

Abstract
Fetal events and obstetric complications are associated with schizophrenia. Here we report the results of a family-based candidate-gene study that assesses the role of maternal-fetal genotype incompatibility at the RHD locus in schizophrenia. We adapted the case-parent–trio log-linear modeling approach to test for RHD maternal-fetal genotype incompatibility and to distinguish this effect from a high-risk allele at or near the RHD locus and from a direct maternal effect alone. Eighty-eight patient-parent trios, 72 patient-mother pairs, and 21 patient-father pairs were genotyped at the RHD locus. Of the 181 patients, 62% were male and 81% were second born or later. Only three patients were born after prophylaxis against maternal isoimmunization had become common practice. There was significant evidence for an RHD maternal-fetal genotype incompatibility, and the incompatibility parameter was estimated at 2.6. There was no evidence to support linkage/association with schizophrenia at or near the RHD locus nor any evidence to support the role of maternal genotype effect alone. Our results replicate previous findings that implicate the RHD locus in schizophrenia, and the candidate-gene design of this study allows the elimination of alternative explanations for the role of this locus in disease. Thus, the present study provides increasing evidence that the RHD locus increases schizophrenia risk through a maternal-fetal genotype incompatibility mechanism that increases risk of an adverse prenatal environment (e.g., Rh incompatibility) rather than through linkage/association with the disorder, linkage disequilibrium with an unknown nearby susceptibility locus, or a direct maternal effect alone. This is the first candidate-gene study to explicitly test for and provide evidence of a maternal-fetal genotype incompatibility mechanism in schizophrenia.
Introduction
Schizophrenia is a neuropsychiatric disorder that occurs in .5%–1% of the population worldwide. It is characterized by a constellation of features, including delusions, hallucinations, disorganized speech and behavior, flattened affect, and inability to initiate and persist in goal-directed activities (American Psychiatric Association 1994). It is a debilitating chronic disorder with age at onset typically between the late teens and mid-30s (American Psychiatric Association 1994).
There is compelling evidence that both genetic and environmental factors are involved in the etiology of schizophrenia. Furthermore, there is growing evidence that the environmental risk factors for schizophrenia include processes involving the prenatal environment (Cannon 1997; Geddes et al. 1999; Cannon et al. 2000; Cantor-Graae et al. 2000; Hulshoff et al. 2000; Lewis and Lieberman 2000; Cannon et al. 2002). Unfortunately, the causes for prenatal complications are quite heterogeneous and difficult to document reliably, making it difficult to test the role of prenatal environmental insults in the pathogenesis of schizophrenia. Furthermore, although some prenatal environmental insults may have a genetic basis, the mechanism by which the genes act may not appear to follow Mendelian inheritance patterns, creating problems in the identification of susceptibility loci when traditional genetic analyses are used.
Rh incompatibility, a phenotype that exhibits non-Mendelian inheritance, has been implicated as a risk factor for schizophrenia in several studies employing different designs and samples (Hollister et al. 1996; Cannon et al. 2002). Three population studies that investigated the association between Rh incompatibility and schizophrenia were included in a recent meta-analysis (Cannon et al. 2002), yielding an overall significant effect and an estimated overall odds ratio of 2.0. Additional confirmation is provided by a case-control study conducted in a large Danish sample of individuals born between 1959 and 1961 (Hollister et al. 1996). In that study, the proportion of Rh-incompatible male offspring (2.1%) was significantly larger than the proportion of schizophrenics in a cohort of Rh-compatible male offspring (0.8%), yielding a relative risk of 2.78. In addition, in a review of previous research on Rh hemolytic disease of the newborn (HDN) and schizophrenia (Hollister and Kohler 2001) the results of a recent Finnish study (unpublished as yet) are summarized in which the rate of schizophrenia was significantly higher among individuals who survived Rh HDN than among individuals who were not exposed to this obstetric complication (odds ratio 2.0). In all of these studies, Rh phenotype information (Rh positive and Rh negative) from labor, delivery, and birth records formed the basis of analysis. When phenotype information and these study designs are used, however, it is not possible to clarify whether the effect of the RHD locus (MIM 111680) results from a maternal-fetal genotype incompatibility, as we hypothesize; from linkage and association (LD) with a high-risk susceptibility allele at or near the RHD locus; or from the effects of the maternal genotype acting alone.
Rh incompatibility is the result of a maternal-fetal genotype incompatibility at the RHD locus, in which the mother is Rh negative (d/d) and the fetus is Rh positive (D/d). One reason that Rh incompatibility may be harmful to the fetus is that it may lead to hypoxia, an obstetric complication found to be associated with schizophrenia (Cannon et al. 1993, 2000; Cannon 1997; Zornberg et al. 2000; Dalman et al. 2001). Rh incompatibility also can lead to an increase in unconjugated bilirubin, a neurotoxin (Hansen 2000, 2001) to which undifferentiated glial cells are sensitive (Amit and Brenner 1993; Rhine et al. 1999). Glial cell abnormalities also have been associated with schizophrenia (Cotter et al. 2001; Moises et al. 2002). Thus, one biological mechanism that can create an adverse prenatal environment is caused by a maternal-fetal genotype combination that adversely affects the developing fetus by inducing a maternal immunological attack, which then increases susceptibility to schizophrenia. This mechanism, which we refer to as a “maternal-fetal genotype incompatibility,” is consistent with the teratogenic antibody hypothesis of disease, which posits that a pregnant female can develop antibodies in response to some antibody-producing stimulus (e.g., contact with paternal antigens) that can interfere with normal fetal neurodevelopment and increase susceptibility to disease, such as schizophrenia (Laing et al. 1995). Of particular importance is the fact that maternal-fetal genotype incompatibilities that initiate adverse prenatal events (at RHD or at other relevant loci), have the potential to be detected in genetic analyses of existing data sets, even years after the adverse event has occurred.
We report the results of a family-based candidate-gene study that assesses the role of maternal-fetal genotype incompatibility at the RHD locus in schizophrenia. We adapted the case-parent–trio log-linear modeling approach (Weinberg et al. 1998; Wilcox et al. 1998) to develop a maternal-fetal genotype incompatibility (MFG) test (Sinsheimer et al., in press), which is sensitive to the effects of RHD maternal-fetal genotype incompatibility and can distinguish these effects from a high-risk allele at or near the RHD locus in the affected child’s genotype or the maternal genotype.
Material and Methods
Subjects
Subjects were ascertained from a larger well-described and characterized Finnish schizophrenia sample (Hovatta et al. 1999; Ekelund et al. 2000, 2001; Paunio et al. 2001). Probands in the larger sample were identified through nationwide health and population registers. First-degree relatives subsequently were invited to participate through permission given by the proband. The research was approved by the Ministry of Social Affairs and Health (Finland) and the appropriate institutional review boards, and informed consent was obtained from subjects. DSM-IV (American Psychiatric Association 1994) best-estimate lifetime diagnoses were made independently by two psychiatrists or psychiatric residents from all available inpatient and outpatient records for probands and their relatives. For the current study, we selected the youngest individual in each independent nuclear family with a diagnosis of schizophrenia, schizoaffective psychosis disorder, or schizophrenia spectrum disorder (i.e., paranoid personality, schizoid personality, and schizotypy) and at least one parent available for genotyping. This selection strategy was used to maximize the likelihood of detecting an RHD maternal-fetal genotype incompatibility effect, since second-born or later babies are more likely to be at risk for the immunological sequelae of RHD maternal-fetal genotype incompatibility (Guyton 1981).
The study sample of 450 Finnish individuals was composed of 88 patient-parent trios, 72 patient-mother pairs, and 21 patient-father pairs. Of the 181 patients, 112 were male (62%), 147 were second born or later (81%), 145 had received a diagnosis of schizophrenia (80%), 25 had received a diagnosis of schizoaffective psychosis disorder (14%), and 11 had received a schizophrenia spectrum diagnosis (6%). Approximately 12% of mothers and 6% of fathers fell into one of those three diagnostic categories. Patients were born during the period 1937–1973, with a median birth year of 1957. Prophylaxis against maternal isoimmunization was not available for the vast majority of the patients in the study, since only three patients were born after it had become common practice in Finland—that is, after 1969 (Eklund and Nevanlinna 1986). Of the 181 patients, 12 had the genotype D/d (Rh positive) when the maternal genotype was d/d (Rh negative) and were at risk for the sequelae of RHD maternal-fetal genotype incompatibility. An additional nine D/d patients who were missing maternal genotypes also were potentially at risk for sequelae of RHD maternal-fetal genotype incompatibility. The number of older heterozygous siblings of these 21 patients ranged from 0 to 5. There was at least one older sibling with the D/d genotype for 11 of the 12 known at-risk patients and 3 of the 9 potential at-risk patients, thereby increasing the chance that maternal isoimmunization had occurred prior to the pregnancy with these at-risk patients.
Genotyping Methods
DNA was extracted from EDTA blood, according to a standard procedure (Blin and Stafford 1976). A PCR-RFLP was used to detect the RHD genotype (Wagner and Flegel 2000). This method identifies all three genotypes (D/D, D/d, and d/d). PCR was performed using primers rez7 (5′-CCTGTCCCCATGATTCAGTTACC-3′) and rnb1 (5′-CCTTTTTTTGTTTGTTTTTGGCGGTGC-3′) and the expand high-fidelity PCR system (DyNAzyme EXT DNA Polymerase). Annealing was at 65°C and extension was for 3 min at 72°C. PCR products were digested with PstI for 3 h at 37°C, and fragments were visualized using a 1.5% agarose gel. Genotyping was conducted blind to diagnostic status. Genotype data were checked for errors through use of PEDCHECK (O'Connell and Weeks 1998) and MENDEL 4.0 (Douglas et al. 2000; Lange 2002; Sobel et al. 2002). Of the 269 genotyped parents, 10.4% were d/d, 44.2% were D/d, and 45.3% were D/D. Thus, the frequency of the D allele was .67 and the frequency of the d allele was .33.
Four microsatellite markers flanking the RHD locus (D1S368 [17.1 cM from RHD], D1S552 [6.9 cM from RHD], D1S1622 [5.1 cM from RHD], and D1S513 [8.5 cM from RHD]) were obtained and genotyped as described elsewhere (Ekelund et al. 2001).
Statistical Analyses
Tests of (i) RHD maternal-fetal genotype incompatibility, (ii) a high-risk allele at or near the RHD locus in the affected child’s genotype, or (iii) a high-risk allele at or near the RHD locus in the maternal genotype were performed using the MFG test (Sinsheimer et al., in press), an adaptation of the log-linear model for case-parent trios (Weinberg et al. 1998; Wilcox et al. 1998; Umbach and Weinberg 2000). The direct effect of the affected child’s genotype (or maternal genotype) and the effect of maternal-fetal genotype incompatibility are modeled jointly in the MFG test, to remove potential confounding of these effects. The joint model is essential to rule out the existence of any susceptibility loci of minor effects that may be in LD with a putative incompatibility locus or to rule out the effect of maternal genotype alone on the fetal environment.
Because the RHD locus is biallelic, each genotyped patient-parent trio falls into one of 15 possible categories (see table 1). As in the study by Weinberg et al. (1998), we assume that mating-type frequencies are symmetric; thus, the 15 trio combinations in table 1 correspond to six different mating types. No assumption of Hardy-Weinberg equilibrium is made. Under a model where disease risk is increased under maternal-fetal genotype incompatibility or because of a high-risk allele at or near the RHD locus in the affected child’s genotype, the expected frequencies of the genotyped trio categories depend on the mating type parameters, γj (j=1,…,6), the relative risk of disease to a child carrying two d alleles, R2; the relative risk of disease to a child carrying one d allele, R1; and the relative risk of disease to a child due to a maternal-fetal genotype incompatibility at the RHD locus, M (see table 1). If there is an incompatibility effect, one would expect to see a greater number of incompatible patient-parent trios than their counterpart compatible trios within a mating type. For hypotheses about the RHD maternal-fetal genotype incompatibility parameter, the number in cell 7 should be greater than that in cell 6, and the number in cell 13 should be greater than that in cell 11 (see table 1).
Table 1.
Description of Trio Categories, Expected Values, and Distribution of Data Under the Model with RHD Maternal-Fetal Genotype Incompatibility and LD[Note]
|
Genotype |
|||||
| CellNumber | Maternal | Paternal | AffectedChild | Expected Valuea | Nb |
| 1 | D/D | D/D | D/D | R2γ1 | 44.3 |
| 2 | D/D | D/d | D/D | R2γ2 | 16.0 |
| 3 | D/D | D/d | D/d | R1γ2 | 16.7 |
| 4 | D/d | D/D | D/D | R2γ2 | 15.6 |
| 5 | D/d | D/D | D/d | R1γ2 | 16.8 |
| 6 | D/D |
d/d |
D/d |
R1γ3 |
3.2 |
| 7 | d/d |
D/D |
D/d |
MR1γ3 |
7.8 |
| 8 | D/d | D/d | D/D | R2γ4 | 13.0 |
| 9 | D/d | D/d | D/d | 2R1γ4 | 15.4 |
| 10 | D/d | D/d | d/d | γ4 | 12.1 |
| 11 | D/d |
d/d |
D/d |
R1γ5 |
2.7 |
| 12 | D/d | d/d | d/d | γ5 | 3.7 |
| 13 | d/d |
D/d |
D/d |
MR1γ5 |
6.4 |
| 14 | d/d | D/d | d/d | γ5 | 2.4 |
| 15 | d/d | d/d | d/d | γ6 | 4.8 |
Note.— Trio categories at risk for incompatibility and their counterpart compatible trio categories are underlined.
M is the relative risk to a child due to a maternal-fetal genotype incompatibility at the RHD locus, R1 and R2 are the relative risk of disease to a child carrying one and two d alleles, respectively, and γj (j=1,…,6) are the mating-type parameters.
Number of trios based on the final iteration of the expectation step from the EM algorithm (Weinberg 1999), using data on 88 patient-parent trios, 72 patient-mother pairs, and 21 patient-father pairs.
Under this scenario and under the assumption of Poisson-distributed counts, the natural logarithm of the expected number in the ith trio combination (i=1,…,15) can be summarized as
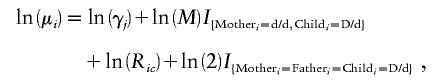 |
where Ric denotes the relative risk of schizophrenia given that the child in combination i has c=0, 1, or 2 copies of the d allele (where R0=1), and I is the indicator function. When associated with ln(M), the indicator variable takes on a value of 1 for the maternal-fetal genotype combination in which the mother is d/d (Rh negative) and the child is D/d (Rh positive), and a value of 0 otherwise. When associated with the constant ln(2), the indicator variable takes on a value of 1 when mother, father, and child are all heterozygous, and a value of 0 otherwise.
Under a model in which disease risk is increased under maternal-fetal genotype incompatibility or because of a high-risk allele at or near the RHD locus in the maternal genotype, the expected frequencies of the trio categories depend on these same mating type parameters, γj (j=1,…,6); the relative risk of disease to a child whose mother has two d alleles, S2; the relative risk of disease to a child whose mother has one d allele, S1; and the relative risk of disease, to a child, that is due to a maternal-fetal genotype incompatibility at the RHD locus, M.
Under this scenario, the natural logarithm of the expected number in the ith trio combination (i=1,…,15) can be summarized as
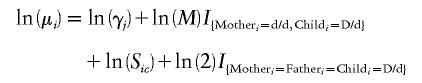 |
In this case, Sic denotes the relative risk of schizophrenia to the child, given that the mother in combination i has c=0, 1, or 2 copies of the d allele (where S0=1).
To use patient-mother and patient-father pairs, the observed log likelihood must incorporate incomplete data—for example, trios missing one parental genotype. The observed log likelihood is analogous to the one presented by Weinberg (1999). A test of the parameter(s) of interest is performed by computing a likelihood-ratio test statistic (LRT) from the log likelihoods obtained by maximizing the fit of the data under specified null and alternative models. The observed data log likelihood can be maximized directly through use of a recursive quadratic programming routine (Lange 1991) or an expectation-maximization (EM) algorithm (Weinberg 1999). Both approaches treat the partial information from incompletely genotyped trios in an unbiased manner and yield the same parameter estimates and observed data log-likelihood maxima. In addition to obtaining parameter estimates from the EM algorithm, we also calculated the number of patient-parent trios that are expected to fall into each of the 15 categories from the final expectation step. We calculated the asymptotic SEs for the parameters through use of the information matrix obtained from the direct maximization of the observed data log likelihood. The LRT is asymptotically distributed as χ2. When we test for the incompatibility effect in the possible presence of LD, the LRT has 1 df. When we test for LD effects in the possible presence of an incompatibility effect (or for maternal effects in the possible presence of an incompatibility effect), the LRT has 2 df. Analyses based on the MFG test were performed using MISMATCH (Sinsheimer 2002).
Tests of linkage and association at flanking microsatellite markers were performed using an extension of the transmission/disequilibrium test (TDT) (Lazzeroni and Lange 1998) in MENDEL 4.0 (Lange 2002). These analyses included 155 affected siblings of the 181 patients.
Results
We tested the primary hypothesis of an RHD maternal-fetal genotype incompatibility effect in the possible presence of LD by comparing the unconstrained model in which ln(M) and the two LD parameters, ln(R1) and ln(R2), are estimated, to a constrained model in which ln(R1) and ln(R2) are estimated but ln(M) is constrained to its null value (0). This comparison produced significant evidence for an RHD maternal-fetal genotype incompatibility (χ2=3.72; 1 df; [one-sided] P=.027). In this analysis, a one-sided test is used, because we are replicating a number of previous findings that suggest that an RHD maternal-fetal genotype incompatibility increases risk to the developing fetus and it is biologically implausible that RHD maternal-fetal genotype incompatibility would decrease the risk of schizophrenia.
Estimates of the three parameters of the unconstrained model were obtained by maximizing the log likelihood. Table 1 provides the number of patient-parent trios falling into the 15 categories on which these estimates are based. There are fractional numbers because patient-mother–only and patient-father–only data are probabilistically apportioned across several trio categories in an unbiased manner. The estimate of ln(M) is 0.96 (SE=0.51; 90% CI 0.12–1.80), ln(R1) is −0.13 (SE=0.13; 90% CI −0.34–0.08), and ln(R2) is 0.22 (SE=0.37; 90% CI −0.39–0.83). For ease of interpretation, we can consider M, R1, and R2, which have values of 2.6, 0.87, and 1.25, respectively, rather than ln(M), ln(R1), and ln(R2). Looking at table 1, it is apparent that the number of trios falling into the RHD maternal-fetal genotype incompatible categories (cells 7 and 13) is larger than the number of trios falling into their compatible counterparts (cells 6 and 11). Furthermore, the maximum-likelihood estimate of M can be understood intuitively by forming the ratio of the sum of the two incompatible categories to the sum of their compatible counterparts; this produces a value of 2.4, which is consistent with the maximum-likelihood estimate.
To rule out the presence of a high-risk allele at or near the RHD locus in the affected child’s genotype, we then conducted two different analyses. First, we used the log-linear case-parent–trio model and compared the unconstrained three-parameter model (ln[M], ln[R1], ln[R2]) to a model in which ln(M) is estimated and ln(R1) and ln(R2) are constrained to 0 (null value). There was no evidence to support linkage/association with schizophrenia at or near the RHD locus (χ2=1.1; 2 df; [two-sided] P=.58). Because there is no biological justification for specifying the direction of an effect for the linkage/association parameters under the alternative hypothesis, a two-sided level of significance is used in this analysis.
Next, we tested for linkage or association between schizophrenia and the RHD locus, as well as several markers near the RHD locus (D1S368, D1S552, RHD, D1S1622, and D1S513), using an extension of the TDT. There was no evidence for linkage or association with schizophrenia at any of these loci (all P values >.05). This is consistent with results of similar analyses performed in the entire Finnish sample (221 families, 557 affected individuals) at the four flanking microsatellite markers, which also were not significant (Ekelund et al. 2001 [and their online supplementary material]). Using formulas described by Knapp (1999), we determined that the current study sample size had >0.93 power to detect linkage at the RHD locus with λs=1.2 under dominant, recessive, additive, and multiplicative models. Together, these analyses provide further evidence that the effect demonstrated at the RHD locus represents the result of a maternal-fetal genotype incompatibility, rather than the direct effect of a high-risk allele at or near the RHD locus in the affected child’s genotype.
Finally, we assessed whether the maternal genotype alone creates an adverse prenatal environment, by testing the null hypothesis of no association between the disease and the maternal genotype at or near the RHD locus. For this analysis, we compared the model in which ln(M), ln(S1), and ln(S2) are estimated to a model in which ln(M) is estimated and ln(S1) and ln(S2) are constrained to their null value (0). This comparison produced no evidence to support that a maternal genotype effect alone is associated with schizophrenia, as measured by association with a high-risk allele at or near the RHD locus (χ2=0.56; 2 df; [two-sided] P=.76).
Discussion
These results replicate previous findings that implicate the RHD locus in schizophrenia, and the candidate-gene design of the present study allows the elimination of alternative explanations of the role of this locus in schizophrenia. Thus, this study provides additional evidence that the RHD locus increases schizophrenia risk through a maternal-fetal genotype incompatibility mechanism that increases risk of an adverse prenatal environment rather than through linkage/association with the disorder, through LD with an unknown nearby susceptibility locus, or through a direct maternal effect alone. This is the first candidate-gene study to explicitly test for and provide evidence of a maternal-fetal genotype incompatibility mechanism in schizophrenia.
The presence of an RHD maternal-fetal genotype incompatibility effect suggests that susceptibility to schizophrenia may be increased by production of maternal anti-D antibodies. This biological mechanism is consistent with the teratogenic antibody hypothesis of interference with normal fetal neurodevelopment (Laing et al. 1995). Because the effects of an RHD maternal-fetal genotype incompatibility occur during pregnancy, this biological mechanism is also consistent with repeated findings that obstetric complications occur more frequently among births of individuals with schizophrenia (Marcelis et al. 1998; Cannon et al. 2000; Zornberg et al. 2000; Dalman et al. 2001) and are associated with structural brain abnormalities in schizophrenia (McNeil et al. 2000).
The estimate of the incompatibility parameter in this study (2.6) is consistent with an odds ratio of 2.0 (Cannon et al. 2002), as well as with an estimate of the incompatibility parameter (1.8) that can be derived from the data presented by Hollister et al. (1996) and an odds ratio of 2.0 summarized in Hollister (2001). This consistency is remarkable, given differences in study design (population, case-control, and family-based studies) and population cohort (United Kingdom/Scotland, Denmark, and Finland). Such consistency across studies suggests that our result is substantively significant and that the statistical significance should be interpreted in light of the moderate sample size. It also suggests that replication studies should be designed to have sufficient power to detect an effect size of ∼2.5. Using RHD genotype frequencies as observed in the Finnish data collection, we calculate that a sample of 130 patient-parent trios would be needed to obtain 0.80 power to detect an incompatibility effect of this magnitude in a one-sided test with α=0.05.
The relative risk associated with RHD maternal-fetal genotype incompatibility acting alone is computed by multiplying the estimates of the incompatibility parameter (M=2.6) and the parameter measuring the effect of a single copy of the d allele in the affected child’s genotype (R1=.87), because both parameters contribute to the at-risk trio category. With a relative risk of 2.26, the risk of schizophrenia due to RHD maternal-fetal genotype incompatibility acting alone is rather small; and, in fact, many individuals who have an RHD genotype incompatibility with their mother do not have schizophrenia. However, because this relative risk represents an average, some individuals will be at higher risk due to an RHD maternal-fetal genotype incompatibility, and some individuals will be at lower risk. Previous research has suggested that there is an association between certain classes of obstetric complications (such as hypoxia) and familial risk for schizophrenia (Zornberg et al. 2000). Because Rh incompatibility can lead to fetal hypoxia, it is possible that an RHD maternal-fetal genotype incompatibility co-occurs with one or more susceptibility genes elsewhere in the genome, to ultimately give rise to the disorder. Stratifying samples by RHD maternal-fetal genotype incompatibility may help to identify other schizophrenia susceptibility genes.
There has been no indication from recent genome scans that a schizophrenia susceptibility locus is located in the region of chromosome 1p that contains the RHD locus (Waterwort et al. 2002). However, this should not be interpreted as lack of evidence for involvement of the RHD locus in schizophrenia. Negative genome scan results in that region are not surprising, given that those statistical methods typically evaluate for the presence of linkage or association between a high-risk allele and disease and do not adequately model the maternal-fetal genotype incompatibility mechanism. We demonstrated that, even in the current sample, there is no evidence for the direct effect of a high-risk allele at or near the RHD locus, although there is evidence for RHD maternal-fetal genotype incompatibility. Thus, “negative” genome scan results for the 1p region do not indicate a failure to replicate our results, because such tests are not sufficiently sensitive to the incompatibility mechanism to reveal the role of RHD in schizophrenia (J.S.S., C.G.S.P., and J.A.W., unpublished data).
An assumption of the MFG test is that the probability of an affected individual surviving to clinical detection does not depend on the RHD genotypes of the mother, father, and child (Umbach and Weinberg 2000). When assessing prenatal risk factors, this assumption becomes particularly relevant. In the case of the RHD locus, the magnitude of fetal loss due to an incompatibility may not be large enough to violate this assumption, because most children survive the sequelae of Rh incompatibility without prophylaxis (Guyton 1981; Chavez 1991). Even if the assumption is violated, the effect of fetal loss due to RHD maternal-fetal genotype incompatibility would be to reduce the number of observed incompatibilities in the patient-parent trios, thereby yielding a conservative test that would be less likely to reject the null hypothesis of no incompatibility.
The present study provides evidence for an RHD maternal-fetal genotype incompatibility effect in a sample drawn from a population born before the widespread use of prophylaxis. Even in the presence of prophylaxis, there are several reasons why an RHD maternal-fetal genotype incompatibility remains a risk factor for schizophrenia. First, active production of maternal anti-D antibodies still occurs in some at-risk pregnancies, either because prophylaxis is not used (Chavez 1991; Bowman 1998) or because its use is not 100% effective at preventing maternal sensitization (de Silva et al. 1985; Thornton et al. 1989). Second, anti-D antibodies produced through passive administration (i.e., prophylaxis) can cross the placenta and are measurable in fetal antibody titres (Maayan-Metzger et al. 2001) and, thus, may produce difficult-to-document transient hemolytic effects that contribute to an increased risk for schizophrenia. Future research is needed to determine the extent to which the availability of prophylaxis has reduced the relative risk of schizophrenia due to RHD maternal-fetal genotype incompatibility, before prophylaxis can be viewed as playing an important role in the prevention of some cases of schizophrenia.
Candidate-gene studies of schizophrenia typically have focused on genes involved in neurochemical pathways (Jurewicz et al. 2001; Waterwort et al. 2002), and the present study suggests that genes that relate to immunological pathways could also be involved in the development of this neuropsychiatric disease. One such immunological pathway produces fetal hypoxia and hyperbilirubinemia; therefore, other genes that could produce one or both of these conditions when present as maternal-fetal genotype incompatibilities (e.g., ABO [MIM 110300]) or when present simply as high-risk alleles in the child’s genotype (e.g., FV Leiden mutation [MIM 227400.0001]) should be investigated as candidate genes for schizophrenia. Another potential immunological pathway is the intolerance that results from maternal-fetal human leukocyte antigen similarity, whereby the mother fails to stimulate blocking antibodies that normally protect the fetus from the mother’s immune response (Stubbs et al. 1985; Van Gent et al. 1997; Ober 1998). Research directed at elucidating the effect that an RHD maternal-fetal genotype incompatibility has on fetal neurodevelopment may identify an important pathway in the development of schizophrenia and could shed light on other risk factors that share the same pathway.
Acknowledgments
The authors thank the families that participated in this research, as well as two anonymous reviewers, for helpful suggestions regarding the manuscript. This research was supported, in part, by United States Public Health Service grants MH066001 and MH59490.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- Department of Molecular Medicine, National Public Health Institute, Helsinki, Publications Online, http://www.ktl.fi/lmgo/lmgo_wwwpub.htm (for supplementary material to Ekelund et al. [2001])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RHD [MIM 111680], ABO [MIM 110300], and FV Leiden [MIM 227400.0001])
References
- American Psychiatric Association (1994) Diagnostic and statistical manual, 4th ed. American Psychiatric Association, Washington, DC [Google Scholar]
- Amit Y, Brenner T (1993) Age-dependent sensitivity of cultured rat glial cells to bilirubin toxicity. Exp Neurol 121:248–255 [DOI] [PubMed] [Google Scholar]
- Blin N, Stafford DW (1976) A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acids Res 3:2303–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman JM (1998) RhD hemolytic disease of the newborn. N Engl J Med 339:1775–1777 [DOI] [PubMed] [Google Scholar]
- Cannon M, Jones PB, Murray RM (2002) Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 159:1080–1092 [DOI] [PubMed] [Google Scholar]
- Cannon TD (1997) On the nature and mechanisms of obstetric influences in schizophrenia: a review and synthesis of epidemiological studies. Int Rev Psychiatry 9:387–397 [Google Scholar]
- Cannon TD, Mednick S, Parnas J, Schulsinger F, Praestholm J, Vestergaard A (1993) Developmental brain abnormalities in the offspring of schizophrenic mothers: I. Contributions of genetic and perinatal factors. Arch Gen Psychiatry 50:551–564 [DOI] [PubMed] [Google Scholar]
- Cannon TD, Rosso IM, Hollister JM, Bearden CE, Sanchez LE, Hadley T (2000) A prospective cohort study of genetic and perinatal influences in the etiology of schizophrenia. Schizophr Bull 26:351–366 [DOI] [PubMed] [Google Scholar]
- Cantor-Graae E, Ismail B, McNeil TF (2000) Are neurological abnormalities in schizophrenic patients and their siblings the result of perinatal trauma? Acta Psychiatr Scand 101:142–147 [DOI] [PubMed] [Google Scholar]
- Chavez GF (1991) Epidemiology of Rh hemolytic disease of the newborn in the United States. JAMA 265:3270–3274 [DOI] [PubMed] [Google Scholar]
- Cotter DR, Pariante CM, Everall IP (2001) Glial abnormalities in major psychiatric disorders: the evidence and implications. Brain Res Bull 55:585–595 [DOI] [PubMed] [Google Scholar]
- Dalman C, Hollie VT, David AS, Gentz J, Lewis G, Allebeck P (2001) Signs of asphyxia at birth and risk of schizophrenia. Br J Psychiatry 179:403–408 [DOI] [PubMed] [Google Scholar]
- de Silva M, Contreras M, Mollison PL (1985) Failure of passively administered anti-Rh to prevent secondary Rh responses. Vox Sang 48:178–180 [DOI] [PubMed] [Google Scholar]
- Douglas JA, Boehnke M, Lange K (2000) A multipoint method for detecting genotyping errors and mutations in sibling-pair linkage data. Am J Hum Genet 66:1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekelund J, Hovatta I, Parker A, Paunio T, Varilo T, Martin R, Suhonen J, Ellonen P, Chan G, Sinsheimer JS, Sobel E, Juvonen H, Arajärvi R, Partonen T, Suvisaari J, Lönnqvist J, Meyer J, Peltonen L (2001) Chromosome 1 loci in Finnish schizophrenia families. Hum Mol Genet 15:1611–1617 [DOI] [PubMed] [Google Scholar]
- Ekelund J, Lichtermann D, Hovatta I, Ellonen P, Suvisaari J, Terwilliger JD, Juvonen H, Varilo T, Arajärvi R, Kokko-Sahin ML, Lönnqvist J, Peltonen L (2000) Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet 9:1049–1057 [DOI] [PubMed] [Google Scholar]
- Eklund J, Nevanlinna HR (1986) Perinatal mortality from Rh(D) hemolytic disease in Finland, 1975-1984. Acta Obstet Gynecol Scand 65:787–789 [DOI] [PubMed] [Google Scholar]
- Geddes JR, Verdoux H, Takei N, Lawrie SM, Bovet P, Eagles JM, Heun R, McCreadie RG, McNeil TF, O'Callaghan E, Stober G, Willinger U, Murray RM (1999) Schizophrenia and complications of pregnancy and labor: an individual patient data meta-analysis. Schizophr Bull 25:413–423 [DOI] [PubMed] [Google Scholar]
- Guyton AC (1981) Textbook of medical physiology. W. B. Saunders Company, Philadelphia [Google Scholar]
- Hansen TW (2000) Bilirubin oxidation in brain. Mol Genet Metab 71:411–417 [DOI] [PubMed] [Google Scholar]
- ——— (2001) Bilirubin brain toxicity. J Perinatol 21:S48–S51 [DOI] [PubMed] [Google Scholar]
- Hollister JM, Kohler C (2001) Schizophrenia: a long-term consequence of hemolytic disease of the fetus and newborn? Int J Mental Health 29:38–61 [Google Scholar]
- Hollister JM, Laing P, Mednick SA (1996) Rhesus incompatibility as a risk factor for schizophrenia in male adults. Arch Gen Psychiatry 53:19–24 [DOI] [PubMed] [Google Scholar]
- Hovatta I, Varilo T, Suvisaari J, Terwilliger JD, Ollikainen V, Arajärvi R, Juvonen H, Kokko-Sahin M-L, Vaisanen L, Mannila H, Lonnqvist J, Peltonen L (1999) A genomewide screen for schizophrenia genes in an isolated Finnish subpopulation, suggesting multiple susceptibility loci. Am J Hum Genet 65:1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshoff HE, Hoek HW, Susser E, Brown AS, Dingemans A, Schnack HG, van Haren NEM, Ramos LMP, Gispen-de Wied CC, Kahn RS (2000) Prenatal exposure to famine and brain morphology in schizophrenia. Am J Psychiatry 157:1170–1172 [DOI] [PubMed] [Google Scholar]
- Jurewicz I, Owen RJ, O'Donovan MC, Owen MJ (2001) Searching for susceptibility genes in schizophrenia. Eur Neuropsychopharmacol 11:395–398 [DOI] [PubMed] [Google Scholar]
- Knapp M (1999) A note on power approximations for the transmission/disequilibrium test. Am J Hum Genet 64:1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing P, Knight J, Wright P, Irving W (1995) Disruption of fetal brain development by maternal antibodies as an etiological factor in schizophrenia. In: Mednick SA, Hollister JM (eds) Neural development and schizophrenia: theory and research. Plenum, New York, pp 215–245 [Google Scholar]
- Lange K (1991) SEARCH release 3.0, Los Angeles, CA [Google Scholar]
- ——— (2002) MENDEL release 4.0, Los Angeles, CA [Google Scholar]
- Lazzeroni LC, Lange K (1998) A conditional inference framework for extending the transmission/disequilibrium test. Hum Hered 48:67–81 [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA (2000) Catching up on schizophrenia: natural history and neurobiology. Neuron 28:325–334 [DOI] [PubMed] [Google Scholar]
- Maayan-Metzger A, Schwartz T, Sulkes J, Merlob P (2001) Maternal anti-D prophylaxis during pregnancy does not cause neonatal haemolysis. Arch Dis Child Fetal Neonatal Ed 84:F60–F62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelis M, van Os J, Sham P, Jones P, Gilvarry C, Cannon M, McKenzie K, Murray R (1998) Obstetric complications and familial morbid risk of psychiatric disorders. Am J Med Genet Neuropsychiatr Genet 81:29–36 [DOI] [PubMed] [Google Scholar]
- McNeil TF, Cantor-Graae E, Ismail B (2000) Obstetric complications and congenital malformation in schizophrenia. Brain Res Rev 31:166–178 [DOI] [PubMed] [Google Scholar]
- Moises HW, Zoega T, Gottesman II (2002) The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2:8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C (1998) HLA and pregnancy: the paradox of the fetal allograft. Am J Hum Genet 62:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, Rinard K, Foti A, Terwilliger JD, Juvonen H, Suvisaari J, Arajarvi R, Suokas J, Partonen T, Lonnqvist J, Meyer J, Peltonen L (2001) Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet 10:1611–1617 [DOI] [PubMed] [Google Scholar]
- Rhine WD, Schmitter SP, Yu AC, Eng LF, Stevenson DK (1999) Bilirubin toxicity and differentiation of cultured astrocytes. J Perinatol 19:206–211 [DOI] [PubMed] [Google Scholar]
- Sinsheimer JS (2002) MISMATCH release 1.0, Los Angeles, CA [Google Scholar]
- Sinsheimer JS, Palmer CGS, Woodward JA. The maternal-fetal genotype incompatibility test: detecting genotype combinations that increase risk for disease. Genet Epidemiol (in press) [DOI] [PubMed] [Google Scholar]
- Sobel E, Papp JC, Lange K (2002) Genotype error detection and integration. Am J Hum Genet 70:496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs EG, Ritvo ER, Mason-Brothers A (1985) Autism and shared parental HLA antigens. J Am Acad Child Psychiatry 24:182–185 [DOI] [PubMed] [Google Scholar]
- Thornton JG, Page C, Foote G, Arthur GR, Tovey LAD, Scott JS (1989) Efficacy and long term effects of antenatal prophylaxis with anti-D immunoglobulin. Br Med J 298:1671–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach DM, Weinberg CR (2000) The use of case-parent trials to study joint effects of genotype and exposure. Am J Hum Genet 66:251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gent T, Heijnen CJ, Treffers PDA (1997) Autism and the immune system. J Child Psychol Psychiatry 38:337–349 [DOI] [PubMed] [Google Scholar]
- Wagner FF, Flegel WA (2000) RHD gene deletions occurred in the Rhesus box. Blood 95:3662–3668 [PubMed] [Google Scholar]
- Waterwort DM, Bassett AS, Brzustowicz LM (2002) Recent advances in the genetics of schizophrenia. Cell Mol Life Sci 59:331–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg CR (1999) Allowing for missing parents in genetic studies of case-parent triads. Am J Hum Genet 64:1186–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT (1998) A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet 62:969–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Lie RT (1998) Distinguishing the effects of maternal and offspring genes through studies of “case-parent triads.” Am J Epidemiol 148:893–901 [DOI] [PubMed] [Google Scholar]
- Zornberg GL, Buka SL, Tsuang MT (2000) At issue: the problem of obstetrical complications and schizophrenia. Schizophr Bull 26:249–256 [DOI] [PubMed] [Google Scholar]