alleles), Kazakhstan (80), Japan (118), Ivory Coast (156), Zimbabwe (138), and Kenya (84). These samples were selected to provide coverage of Europe, Central and East Asia, and three geographically separated regions of Africa. Among the 780 alleles, 297 different alleles defined by unique patterns of variant-repeat distributions were identified, ranging in size from 17 to 630 repeats. Pairwise comparisons of the structures of different alleles allowed them to be assigned to distinct lineages and sublineages (see the “Material and Methods” section). In total, 22 different lineages of clearly related alleles were identified. All 22 lineages were found in the African populations. In contrast, the populations from the United Kingdom, Kazakhstan, and Japan shared the same very small subset of just 3 of the 22 lineages found in Africans, namely, lineages I, IIIA, and IIIB. This major difference in lineage composition between Africans and non-Africans is reflected by estimates of gene diversity (Nei 1987), which, when based on minisatellite lineages (groups of alleles with similar structures) rather than specific alleles (unique structures at the minisatellite), generate heterozygosities of 88%–90% in Africans, compared with just 10%–43% in non-Africans (table 1). Lineage frequencies in the six populations analyzed in the present study are shown in table 2, with examples of MVR codes from specific alleles shown in fig. 1. Codes for all alleles are available at the authors' Web site.
Table 1.
Genetic Diversity at the INS VNTR[Note]
| UnitedKingdom(%) | Kazakhstan(%) | Japan(%) | IvoryCoast(%) | Zimbabwe(%) | Kenya(%) | |
| Minisatellite lineages | 43.4 | 26.7 | 9.8 | 88.4 | 90.1 | 90.4 |
| Specific alleles | 96.2 | 96.0 | 91.7 | 97.9 | 98.8 | 98.4 |
Note.— Nei's gene diversity statistic (Nei 1987) was used to estimate heterozygosity for each population on the basis of either the minisatellite lineages or the identity of specific alleles at the INS VNTR defined by unique MVR codes. Estimates of diversity that were based on specific alleles are high and are similar between all six populations, reflecting the high rate of simple mutation at the minisatellite (Stead and Jeffreys 2000).
Table 2.
Percentage Lineage Frequencies in Six Populations[Note]
| Lineage | UnitedKingdom | Kazakhstan | Japan | IvoryCoast | Zimbabwe | Kenya |
| I | 71.6 | 85.0 | 94.9 | 19.2 | 18.8 | 15.5 |
| IIIA | 23.0 | 11.3 | 4.2 | 5.8 | 1.4 | 7.1 |
| IIIB | 5.4 | 3.8 | .8 | 3.2 | 1.4 | 3.6 |
| F | .7 | |||||
| G | 1.3 | .7 | ||||
| H | 1.4 | 3.6 | ||||
| J | 9.6 | 8.0 | 9.5 | |||
| K | 20.5 | 17.4 | 20.2 | |||
| L | 8.3 | 5.1 | 3.6 | |||
| M | 3.2 | .7 | 3.6 | |||
| N | 2.6 | 5.8 | ||||
| O | 2.6 | |||||
| P | 2.9 | 2.4 | ||||
| Q | 1.9 | 5.8 | 9.5 | |||
| S | 4.5 | .7 | 3.6 | |||
| T | .6 | 4.3 | 1.2 | |||
| U | 1.4 | |||||
| V | 1.3 | 1.4 | 4.8 | |||
| W | 12.8 | 13.0 | 9.5 | |||
| X | 1.9 | 4.3 | 2.4 | |||
| Y | .6 | 3.6 | ||||
| Z | .7 |
Note.— Blank entries indicate that no alleles were seen.
Figure 1.

Allele structures at the INS VNTR. Selected examples of allele codes defined by MVR-PCR analysis are presented in 5′-to-3′ orientation, with the INS gene to the right. Although alleles as long as 630 repeats have been analyzed, for convenience only alleles shorter than 90 repeats are shown. All of these non-African alleles are from a single lineage, whereas there are 11 lineages (arranged in groups) in Africans within this size range. Each square represents a single repeat unit, with different colors representing different variant repeats as follows: green = A, red = B, dark blue = C, pale blue = E, yellow = F, and pink = H. Some repeats (blackened squares) were not amplifiable, because of the presence of additional unknown sequence variants. Gaps were introduced by hand to facilitate allele alignments. All allele codes are available at the authors' Web site.
This difference in genetic diversity between Africans and non-Africans is unusually large. For example, most nuclear polymorphisms produce Fst values of 0.1–0.15, even between Africans and non-Africans, indicating that only 10%–15% of genetic variation is due to differences between population groups (Jorde et al. 1998). However, the INS VNTR lineage data generate Fst estimates between Africans and non-Africans as high as 0.45 (table 3), higher than estimates from most genomic regions (although such values are not unprecedented; e.g., see Chang et al. 1995). Similarly, hierarchical AMOVA (Excoffier et al. 1992) showed that 27.8% of the total genetic variance was due to differences between Africans and non-Africans, substantially greater than at most other loci (table 4; Barbujani et al. 1997). In contrast, differences between populations within a continent were unusually low, despite their substantial geographical separations (table 4; Barbujani et al. 1997). These substantial differences between Africans and non-Africans would be consistent with differential and directional selection operating on the two population groups (Cavalli-Sforza et al. 1994).
Table 3.
Fst Estimates at the INS VNTR[Note]
| Population | UnitedKingdom | Kazakhstan | Japan | Ivory Coast | Zimbabwe | Kenya |
| United Kingdom | .051 |
.124 |
.231 |
.238 |
.263 |
|
| Kazakhstan | .016 | .271 |
.271 |
.312 |
||
| Japan | .372 |
.376 |
.448 |
|||
| Ivory Coast | .001 | .000 | ||||
| Zimbabwe | .001 | |||||
| Kenya |
Note.— Global Fst=0.209. Fst values were estimated from minisatellite lineage data for each pair of populations following division of alleles into 22 lineages. Underlined values were significant (P<.01). All estimations were performed with Arlequin v. 2.000 (Schneider et al. 2000).
Table 4.
AMOVA at the INS VNTR[Note]
|
Average Variance Components(%) |
|||
| Polymorphism | Within Samples | Among SamplesWithin Groups | Among Groups |
| INS VNTR | 70.5 | 1.6 | 27.8 |
| 109 RFLPs/ microsatellites | 84.5 | 4.7 | 10.8 |
Note.— For comparison, data from Barbujani et al. (1997) are presented that show the mean molecular variance at 109 DNA polymorphisms within and between four or five continents, including Africa.
Despite this major divergence between Africans and non-Africans, the three universally present allele lineages (I, IIIA, and IIIB) are structurally similar in all populations. For example, lineage I divides into four sublineages (IC, ID+, ID−, and IE), on the basis of MVR structure and flanking haplotype. Three of these sublineages are present in every population (fig. 2). Although sublineage IE is African specific, it is uncommon (frequency 0.8%) and could be of recent origin, possibly after the out-of-Africa migration. The observation that three of the four sublineages of lineage I identified in Africans are also present in all non-African populations is striking, given the massive overall reduction in lineage diversity in non-Africans; this suggests a possible enrichment of lineage I within non-Africans, consistent with positive selection.
Figure 2.

Sublineages within lineage I. Alleles from each sublineage of lineage I are presented, as described in figure 1. The relative frequencies of each sublineage within lineage I are shown for the combined non-African populations (323 lineage I chromosomes) and the combined African populations (69 lineage I chromosomes). Lineage I alleles in Africans can be divided by MVR code into three sublineages, IC, ID, and IE, with ID alleles further dividing into sublineages ID+ and ID− (Stead et al. 2000). Each sublineage is distinguished by variation, both at the minisatellite and in the flanking haplotype (authors' unpublished data).
To test whether the presence of lineages I, IIIA, and IIIB in all populations could be due, in part, to recent admixture, we analyzed the degree of sharing of specific alleles, rather than lineages, between populations (table 5). This showed that the frequencies of specific alleles varied substantially between populations and that allele sharing between populations was very low, especially between Africans and non-Africans. For example, the most common allele in the United Kingdom (ID42.4, frequency 14.2%) was seen only once in the Japanese (0.8%) and was absent from all three African populations. Furthermore, analysis of haplotypes surrounding the minisatellite (authors' unpublished data) showed that all non-African IIIA haplotypes (23% of all U.K. haplotypes) differed from all African IIIA haplotypes by variation at the SNP +1355C/T (Julier et al. 1991). Only one allele (ID40.2) was identified in all six populations. These data suggest a very low level of admixture and establish that the presence of the three non-African lineages in Africa represents a true component of indigenous African diversity.
Table 5.
Sharing of Identical Alleles between Populations[Note]
| Population (No. of Different Alleles) | UnitedKingdom | Kazakhstan | Japan | IvoryCoast | Zimbabwe | Kenya |
| United Kingdom (88) | 14 | 15 | 6 | 3 | 4 | |
| Kazakhstan (44) | 14 | 4 | 3 | 4 | ||
| Japan (39) | 5 | 3 | 3 | |||
| Ivory Coast (85) | 21 | 13 | ||||
| Zimbabwe (76) | 19 | |||||
| Kenya (52) |
Note.— Data are the number of different alleles (defined by unique MVR codes).
Diversity and Mutation
Most differences between alleles within each of the 22 lineages are due to the simple gain or loss of a few repeats. As a result, alleles within a lineage fall within a narrow range of allele sizes (fig. 3). This tight correlation between lineage and size, together with restricted diversity in non-Africans, creates the bimodal allele-size distribution seen in these populations. Although lineage size and diversity is far greater in Africans, allele lengths do approximate a trimodal distribution, from which the original definition of class I, II, and III alleles is derived (fig. 3; Bell et al. 1981). The low diversity within a lineage is consistent with previous studies showing that most germline mutations at the INS VNTR result in small changes in repeat distribution, perhaps driven by polymerase slippage during DNA replication or repair (Stead and Jeffreys 2000). Rough estimates of mutation rates within each lineage were obtained, through Ewens's distribution, by comparing the number of different alleles in a lineage with the total number of alleles in that lineage (Ewens 1972). This showed that the estimated mutation rate increases significantly with allele size (0%–1.1% per gamete [fig. 4]), as might be expected for mutation by polymerase slippage. Interestingly, >80% of repeats in every lineage contained a potentially mutagenic polymerase-α arrest site (TGRRGA) (Krawczak and Cooper 1991; Todorova and Danieli 1997; Templeton et al. 2000).
Figure 3.

Lineage size and frequency distributions. Relative frequencies and size distribution of the 22 lineages defined by MVR-PCR are presented for each of the six populations analyzed.
Figure 4.

Relationship between lineage size and estimated mutation rate. Mutation rates were estimated from the combined African data by use of Ewens's distribution (Ewens 1972) for lineages that each contained at least five alleles and at least one instance of duplicate examples of the same allele. Lineages IIIA (∼150 repeats) and Y (∼450 repeats) were thus excluded. The positive correlation between estimated mutation rate and allele size is significant (r=0.77, P<.001).
The INS VNTR mutates not only by simple gain and loss of small numbers of repeats but also by rarer meiotic recombination events that can result in major restructuring of alleles (Stead and Jeffreys 2000). However, even the most complex mutants can be aligned to the progenitor alleles along at least part of their lengths (Stead and Jeffreys 2000). In contrast, it is generally impossible to align any of the 22 different lineages to each other, suggesting that they have diverged through multiple mutations, probably including complex rearrangements. This indicates a deep and ancient divergence between different minisatellite lineages. The apparent lack of any structural intermediates between the 22 lineages suggests that even within Africans there has been a substantial loss of genetic diversity, consistent with other studies which suggest the existence of a population bottleneck within the ancestral African population (Li and Sadler 1991; Crouau-Roy et al. 1996; Kaessmann et al. 1999).
Comparisons of Humans and Nonhuman Primates at the Minisatellite
In an attempt to identify which lineages within extant human populations are closest to the ancestral state, we analyzed INS VNTR alleles in one chimpanzee, one gorilla, and one orangutan (fig. 5A). Each great ape was homozygous, with allele lengths of 19, 11, and 3 repeats, respectively, indicating a trend toward an increase in allele size with phylogenetic proximity to humans. The minisatellite is probably polymorphic in chimpanzees, since the allele analyzed here has a different structure from a previously published allele (Seino et al. 1992) (fig. 5B). None of the ape alleles bore any structural similarity to any of the human lineages, preventing the identification of the most ancestral lineage. As noted elsewhere (Seino et al. 1992), the most common variant repeat in humans (A) is absent from each of the primates. Indeed, only the B- and C-variant repeats were found in apes, with the orangutan showing only B-type repeats.
Figure 5.

The INS VNTR in primates. A, MVR-PCR–typed alleles from one chimpanzee, one gorilla, and one orangutan (as in fig. 1). B, A single chimpanzee allele characterized elsewhere, using sequencing (Seino et al. 1992), with the sequence converted in silico into a “pseudo-MVR” code. The 3′ terminal repeat is a null repeat undetectable by the PCR primers used in MVR-PCR; such terminal null repeats could exist in other great ape alleles. C, Alleles from the five human lineages containing the shortest alleles.
Discussion
Variant-repeat analysis at the INS VNTR in six human populations identified a total of 22 highly diverged lineages, including alleles ranging from 17 to 630 repeats in length. The presence of massive structural differences between lineages suggests that they have diverged through multiple mutations, probably including complex rearrangements and indicating a deep-rooted phylogeny. Furthermore, the apparent loss from extant populations of structural intermediates between lineages suggests a population bottleneck in the ancestry of all humans. Similar conclusions have come from studies on mtDNA and autosomal markers that demonstrated a marked reduction in genetic diversity within modern human populations compared with other species (Li and Sadler 1991; Morin et al. 1994; Crouau-Roy et al. 1996).
All 22 lineages were identified in African populations, whereas a small subset of only three lineages, which are shared by all populations analyzed to date, were identified in non-Africans. Although these data are consistent with a range of nuclear genetic markers—including protein polymorphisms, RFLPs, and microsatellites—that suggest a common out-of-Africa origin for all modern non-African populations (Bowcock et al. 1994; Deka et al. 1995; Jorde et al. 1995, 1997), both Fst (table 3) and AMOVA (table 4) analyses reveal the difference between Africans and non-Africans at the INS VNTR to be unusually large. Although marked differences in diversity between Africans and non-Africans have similarly been reported for mtDNA (Vigilant et al. 1991; Ingman et al. 2000), these sequences will be more prone to genetic drift because of the smaller effective population sizes of uniparentally inherited loci (Fay and Wu 1999). One explanation for this African/non-African divergence is that minisatellite lineage data are not directly comparable with data from other polymorphisms because of differences in the rates and mechanisms of mutation, as well as differences in how an allele or lineage is defined. Nevertheless, two other studies that analyzed minisatellites by MVR-PCR in different populations did not find such a striking difference in lineage diversity. Minisatellite MS205 showed a less-than-threefold difference in lineage diversity between Africans and non-Africans, whereas analysis of the HRAS1 minisatellite in populations from Iberia and southeastern Africa identified five lineages, only one of which was restricted to Africa (Vega et al. 2001), very different from the 22-to-3 reduction in lineage diversity seen at the INS VNTR. This major decrease in insulin-lineage diversity shared by all non-African populations implies that an unusually intense population bottleneck in their common founding population has operated on this genomic region.
In contrast with the massive divergence between Africans and non-Africans, all three African populations are remarkably similar (table 3). Low divergence between populations from Kenya and Zimbabwe is perhaps unsurprising, given that both populations would have been affected by the Bantu expansion occurring within the last 2,500 years (Cavalli-Sforza et al. 1994). However, the population from the Ivory Coast should not have been affected by this event, so the reason for the similarity between all three populations remains unclear.
Curiously, although the level of lineage diversity is massively reduced in non-Africans, they still retain three of the four African lineage I sublineages (IC, ID+, ID−, and IE) (fig. 2). These sublineages are unlikely to have arisen independently in different populations, because they are each defined by variation both at the minisatellite and in the flanking haplotype, and this variation is shared across all populations (authors' unpublished data). Computer simulations with the use of present African lineage frequencies show that it is unlikely that a bottleneck would purge all lineages except I, IIIA, and IIIB, yet retain three of the four lineage I sublineages. Thus, 0.17% of simulations in which diversity was reduced to three lineages yielded a derived population containing only lineages I, IIIA, and IIIB, as seen in non-Africans; this low likelihood simply reflects the low frequency of lineages IIIA and IIIB in Africans (table 2). Of these successful simulations, only 3% retained at least three of the four lineage I sublineages; these simulations resulted from the sampling of only five to eight chromosomes (P>.95), again pointing to an intense bottleneck. Different lineage I chromosomes therefore appear to be significantly overrepresented in non-Africans and could reflect positive selection for lineage I chromosomes acting specifically in ancestral or extant non-African populations. It is therefore interesting that a recent study of three non-African populations identified transmission-ratio distortion at the INS VNTR, with lineage I chromosomes being overtransmitted to offspring at a frequency of 0.54 (Eaves et al. 1999). Similar studies have not been conducted in African populations.
These findings have important implications for disease-association studies. Most analyses of the INS VNTR have focused on populations of European descent (Bennett and Todd 1996; Waterworth et al. 1997; Dunger et al. 1998; Huxtable et al. 2000). Although different associations have been described for each of the three non-African INS VNTR lineages (Bennett et al. 1995), low lineage diversity and tight linkage disequilibrium surrounding the minisatellite in these populations have prevented the basis of these associations from being fully resolved (Doria et al. 1996; Stead et al. 2000). The present study indicates that, although similar problems could arise when analyzing any non-African population, increased diversity in Africans (both at the minisatellite and in the flanking haplotype) could help to distinguish between putative etiological variants. However, because of this diversity, future disease-association studies in Africans would require analysis of very large cohorts that could not be readily typed by MVR-PCR because of the cost, labor intensiveness, and technical difficulty of the technique. Furthermore, the frequencies of many African lineages would be too low for lineage-specific associations to be detected. Different phylogenetically related lineages would therefore need to be pooled prior to association analysis. Although the occasional common short motif can be detected between some structurally diverged lineages (data not shown), this sharing of motifs might not necessarily reflect lineage relationships but, instead, could simply be the result both of mutational convergence and of recombination between lineages which can be elevated at minisatellites (Jeffreys et al. 1998). The only obvious criterion for pooling lineages is therefore allele size, which could lead to highly divergent lineages being erroneously combined, with attendant loss of signal from the true etiological variants.
Without structural intermediates between minisatellite lineages, the number of mutational steps separating them cannot be determined from MVR data, preventing the estimation of phylogenetic relationships between lineages or of their divergence times. However, low diversity within each minisatellite lineage implies a monophyletic origin, whereas major structural differences between lineages suggests a deep divergence. If this is the case, then SNPs should have accumulated in the DNA flanking the minisatellite in each lineage and, in the absence of recombination, should have remained associated with the minisatellite lineage. Such lineage-restricted SNPs, which have already been identified for the three U.K. lineages (Bennett et al. 1995; Stead et al. 2000), would not only serve as easily typed surrogate markers of minisatellite lineage but could also be used to confirm monophyly within a lineage and to reconstruct phylogenetic relationships between lineages. This would provide a framework for a cladistic, high-throughput approach to disease-association studies within diverse population cohorts. Such lineage-restricted SNPs are currently under investigation.
Acknowledgments
We are very grateful to J. Clegg and Y. Dubrova, for kindly providing DNA samples; to M. Hurles, M. Jobling, and colleagues, for invaluable discussions; and to two anonymous reviewers, for helpful comments on the manuscript. This work was supported by Wellcome Trust grant 061869/Z/00/Z and by a Royal Society grant (both to A.J.J.).
Electronic-Database Information
The accession number and URLs for data presented herein are as follows:
- Variant Repeat Mapping at the Insulin Minisatellite, http://www.leicester.ac.uk/genetics/ajj/insulin/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for INS VNTR [MIM 125852])
References
- Alonso S, Armour JA (2001) A highly variable segment of human subterminal 16p reveals a history of population growth for modern humans outside Africa. Proc Natl Acad Sci USA 98:864–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour JA, Anttinen T, May CA, Vega EE, Sajantila A, Kidd JR, Kidd KK, Bertranpetit J, Paabo S, Jeffreys AJ (1996) Minisatellite diversity supports a recent African origin for modern humans. Nat Genet 13:154–160 [DOI] [PubMed] [Google Scholar]
- Barbujani G, Magagni A, Minch E, Cavalli-Sforza LL (1997) An apportionment of human DNA diversity. Proc Natl Acad Sci USA 94:4516–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GI, Karam JH, Rutter WJ (1981) Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc Natl Acad Sci USA 78:5759–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F, Nerup J, Bouzekri N, Cambon-Thomsen A, Ronningen KS, Barnett AH, Bain SC, Todd JA (1995) Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet 9:284–292 [DOI] [PubMed] [Google Scholar]
- Bennett ST, Todd JA (1996) Human type 1 diabetes and the insulin gene: principles of mapping polygenes. Annu Rev Genet 30:343–370 [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL (1994) High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457 [DOI] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325:31–36 [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A (1994) History and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]
- Chang F, Kidd JR, Livak KJ, Pakstis AJ, Kidd KK (1995) The world-wide distribution of allele frequencies at the human dopamine D4 receptor locus. Hum Genet 98:91–101 [DOI] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, Sing CF (1998) Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouau-Roy B, Service S, Slatkin M, Freimer N (1996) A fine-scale comparison of the human and chimpanzee genomes: linkage, linkage disequilibrium and sequence analysis. Hum Mol Genet 5:1131–1137 [DOI] [PubMed] [Google Scholar]
- Deka R, Shriver MD, Yu LM, Ferrell RE, Chakraborty R (1995) Intra- and inter-population diversity at short tandem repeat loci in diverse populations of the world. Electrophoresis 16:1659–1664 [DOI] [PubMed] [Google Scholar]
- Doria A, Lee J, Warram JH, Krolewski AS (1996) Diabetes susceptibility at IDDM2 cannot be positively mapped to the VNTR locus of the insulin gene. Diabetologia 39:594–599 [DOI] [PubMed] [Google Scholar]
- Dunger DB, Ong KK, Huxtable SJ, Sherriff A, Woods KA, Ahmed ML, Golding J, Pembrey ME, Ring S, Bennett ST, Todd JA (1998) Association of the INS VNTR with size at birth. Nat Genet 19:98–100 [DOI] [PubMed] [Google Scholar]
- Eaves IA, Bennett ST, Forster P, Ferber KM, Ehrmann D, Wilson AJ, Bhattacharyya S, Ziegler AG, Brinkmann B, Todd JA (1999) Transmission ratio distortion at the INS-IGF2 VNTR. Nat Genet 22:324–325 [DOI] [PubMed] [Google Scholar]
- Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Popul Biol 3:87–112 [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu C-I (1999) A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol 16:1003–1005 [DOI] [PubMed] [Google Scholar]
- Feldman MW, Bergman A, Pollock DD, Goldstein DB (1997) Microsatellite genetic distances with range constraints: analytic description and problems of estimation. Genetics 145:207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Bond J, Schneider JA, Hamilton B, Harding RM, Boyce AJ, Clegg JB (2000) Polymorphism and divergence in the beta-globin replication origin initiation region. Mol Biol Evol 17:179–188 [DOI] [PubMed] [Google Scholar]
- Gilad Y, Rosenberg S, Przeworski M, Lancet D, Skorecki K (2002) Evidence for positive selection and population structure at the human MAO-A gene. Proc Natl Acad Sci USA 99:862–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Poulter M, Zvarik M, Ferak V, Krause A, Jenkins T, Saha N, Kozlov AI, Swallow DM (2001) Lactase haplotype diversity in the Old World. Am J Hum Genet 68:160–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable SJ, Saker PJ, Haddad L, Walker M, Frayling TM, Levy JC, Hitman GA, O'Rahilly S, Hattersley AT, McCarthy MI (2000) Analysis of parent-offspring trios provides evidence for linkage and association between the insulin gene and type 2 diabetes mediated exclusively through paternally transmitted class III variable number tandem repeat alleles. Diabetes 49:126–130 [DOI] [PubMed] [Google Scholar]
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408:708–713 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, MacLeod A, Tamaki K, Neil DL, Monckton DG (1991) Minisatellite repeat coding as a digital approach to DNA typing. Nature 354:204–209 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neil DL, Neumann R (1998) Repeat instability at human minisatellites arising from meiotic recombination. EMBO J 17:4147–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R, Wilson V (1990) Repeat unit sequence variation in minisatellites: a novel source of DNA polymorphism for studying variation and mutation by single molecule analysis. Cell 60:473–485 [DOI] [PubMed] [Google Scholar]
- Jorde LB, Bamshad M, Rogers AR (1998) Using mitochondrial and nuclear DNA markers to reconstruct human evolution. Bioessays 20:126–136 [DOI] [PubMed] [Google Scholar]
- Jorde LB, Bamshad MJ, Watkins WS, Zenger R, Fraley AE, Krakowiak PA, Carpenter KD, Soodyall H, Jenkins T, Rogers AR (1995) Origins and affinities of modern humans: a comparison of mitochondrial and nuclear genetic data. Am J Hum Genet 57:523–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Rogers AR, Bamshad M, Watkins WS, Krakowiak P, Sung S, Kere J, Harpending HC (1997) Microsatellite diversity and the demographic history of modern humans. Proc Natl Acad Sci USA 94:3100–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ, Dixon ME, Ricker CE, Seielstad MT, Batzer MA (2000) The distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet 66:979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julier C, Hyer RN, Davies J, Merlin F, Soularue P, Briant L, Cathelineau G, Deschamps I, Rotter JI, Froguel P, Boitard C, Bell JI, Lathrop GM (1991) Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature 354:155–159 [DOI] [PubMed] [Google Scholar]
- Kaessmann H, Wiebe V, Paabo S (1999) Extensive nuclear DNA sequence diversity among chimpanzees. Science 286:1159–1162 [DOI] [PubMed] [Google Scholar]
- Krawczak M, Cooper DN (1991) Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet 86:425–441 [DOI] [PubMed] [Google Scholar]
- Le Stunff C, Fallin D, Bougneres P (2001) Paternal transmission of the very common class I INS VNTR alleles predisposes to childhood obesity. Nat Genet 29:96–99 [DOI] [PubMed] [Google Scholar]
- Li WH, Sadler LA (1991) Low nucleotide diversity in man. Genetics 129:513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu E, Calafell F, Lao O, Bonne-Tamir B, Kidd JR, Pakstis A, Kidd KK, Bertranpetit J (2001) Worldwide genetic analysis of the CFTR region. Am J Hum Genet 68:103–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PA, Moore JJ, Chakraborty R, Jin L, Goodall J, Woodruff DS (1994) Kin selection, social structure, gene flow, and the evolution of chimpanzees. Science 265:1193–1201 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Jorde LB, Ishigami T, Umemura S, Emi M, Lalouel JM, Inoue I (2002) Nucleotide diversity and haplotype structure of the human angiotensinogen gene in two populations. Am J Hum Genet 70:108–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauta MJ, Weissing FJ (1996) Constraints on allele size at microsatellite loci: implications for genetic differentiation. Genetics 143:1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York [Google Scholar]
- Relethford JH, Jorde LB (1999) Genetic evidence for larger African population size during recent human evolution. Am J Phys Anthropol 108:251–260 [DOI] [PubMed] [Google Scholar]
- Schneider S, Roessli D, Excoffier L (2000) Arlequin. Genetics and Biometry Laboratory, Geneva, University of Geneva [Google Scholar]
- Seielstad M, Bekele E, Ibrahim M, Toure A, Traore M (1999) A view of modern human origins from Y chromosome microsatellite variation. Genome Res 9:558–567 [PMC free article] [PubMed] [Google Scholar]
- Seino S, Bell GI, Li WH (1992) Sequences of primate insulin genes support the hypothesis of a slower rate of molecular evolution in humans and apes than in monkeys. Mol Biol Evol 9:193–203 [DOI] [PubMed] [Google Scholar]
- Stead JD, Jeffreys AJ (2000) Allele diversity and germline mutation at the insulin minisatellite. Hum Mol Genet 9:713–723 [DOI] [PubMed] [Google Scholar]
- Stead JD, Buard J, Todd JA, Jeffreys AJ (2000) Influence of allele lineage on the role of the insulin minisatellite in susceptibility to type 1 diabetes. Hum Mol Genet 9:2929–2935 [DOI] [PubMed] [Google Scholar]
- Templeton AR (2002) Out of Africa again and again. Nature 416:45–51 [DOI] [PubMed] [Google Scholar]
- Templeton AR, Clark AG, Weiss KM, Nickerson DA, Boerwinkle E, Sing CF (2000) Recombinational and mutational hotspots within the human lipoprotein lipase gene. Am J Hum Genet 66:69–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova A, Danieli GA (1997) Large majority of single-nucleotide mutations along the dystrophin gene can be explained by more than one mechanism of mutagenesis. Hum Mutat 9:537–547 [DOI] [PubMed] [Google Scholar]
- Vega A, Salas A, Costas J, Barros F, Carracedo A (2001) Length variability and interspersion patterns of the HRAS1 minisatellite: a new approach for the reconstruction of human population relationships. Ann Hum Genet 65:351–361 [DOI] [PubMed] [Google Scholar]
- Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC (1991) African populations and the evolution of human mitochondrial DNA. Science 253:1503–1507 [DOI] [PubMed] [Google Scholar]
- Waterworth DM, Bennett ST, Gharani N, McCarthy MI, Hague S, Batty S, Conway GS, White D, Todd JA, Franks S, Williamson R (1997) Linkage and association of insulin gene VNTR regulatory polymorphism with polycystic ovary syndrome. Lancet 349:986–990 [DOI] [PubMed] [Google Scholar]