

Significance
Protein–protein interactions are attractive targets for interfering with processes leading to disease states. Proteins often use folded domains or secondary structures to contact partner proteins. Synthetic molecules that mimic these domains could disrupt protein–protein contacts, thereby inhibiting formation of multiprotein complexes. This article describes protein domain mimetics (PDMs) that modulate interactions between two proteins that control expression of a multitude of genes under hypoxic environments, such as those found inside tumors. The low-oxygen conditions promote angiogenesis—process of formation of new blood vessels—that together with invasion and altered energy metabolism facilitates tumor growth. We find that the PDMs can control expression of target hypoxia-inducible genes in cell culture and reduce tumor burden in mice.
Keywords: helix mimetics, synthetic inhibitors of transcription, rational design, hydrogen bond surrogate helices
Abstract
Selective blockade of gene expression by designed small molecules is a fundamental challenge at the interface of chemistry, biology, and medicine. Transcription factors have been among the most elusive targets in genetics and drug discovery, but the fields of chemical biology and genetics have evolved to a point where this task can be addressed. Herein we report the design, synthesis, and in vivo efficacy evaluation of a protein domain mimetic targeting the interaction of the p300/CBP coactivator with the transcription factor hypoxia-inducible factor-1α. Our results indicate that disrupting this interaction results in a rapid down-regulation of hypoxia-inducible genes critical for cancer progression. The observed effects were compound-specific and dose-dependent. Gene expression profiling with oligonucleotide microarrays revealed effective inhibition of hypoxia-inducible genes with relatively minimal perturbation of nontargeted signaling pathways. We observed remarkable efficacy of the compound HBS 1 in suppressing tumor growth in the fully established murine xenograft models of renal cell carcinoma of the clear cell type. Our results suggest that rationally designed synthetic mimics of protein subdomains that target the transcription factor–coactivator interfaces represent a unique approach for in vivo modulation of oncogenic signaling and arresting tumor growth.
Transcription factors are among the most challenging but attractive targets for drug discovery (1, 2). High-resolution structures of transcription factors in complex with protein partners offer a foundation for rational drug design strategies (3–5). Although many transcription factors exhibit significant intrinsic disorder (6), their complexes with coactivator proteins often feature discrete protein secondary structures, such as α-helices, that contribute significantly to binding and may be used as templates for rational drug design (7, 8). This paper describes design of a stabilized peptide α-helix that can modulate transcription of hypoxia-inducible genes by interfering with interactions of the C-terminal activation domain (C-TAD) of hypoxia-inducible factor-1α (HIF-1α) and the cysteine–histidine rich 1 (CH1) domain of the coactivators p300 or CREB-binding protein (CBP; Fig. 1) (9–12). We have shown that an optimized mimic of HIF-1α C-TAD, HBS 1, a high-affinity ligand of CH1, can down-regulate target genes under hypoxic conditions without affecting the endogenous levels of HIF-1α. HBS 1 does not adversely affect cell growth at high concentrations, which suggests that the compound is generally nontoxic to normoxic cells. This constrained α-helix retains significant activity in mouse plasma compared with its unconstrained peptide analog (peptide 3), highlighting the ability of stabilized helices to evade serum proteases. We compared the genome-wide effects of HIF-1α C-TAD mimic 1 and a negative control (HBS 2) using gene expression profiling. The results show that HBS 1 modulates expression of a select set of genes, many of which are of direct relevance to the predicted pathways. Last, the ability of HBS 1 to control tumor progression in a mouse tumor xenograft model was examined. We find that the synthetic helix provides rapid and effective regression of tumor growth. These results support the hypothesis that functional mimics of protein subdomains that mediate interactions between partner proteins offer an attractive strategy for inhibitor design (13, 14). Such rationally designed molecules, whose cellular targets and function may be envisioned from structures of parent complexes, could offer specific tools to probe cellular signaling networks in a predictable manner.
Fig. 1.

(A) Transcription of hypoxia-inducible genes is controlled by the interaction of HRE-bound HIF-1α/ARNT heterodimer with transcriptional coactivator p300/CBP. (B) Protein domain mimetics should competitively inhibit the interaction and associated gene expression. (C) The C-TAD793–826 domain of HIF-1α uses helical motifs to target the CH1 region of p300/CBP. HIF-1α is shown in gold and p300/CBP in gray [Protein Data Bank (PDB) ID code 1L8C].
Results
Design and Synthesis of Stabilized α-Helices.
HIF-1α forms a heterodimer with its β-subunit, aryl hydrocarbon receptor nuclear translocator (ARNT), to recognize hypoxia response element (HRE) and up-regulate expression of hypoxia-inducible genes, which are important contributors to tumor progression (11, 12, 15, 16). Pyrrole–imidazole polyamides, which are programmable DNA-binding small molecules (17), have been shown to regulate transcription of hypoxia-inducible genes by binding to the HRE (18, 19). Initiation of HIF-mediated transcription also requires complex formation between the CH1 domain of the coactivator protein p300 (or the homologous CBP) and the C-TAD786–826 of HIF-1α (Fig. 1A) (20, 21). This transcription factor–coactivator interaction represents an alternative target for controlling hypoxia signaling. Structural studies provide a molecular basis for this interaction and identify two short α-helical domains, αA and αB, from HIF-1α as key determinants for its recognition by p300 (Fig. 1C) (20, 21). Both αA and αB subdomains of HIF-1α C-TAD contain residues that contribute significantly to the complex formation, as shown by experimental mutagenesis studies (20–23). In earlier work (9), we stabilized the αA peptide sequence using the hydrogen bond surrogate (HBS) approach (24), which uses a carbon–carbon bond in place of the intramolecular hydrogen bond in α-helices. HBS helices have been shown to disrupt intracellular protein–protein interactions with high affinity and specificity (9, 25, 26). The αA mimetic was shown to down-regulate mRNA levels of VEGF and GLUT1, two genes under the control of HIF-1α, whereas the linear peptide mimic of αA remained inactive. Importantly, the compound did not display significant toxicity compared with chetomin (27), a small molecule known to target the same interaction. The HBS αA mimic provided a strong proof of principle that mimics of HIF-1α C-TAD helices can down-regulate transcription of hypoxia-inducible genes. However, despite its efficacy in cell-free and cell culture assays, we excluded this molecule from further studies due to the concern about the stability and reactivity of Cys800 residue present in this sequence that forms a key contact with the CH1 domain of p300, and hence was required for function (22). In the present study we explored the ability of αB mimics to inhibit the target interaction control gene expression in cell culture and test its efficacy in murine tumor xenograft models.
A key premise of rational design is that, unlike high-throughput screening efforts, a handful of molecules that fit certain criteria need to be designed de novo. In an ideal scenario, these predictions would lead to both a potent ligand for the target receptor and a compound serving as a negative control, featuring minor alterations but binding the same protein with reduced affinity. Such a result would confirm the fundamental design principles while allowing the specificity of designed compounds to be evaluated. Accordingly, we conceived two stabilized helices based on the wild-type sequence, along with the unconstrained control (Fig. 2). HBS 1 is a direct mimic of HIF-1α817–824 with the exception of Leu819, which was changed to an alanine residue to streamline synthetic effort; coupling of an N-alkyl alanine to the next residue is more efficient than N-alkyl leucine. Computational alanine scanning mutagenesis analysis suggests that Leu819 is not a significant contributor to binding energy as opposed to Leu818, Leu822, Asp823, and Gln824 (SI Appendix, Table S1). HBS 2 was designed to be a specificity control in which the critical Leu822 residue is replaced with an alanine; based on computational data, HBS 2 would be expected to bind CH1 with an order of magnitude weaker binding affinity than 1. Peptide 3 is an unconstrained analog of HBS 1, allowing us to evaluate the effect of helix stabilization on the activity of the compounds. The HBS helices were synthesized, purified, and characterized as described (SI Appendix, Figs. S1 and S2) (28). The constrained peptides showed characteristic α-helical circular dichroism spectroscopy signatures in aqueous buffers compared with the unconstrained derivative, which displays no discernible helicity, as expected for a very short peptide (SI Appendix, Fig. S3) (24).
Fig. 2.

(A) Structures of stabilized helices and linear peptide. HBS 1 mimics the αB domain of HIF-1α and features four residues that contribute significantly to binding (L818, L822, D823, and Q824). HBS 2 was designed to be a specificity control; this compound is identical to 1 with the exception of L822, which was mutated to an alanine group. (B) Peptide 3 is an unconstrained negative control with the amino acid sequence that repeats that of 1.
Designed Ligands Target p300-CH1 in a Predictive Manner.
The CH1 domain of p300/CBP is stabilized by three zinc ions. Prior NMR structural studies have shown that the purified protein can rapidly aggregate in a buffer with excess or deficient Zn2+ (29, 30). Attempts to evaluate binding of compounds with this protein repeatedly resulted in protein aggregation and precipitation, even at low micromolar protein concentrations. The difficulty in working with this protein is directly correlated with its expression protocol, and slight changes in the concentrations of Zn2+ in the bacterial growth media, supplemented with ZnSO4, could lead to purified protein samples that bind with different binding affinities (Kd ∼30 nM to 2 μM) to HIF-1α C-TAD786–826. To overcome this variability, we prepared 15N-labeled protein and monitored peak dispersion (and protein folding) by 1H-15N heteronuclear single-quantum coherence (HSQC) NMR experiments (SI Appendix, Fig. S4). This 15N-labeled, properly folded protein with the optimal levels of zinc shows a diminished tendency to aggregate and was used for binding assays.
The affinity of peptides for the 15N-labeled p300 CH1 domain was evaluated using tryptophan fluorescence spectroscopy. The intrinsic fluorescence intensity of Trp403 has been shown to be a sensitive probe for CH1 folding (29). Significantly, this tryptophan lies in the αB binding pocket of p300/CBP, providing a unique probe for interrogating direct binding of αB mimics (SI Appendix, Fig. S5A). Using this fluorescence method, we calculate that HBS 1 binds to p300-CH1 with a Kd of 690 ± 25 nM (Fig. 3A and SI Appendix, Fig. S5B). For comparison, HIF-1α C-TAD786–826 binds p300-CH1 with a Kd of 38 ± 0.14 nM under these conditions. The binding affinity of HIF-1α C-TAD to CH1 in this assay is consistent with that obtained from a fluorescence polarization assay using fluorescein-labeled HIF-1α C-TAD (SI Appendix, Fig. S6) and those reported in the literature with isothermal titration microcalorimetry (21). The designed specificity control, HBS 2, targets CH1 with fourfold weaker binding affinity (Kd = 2,820 ± 140 nM), supporting the computational predictions. Peptide 3 is an unconstrained analog of HBS 1 and binds CH1 domain with a Kd of 6,060 ± 320 nM. The results indicate that stabilization of the peptide conformation offers a ninefold increase in binding affinity.
Fig. 3.

HBS 1 targets p300-CH1 with high affinity and inhibits its binding to HIF-1α C-TAD786–826. (A) The affinity of 1–3 and HIF-1α C-TAD786–826 for CH1 domain was determined by tryptophan fluorescence spectroscopy. (B) Molecular model that depicts the results of a 1H-15N HSQC NMR titration experiment. The p300-CH1 residues undergoing chemical shift perturbations upon addition of HBS 1 are color-mapped, matching the magnitude of the chemical shift changes. HIF-1α helix B is shown in gold. The model was refined from the NMR structure of the HIF-1α/CH1 complex (PDB ID code 1L8C). (C) Results of fluorescence anisotropy experiments, showing the ability of HBS 1 to inhibit CH1−FluHIF C-TAD786–826 complex formation.
To further characterize the interaction of HBS 1 with the CH1 domain, we performed 1H-15N HSQC NMR titration experiments with uniformly 15N-labeled CH1. Addition of HBS 1 to 69 μM CH1 in CH1:HBS 1 ratios of 1:1, 1:3, 1:5, and 1:10 resulted in a concentration-dependent shift in resonances of several CH1 residues (Fig. 3B and SI Appendix, Fig. S7). Specifically, addition of HBS 1 leads to shifts in resonances of residues corresponding to the cleft into which the αB helix of HIF binds. This cleft includes Trp403, and chemical shift perturbations observed for this residue support the results of the fluorescence titration experiments. The CH1 domain binds to numerous proteins and has been termed a scaffold for protein folding (20, 31). Earlier NMR studies have suggested that Zn2+-bound CH1 has a relatively rigid structure (30), although evidence of plasticity in CH1 has also been discussed (20, 29). Our HSQC titration experiment with HBS 1 supports the view that CH1 has a stable conformation that does not reorganize substantially, at least upon binding of small ligands. Titration of HBS 1 to zinc-bound CH1 led to a relatively large shift in the side-chain indole NH of W403 compared with the backbone amide proton of this residue, suggesting that side-chain repacking governs binding of these partners (Fig. 3B and SI Appendix, Fig. S7) (30).
HBS 1 Disrupts the HIF-1α/p300-CH1 Complex in Vitro.
A fluorescence polarization assay was used to evaluate the ability of HBS 1 to inhibit the binding of fluorescein-labeled HIF-1α C-TAD786–826 domain to p300-CH1 (32, 33). Addition of 1 to the preformed protein complex provided a concentration-dependent decrease in fluorescence polarization, with an inhibitory constant, Ki = 3.5 ± 1.2 μM (Fig. 3C). Titration of HBS 2 or peptide 3 did not lead to reproducible inhibition of the complex, as expected from their weaker affinity for CH1 domain.
HBS 1 Down-Regulates Hypoxia-Inducible Gene Expression and VEGF Protein Levels in Hypoxic Cells.
Based on the confirmed ability of HBS 1 to bind purified p300-CH1 and disrupt CH1/HIF-1α C-TAD786–826 complex formation, we set out to evaluate its potential to down-regulate the hypoxia-inducible promoter activity in a luciferase-based reporter gene system. A construct containing five tandem repeats of the HRE consensus sequence found in the VEGF promoter (TACGTGGG), cloned upstream of the human CMV minimal promoter was used to drive expression of firefly luciferase (34). This construct was stably transfected into a triple-negative breast cancer cell MDA-MB-231 that does not express estrogen or progesterone receptors or exhibit human epidermal growth factor receptor 2/Neu (Her2/Neu) amplification. The cells were subsequently treated with the peptides; hypoxia was mimicked by placing cells into a GasPak EZ pouch. Under these conditions, treatment with HBS 1 at a concentration of 50 µM reduced luciferase expression by 25% (SI Appendix, Fig. S8). At the same concentrations, specificity control HBS 2 and unconstrained peptide 3 were found to be less effective. Despite the moderate extent of inhibition of the promoter activity, we were encouraged by these results because MDA-MB-231 cells are aggressive and under hypoxia conditions exhibit confluence-dependent resistance to some anticancer drugs (35). Our luciferase reporter assays suggest that treatment with HBS 1 results in a statistically significant down-regulation of HIF-1α inducible transcription in this cell line.
To exclude the possibility that the observed down-regulation in the expression of hypoxia-inducible genes was due to a change in the levels of HIF1α protein itself, we performed a Western blot analysis of HIF-1α in hypoxic cells treated with HBS 1. HIF-1α protein was not detectable under normoxia, but is strongly induced under hypoxia mimetic conditions. As expected, the levels of induced HIF-1α protein were unaffected by the treatment with HBS 1 (SI Appendix, Fig. S9).
The ability of HBS 1 and 2 to inhibit hypoxia-induced transcription of target genes (VEGFA, SLC2A1/GLUT-1, and LOX) was evaluated using real-time quantitative RT-PCR (qRT-PCR) assays (SI Appendix). The data from qRT-PCR experiments are presented in Fig. 4. HBS 1 reduced expression levels of VEGF by 50% at 10 µM and greater than 60% at 50 µM, showing marked dose dependence. In contrast, HBS 2 reduced expression levels of this gene by only 10% at 50 µM, and peptide 3 was completely ineffective even at 50-µM concentration (Fig. 4A). Next, we determined if this inhibition could be observed for other therapeutically relevant hypoxia-inducible genes. We examined the expression of the SLC2A1 (GLUT1) gene (36), one of the markers of glycolysis in tumors, and LOX (37), the hypoxia-inducible gene that has been shown to promote metastasis. In HeLa cells under hypoxia conditions, HBS 1 showed dose-dependent inhibition of SLC2A1 by 50–60%, comparable to that of VEGF gene in the same cell line (Fig. 4B). Similarly, HBS 1 significantly down-regulated levels of expression of the LOX gene in a dose-dependent manner (55% and 70%, respectively, Fig. 4C); HBS 2 showed no activity in these assays, whereas peptide 3 had reduced activity of 25%. To rule out the possibility that the compounds are only efficacious under deferoxamine (DFO)-mimicked hypoxia, we compared the efficacies of the HBS peptides in down-regulating VEGF gene expression under two different hypoxia mimetic conditions: DFO and prolonged incubation in an anaerobic pouch. Under both conditions, HBS 1 showed dose-dependent inhibition of VEGF expression (Fig. 4D). Next, we assessed the effect of HBS 1 treatment on the levels of secreted VEGF protein. An ELISA shows that HBS 1 down-regulates VEGF protein levels in HeLa cells in a dose-dependent manner (SI Appendix, Fig. S10).
Fig. 4.

HBS down-regulates hypoxia-induced transcription in cell culture. HBS 1 reduced expression levels of (A) VEGFA, (B) SLC2A1 (GLUT1), and (C) LOX genes in dose-dependent manner in HeLa cells under hypoxia conditions as measured by real-time qRT-PCR. Hypoxia was mimicked with DFO (300 µM). HBS 2 and peptide 3 show reduced inhibitory activities at the same concentrations. Error bars are ±SEM of four independent experiments. ***P < 0.0001, **P < 0.01, *P < 0.05, t test. (D) Comparison of the efficacies of HBS 1 in down-regulating expression levels of VEGFA gene in HeLa cells under two different hypoxia-mimetic conditions (DFO and hypoxia bag) as measured by the real-time qRT-PCR. For each experiment under hypoxia-mimetic conditions, mRNA levels were normalized to VEGFA mRNA levels found in the vehicle-treated normoxic cells.
HBS 1 is an efficient modulator of contacts between HIF-1α and p300/CBP. Known inhibitors of this interaction typically function allosterically, by inducing unfolding of p300/CBP through abstraction of zinc ions (27, 33, 38, 39), which could lead to nonspecific abstraction of metal ions from other biomolecules. We predicted that our HIF-1α mimetics should manifest their function in a more specific manner, and should not be generally cytotoxic (9). Cell viability assays confirm this hypothesis; we find that HBS 1 is essentially noncytotoxic within the entire range of tested concentrations (1–100 µM; SI Appendix, Fig. S11). Interestingly, HBS 2 shows a higher level of cytotoxicity than HBS 1, suggesting that this compound may be interacting with a different set of biomolecular targets as seen from gene expression profiling data (vide infra). Thus, HBS 2 may not just be a straightforward lower-affinity analog of 1, as designed.
Gene Expression Profiling.
Proteins p300 and CBP are pleiotropic multidomain coactivators that directly interact with multiple transcription factors (31). One potential limitation of the use of coactivator-targeting ligands to control gene expression is specificity, due to the fact that the compounds could lead to inhibition of large numbers of genes that depend on the function of p300 or CBP. To evaluate the genome-wide effects of HBS 1 and 2 under hypoxia conditions, we used Affymetrix Human Gene ST 1.0 arrays containing oligonucleotide sequences representing over 28,000 transcripts; gene expression levels were normalized to DFO-treated cells.
In hypoxic cells, clustering identified over 5,000 genes that changed in expression levels under one of the specified treatments: DFO, DFO + HBS 1, or DFO + HBS 2 (SI Appendix, Fig. S12). Treatment with HBS 1 affected the expression of 122 transcripts by at least 1.1-fold (P < 0.05), whereas at the same threshold, control HBS 2 affected expression of 155 transcripts (Fig. 5 A and C). Remarkably, only 33 transcripts were overlapping, indicating that the subtle difference in structure between these two compounds results in a significant difference in genome-wide effects. For comparison, DFO treatment alone affected the expression of 368 transcripts. Clustering analysis was performed to identify similarities in the expression profiles between the different treatments (Fig. 5A). The expression profile of cells treated with HBS 1 resembles the profile of cells treated with DFO under the conditions of the analysis and, as mentioned above, is different from the profile of cells treated with HBS 2 despite the structural similarity between the two compounds. As expected, the expression profile of the normoxic cells is significantly different from the other three profiles. Analysis of transcripts affected by both HBS 1 and HBS 2 shows that only 28 and five transcripts are commonly down- and up-regulated, respectively, by at least 1.1-fold (P < 0.05). It is not surprising that there is some overlap in genes affected by both compounds given the complexity of cellular signaling pathways involved in the hypoxic response. We found that DFO induced the expression of 45 transcripts by at least fourfold (P < 0.05; Fig. 5B). Within this dataset, we identified multiple genes that belong to the hypoxia-inducible pathway. HBS 1 and, to some extent HBS 2, affected almost all genes in this set.
Fig. 5.

Results from gene expression profiling obtained with Affymetrix Human Gene ST 1.0 arrays. (A) Hierarchical agglomerative clustering of 368 transcripts induced or repressed twofold or more (one-way ANOVA, P < 0.05) by 300 µM DFO under the three specified conditions: –, no treatment; 1, HBS 1 (50 µM); 2, HBS 2 (50 µM). Clustering was based on a Pearson-centered correlation of intensity ratios for each treatment compared with DFO-induced cells (controls) using average linkage as a distance. Of this DFO-induced set, 92 were inhibited and 30 were induced by HBS 1, whereas 81 were inhibited and 70 induced by HBS 2 (|fold-change| ≥ 1.1, P < 0.05). (B) Clustering of expression changes of the 45 transcripts induced or repressed fourfold or more (P < 0.05) by 300 µM DFO or by the treatments under the designated treatment conditions. Clustering parameters were the same as in A. (C) Venn diagrams representing transcripts down- and up-regulated (|fold-change| ≥ 1.1, P < 0.05) by HBS 1 and HBS 2. Numbers inside the intersections represent DFO-induced transcripts affected by both treatments.
Antitumor Activity of HBS 1 in Mouse Xenograft Models.
We used a mouse xenograft tumor model to assess the in vivo efficacy of HBS 1. We first measured the relative plasma stabilities of HBS 1 and linear peptide 3 in mice. In this experiment, female BALB/c mice were injected with either HBS 1 or peptide 3 at a dose of 1 mg/kg and killed at various time points. Blood was collected and the plasma concentration profiles for HBS 1 and peptide 3 were determined, as shown in SI Appendix, Fig. S13. Though both compounds exhibited a biexponential pattern of decay, HBS 1 was retained in plasma at much higher concentrations compared with peptide 3 during the same time intervals, suggesting that the internally constrained structure of HBS 1 favorably impacts its serum stability. This observation is consistent with the fact that proteases largely bind and cleave peptides in extended conformations (40); the plasma stability of HBS 1 is also consistent with the published stability of hydrocarbon-bridged helices (3).
For efficacy studies we used the CrTac:NCr-Foxn1nu mouse (Taconic, Inc.). We selected 786-O RCC of the clear cell type cell line because of its high HIF levels due to a mutation in the VHL gene. Measurable tumors (∼100 mm3) grew in as little as 2–3 wk after the inoculation of 2 × 106 cells into the either flank of mice. Mice then were separated into two experimental groups; one group was treated with HBS 1 and the second group was not treated (control). We estimated 13 mg/kg as an acceptable dose, based on the concentration of HBS 1 required for >50% VEGF and LOX mRNA down-regulation in cell culture and plasma concentrations of the compounds (vide supra). Tumor sizes were measured in accordance with the literature recommendations (SI Appendix) (41). Throughout the course of the treatment and at the experiment endpoint, mice treated with HBS 1 had smaller tumors with median tumor volume reduction of 53% compared with the mice from the control group (Fig. 6A). Both control group and mice treated with HBS 1 under this regimen showed no signs of distress or weight loss (Fig. 6B). To rule out the possibility that treatment resulted in a reduction of the size of the main tumor but concurrently resulted in an elevated rate of metastasis, as reported with some anti-VEGF therapeutics (42), the animals were injected with IR-783, a near-infrared contrast agent that targets tumors, circulating tumor cells, and metastases (43), and imaged from both sides using a small animal imager. The images show no detectable near-infrared (NIR) signal within the lymph nodes, brain, or other organs, and significantly reduced signal from the main tumor (Fig. 6C).
Fig. 6.

HBS 1 suppresses tumor growth in mouse xenograft models. (A) Box-and-whisker diagram of tumor volumes measured throughout the study with boxes representing the upper and lower quartiles and median and error bars showing maximum and minimum volumes. Tumors from mice treated with HBS 1 were smaller (median volume: 138 mm3) than those of control mice (median: 293 mm3). (B) Results of the weight measurements of control and HBS 1-treated mice throughout the entire duration of the experiments, showing the absence of toxicity-related weight loss. (C) HBS 1 lowers overall tumor burden in mice. Images of mice injected with the tumor-accumulating NIR contrast agent. Mice from the HBS 1-treated group show significantly lower intensity of the NIR signal compared with the control group. The color bar indicates the NIR radiant efficiency (photons s–1 cm–2 sr–1 per μW cm\x{2212}2) × 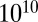 .
.
Discussion
Synthetic inhibitors that block the transactivation domains of transcription factors from contacting their cognate coactivators (3, 44–46), and programmable small molecules that sequence-specifically inhibit DNA–transcription factor interactions, provide powerful strategies for regulating gene expression (17, 47, 48). The blockade of transcription factor interactions can be especially attractive in targeting cellular pathways that promote oncogenic transformation and typically involve a large number of signaling proteins that ultimately converge on a much smaller set of oncogenic transcription factors (2). Given that both CBP and p300 regulate multiple signaling pathways, they provide an intriguing opportunity for an effective modulation of the expression of genes involved in cancer progression and metastasis (31). Our design strategy involves judicious mimicry of transcription factor fragments that contact p300/CBP to rationally develop artificial regulators of transcription (4, 5).
Our results indicate that synthetic helices that mimic protein subdomains bind their p300/CBP target with high affinity and disrupt the HIF-1α C-TAD–p300/CBP complex in vitro. Importantly, the designed compounds bound the target protein in a predictable manner; the single residue mutant HBS 2 shows an expected weaker affinity for CH1 compared with HBS 1. The CH1 binding site for HBS 1 was confirmed by NMR HSQC titration experiments. As anticipated based on fluorescence experiments, HBS 1 causes a concentration-dependent chemical perturbation shift for the side chain ε-NH of Trp403. This result supports our design principle that a locked helix can occupy the binding clefts of individual protein α-helices (3, 25). The in vitro assays showed significant reduction in promoter activity and effective down-regulation of the expression of HIF-1α–inducible genes responsible for promoting angiogenesis, invasion, and glycolysis. In addition, the HBS 1-mediated transcriptional blockade of VEGF correlates with decreased levels of its secreted protein product, suggesting that compensatory cellular stress-response mechanisms, such as internal ribosome entry sites or mechanisms enhancing protein translation, do not affect the observed down-regulation in expression. Therefore, reducing the cellular mRNA levels of HIF-1α target genes with HBS 1 could be an effective means of attenuating hypoxia-inducible signaling in tumors.
Comparative analysis of the genome-wide effects of the HBS 1 and 2 provided additional insights into the ability of the compounds to disrupt transcriptional activity of hypoxia-inducible genes. Despite the similarity in structures, these compounds have a very different impact on the level of expression of hypoxia-inducible genes and show distinct genome-wide effects. Treatment with 1 affects 122 genes (less than 0.5% of the entire transcriptome) at a fixed 1.1-fold threshold, with 92 hypoxia-inducible genes being down-regulated. Despite the fact that HBS 2 has a similar genome-wide impact at the same threshold, it does not affect a majority of hypoxia-inducible genes. Because many biological responses are threshold based, the observed decrease in transcriptional activity of primary hypoxia-inducible genes could have pronounced downstream effects on the levels of protein products of hypoxia-inducible transcription.
To assess the in vivo potential of HBS 1, murine tumor xenografts derived from the RCC of the clear cell type were treated with the compound. After five injections of HBS 1, the median tumor volume was reduced by 53% in the treated group. Importantly, the HBS 1 treatment did not cause measurable changes in animal body weight or other signs of toxicity in tumor-bearing animals, or increase the metastasis rate.
Taken together, the results reported herein support the hypothesis that designed protein domain mimetics can provide valuable tools for probing the mechanisms of transcription. Because the p300/CBP pleiotropic coactivator system interacts with diverse transcription factors, it represents an excellent model system to assess the specificity of designed synthetic ligands in gene regulation. Future designs will address the possibility of targeting a desired region of a general coactivator as a means of selectively modulating genes under the control of a specific group of transcription factors. The strategy described herein provides a foundation for the development of next-generation genomic tools and, potentially, transcription-based therapies.
Methods
Synthesis and characterization of HBS 1 and 2, and peptide 3 can be found in SI Appendix. Detailed protocols for cloning and expression of the p300-CH1-GST protein, binding assays, cell viability assays, and luciferase expression assays are also included. Western blot analysis of HIF-1α levels, VEGF ELISAs, gene expression profiling, and in vivo efficacy are described in SI Appendix.
Supplementary Material
Acknowledgments
The authors thank Maria Yamout (Wright group, The Scripps Research Institute) for advice on p300 expression; Jian-Ching Ren of the University of Southern California for help with plasma stability assays; and Daniela Buccella at New York University (NYU) for the use of the spectrofluorometer. Financial support for this work was provided by US National Science Foundation CAREER Award CHE-1161644 (to B.Z.O.) and National Institutes of Health Grant R01GM073943 (to P.S.A.). The microarray analysis was performed at the Genome Technology Center, NYU School of Medicine, partially supported by NYU Cancer Center Support Grant 5P30CA16087-33.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The gene expression profiling data reported in this manuscript have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE48002).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312473110/-/DCSupplemental.
References
- 1.Ptashne M, Gann A. Genes and Signals. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2002. [Google Scholar]
- 2.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 3.Moellering RE, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rutledge SE, Volkman HM, Schepartz A. Molecular recognition of protein surfaces: High affinity ligands for the CBP KIX domain. J Am Chem Soc. 2003;125(47):14336–14347. doi: 10.1021/ja034508o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhrlage SJ, et al. Amphipathic small molecules mimic the binding mode and function of endogenous transcription factors. ACS Chem Biol. 2009;4(5):335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, et al. Intrinsic disorder in transcription factors. Biochemistry. 2006;45(22):6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullock BN, Jochim AL, Arora PS. Assessing helical protein interfaces for inhibitor design. J Am Chem Soc. 2011;133(36):14220–14223. doi: 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jochim AL, Arora PS. Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors. ACS Chem Biol. 2010;5(10):919–923. doi: 10.1021/cb1001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henchey LK, et al. Inhibition of hypoxia inducible factor 1-transcription coactivator interaction by a hydrogen bond surrogate alpha-helix. J Am Chem Soc. 2010;132(3):941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraisl P, Mazzone M, Schmidt T, Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev Cell. 2009;16(2):167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol. 2006;59(1):15–26. doi: 10.1016/j.critrevonc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 13.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5(3):161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 14.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450(7172):1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 15.O’Rourke JF, Pugh CW, Bartlett SM, Ratcliffe PJ. Identification of hypoxically inducible mRNAs in HeLa cells using differential-display PCR. Role of hypoxia-inducible factor-1. Eur J Biochem. 1996;241(2):403–410. doi: 10.1111/j.1432-1033.1996.00403.x. [DOI] [PubMed] [Google Scholar]
- 16.Ivan M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292(5516):464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 17.Dervan PB, Edelson BS. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr Opin Struct Biol. 2003;13(3):284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 18.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem Biol. 2007;2(8):561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olenyuk BZ, et al. Inhibition of vascular endothelial growth factor with a sequence-specific hypoxia response element antagonist. Proc Natl Acad Sci USA. 2004;101(48):16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freedman SJ, et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA. 2002;99(8):5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002;99(8):5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu J, Milligan J, Huang LE. Molecular mechanism of hypoxia-inducible factor 1alpha-p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem. 2001;276(5):3550–3554. doi: 10.1074/jbc.M009522200. [DOI] [PubMed] [Google Scholar]
- 23.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 24.Patgiri A, Jochim AL, Arora PS. A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation. Acc Chem Res. 2008;41(10):1289–1300. doi: 10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7(9):585–587. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, Lu M, Arora PS. Inhibition of HIV-1 fusion by hydrogen-bond-surrogate-based alpha helices. Angew Chem Int Ed Engl. 2008;47(10):1879–1882. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]
- 27.Kung AL, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Patgiri A, Menzenski MZ, Mahon AB, Arora PS. Solid-phase synthesis of short α-helices stabilized by the hydrogen bond surrogate approach. Nat Protoc. 2010;5(11):1857–1865. doi: 10.1038/nprot.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dial R, Sun ZY, Freedman SJ. Three conformational states of the p300 CH1 domain define its functional properties. Biochemistry. 2003;42(33):9937–9945. doi: 10.1021/bi034989o. [DOI] [PubMed] [Google Scholar]
- 30.De Guzman RN, Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. CBP/p300 TAZ1 domain forms a structured scaffold for ligand binding. Biochemistry. 2005;44(2):490–497. doi: 10.1021/bi048161t. [DOI] [PubMed] [Google Scholar]
- 31.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276(17):13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 32.Block KM, et al. Direct inhibition of hypoxia-inducible transcription factor complex with designed dimeric epidithiodiketopiperazine. J Am Chem Soc. 2009;131(50):18078–18088. doi: 10.1021/ja807601b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubey R, et al. Suppression of tumor growth by designed dimeric epidithiodiketopiperazine targeting hypoxia-inducible transcription factor complex. J Am Chem Soc. 2013;135(11):4537–4549. doi: 10.1021/ja400805b. [DOI] [PubMed] [Google Scholar]
- 34.Shibata T, Giaccia AJ, Brown JM. Development of a hypoxia-responsive vector for tumor-specific gene therapy. Gene Ther. 2000;7(6):493–498. doi: 10.1038/sj.gt.3301124. [DOI] [PubMed] [Google Scholar]
- 35.Fang Y, Sullivan R, Graham CH. Confluence-dependent resistance to doxorubicin in human MDA-MB-231 breast carcinoma cells requires hypoxia-inducible factor-1 activity. Exp Cell Res. 2007;313(5):867–877. doi: 10.1016/j.yexcr.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276(12):9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 37.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 38.Cook KM, et al. Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1alpha (HIF-1alpha) and p300 by a zinc ejection mechanism. J Biol Chem. 2009;284(39):26831–26838. doi: 10.1074/jbc.M109.009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kushal S, Wang H, László CF, Szábo LZ, Olenyuk BZ. Inhibition of hypoxia-inducible transcription factor complex with designed epipolythiodiketopiperazine. Biopolymers. 2011;95(1):8–16. doi: 10.1002/bip.21550. [DOI] [PubMed] [Google Scholar]
- 40.Tyndall JD, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites. Chem Rev. 2005;105(3):973–999. doi: 10.1021/cr040669e. [DOI] [PubMed] [Google Scholar]
- 41.James K, et al. Measuring response in solid tumors: Unidimensional versus bidimensional measurement. J Natl Cancer Inst. 1999;91(6):523–528. doi: 10.1093/jnci/91.6.523. [DOI] [PubMed] [Google Scholar]
- 42.Pàez-Ribes M, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, et al. Near IR heptamethine cyanine dye-mediated cancer imaging. Clin Cancer Res. 2010;16(10):2833–2844. doi: 10.1158/1078-0432.CCR-10-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravindranathan P, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4:1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 45.Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol. 2010;14(3):331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee LW, Mapp AK. Transcriptional switches: Chemical approaches to gene regulation. J Biol Chem. 2010;285(15):11033–11038. doi: 10.1074/jbc.R109.075044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang F, et al. Antitumor activity of a pyrrole-imidazole polyamide. Proc Natl Acad Sci USA. 2013;110(5):1863–1868. doi: 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muzikar KA, Nickols NG, Dervan PB. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc Natl Acad Sci USA. 2009;106(39):16598–16603. doi: 10.1073/pnas.0909192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.