

Significance
A scalable, chemistry-independent, fluid-instability–mediated in-fiber route for fabricating uniformly sized spherical polymeric particles over a wide span of diameters is developed targeting biomedical applications. Both surface functionalization of solid biocompatible polymer particles for protein–protein interactions and volume encapsulation of a biological material in spherical hollow polymer shells are confirmed, in addition to combining both surface and volumetric functionalities in the same polymeric particle.
Abstract
Polymeric micro- and nanoparticles are becoming a mainstay in biomedicine, medical diagnostics, and therapeutics, where they are used in implementing sensing mechanisms, as imaging contrast agents, and in drug delivery. Current approaches to the fabrication of such particles are typically finely tuned to specific monomer or polymer species, size ranges, and structures. We present a general scalable methodology for fabricating uniformly sized spherical polymeric particles from a wide range of polymers produced with complex internal architectures and continuously tunable diameters extending from the millimeter scale down to 50 nm. Controllable access to such a wide range of sizes enables broad applications in cancer treatment, immunology, and vaccines. Our approach harnesses thermally induced, predictable fluid instabilities in composite core/cladding polymer fibers drawn from a macroscopic scaled-up model called a “preform.” Through a stack-and-draw process, we produce fibers containing a multiplicity of identical cylindrical cores made of the polymers of choice embedded in a polymer cladding. The instability leads to the breakup of the initially intact cores, independent of the polymer chemistry, into necklaces of spherical particles held in isolation within the cladding matrix along the entire fiber length. We demonstrate here surface functionalization of the extracted particles for biodetection through specific protein–protein interactions, volumetric encapsulation of a biomaterial in spherical polymeric shells, and the combination of both surface and volumetric functionalities in the same particle. These particles used in distinct modalities may be produced from the desired biocompatible polymer by changing only the geometry of the macroscopic preform from which the fiber is drawn.
Medical applications in diagnostics (1), imaging (2, 3), and drug delivery (4) typically make use of micro- and nanoparticles in two distinct modes: The surfaces may serve as loci for chemical or biological interactions through surface functionalization (5), or, alternatively, the volume may be used to carry cargo (6, 7) for drug delivery and multidimensional imaging modalities (8). Polymeric nanoparticles in particular are steadily gaining importance in medicine because of their long half-life in the blood stream (9, 10) and the versatility by which their composition, size, shape, and physicochemical properties (11) may be tuned via a variety of processing approaches. Recent medical achievements based on the use of polymer particles in specific applications include the targeted delivery of drugs and toxins using surface-conjugated biodegradable polymer particles (12, 13), treatment of antibiotic-resistant bacterial infections (14, 15), and induction of regulatory T cells for treatment of multiple sclerosis (16).
Current bottom-up synthetic approaches produce nanoparticles that typically have considerable size dispersion, cannot reach the microscale, and are sensitively tuned to specific chemical building blocks (17). Top-down processes, on the other hand, such as emulsification (18), the use of templates (19), and microfluidics-based approaches (20–25), exploit polymers in a low-viscosity state or in solution and rely on prefabricated devices to impart form and size to the particles, which are subsequently solidified. In general, the process kinetics in both strategies, bottom-up and top-down, limit each approach to a narrow set of materials, sizes, and structures. Individualized biomedical applications require varying particle sizes and modalities, which, in turn, necessitate different materials-specific fabrication pathways for producing such diverse structures. For example, no single nanoparticle size can reach the different areas of a tumor and the surrounding stroma (26, 27), and particle size was found to be crucial in cell adsorption and internalization (28). These findings suggest the benefit of particle fabrication processes that are not constrained to narrow size spans. The widespread adoption of polymeric particles in current and future biomedical applications would be enhanced by addressing the challenge of developing a scalable process that is sufficiently versatile to bridge the micro- and nanoscales, is compatible with a variety of polymers, and is capable of producing a multiplicity of particle architectures optimized for specific applications.
Here, we describe a scalable top-down process that yields uniformly sized spherical polymer particles with continuously tunable diameters from micro- to nanoscales realized in prescribed structures without recourse to any premade device in which the particles are formed. In lieu of prefabricated templates (19) or microfluidic channels filled with coflowing fluids (25), we rely here on thermal treatment of a core/cladding polymer fiber to fluidize the core and controllably induce at the interfaces the Plateau–Rayleigh capillary instability (PRI) (29–31), the natural tendency of fluid cylinders to break up into spheres. The desired polymer is placed in the core of a scaled-up macroscopic (centimeter-scale) “preform” in the solid state, which is then thermally drawn into an extended fiber that defines the initial conditions for the process (32, 33). The PRI results in an initially intact cylindrical core breaking up into a necklace of uniformly sized spherical particles held stationary in isolation following quenching in the solid cladding matrix along the whole fiber length, to be released when needed using an appropriate solvent (34, 35). Complex particle architectures are achieved by judicious design of the core geometry at the macroscopic preform stage, which is, in turn, inherited by the resulting particles. The scalability of the process stems from stacking a high density of cores inside a single fiber produced rapidly with extended lengths using the traditional process of thermal drawing. Furthermore, we confirm that the PRI-based process is “chemistry-independent” by using it to produce particles from different polymers, some of which have not been used heretofore in particle form despite their useful biochemical, mechanical, and optical properties. Such salutary features combined in this in-fiber fabrication approach (i.e., size control, scalability, compositional, structural control) may enable such polymeric particles to play an important role in biomedical applications, such as recognition of specific cell types through protein binding (36), separation of specific cells from a mixture (37), and delivering biological materials or drugs (38). The large quantities of particles produced may enable applications in cosmetics (39) and emulsions used in coatings and paints, whereas the size uniformity of polymeric particles is crucial in ultrasound imaging (40). We establish critical steps toward achieving these goals via three key demonstrations of biomedical importance: (a) protein binding to the particle surfaces for biosensing, (b) quantitative control over differential surface protein binding, and (c) encapsulation of a biological material in a polymeric shell using collagen as a model material. These demonstrations are carried out using the same biocompatible polymer in two different particle structures, a solid spherical particle for surface functionalization and a hollow spherical particle for volumetric cargo encapsulation, produced by the in-fiber fabrication methodology. We also confirm that both surface and volume functionalities may be combined in the same particle, a result that has wide implications for site-specific drug delivery (41).
Results
The in-fiber particle fabrication procedure may be divided into three stages (35) (Fig. 1A): (i) macroscopic preform preparation (stage I), (ii) thermal fiber drawing (stage II), and (iii) thermally induced in-fiber emulsification via the PRI (stage III). The structure of the particle is determined in the preform fabrication stage, whereas its size, proportional to the fiber core diameter, is set in the fiber-drawing stage. The three processing stages are illustrated in Fig. 1A, where a preform consisting of a 700-μm-diameter cylindrical cyclic olefin polymer (COP) (42) core surrounded by a 35-mm-diameter polysulfone (PSU) cladding (stage I) is thermally drawn into hundreds of meters of a uniform 1-mm-diameter fiber with a 20-μm-diameter core (stage II; Fig. 1B). The drawn fiber is thermally treated under ambient conditions, thereby reducing materials viscosity and inducing the PRI (stage III), whose dynamics are shown in Fig. 1C. The process is seeded by thermodynamic fluctuations that arise at the core/cladding interface upon heating. The classic Tomotika model of the PRI predicts that the fluctuation wavelength with the shortest growth time dominates and its amplitude increases without bound (34, 43), leading to the breakup of the COP core into a necklace of uniformly sized spherical particles held in the PSU cladding. The breakup wavelength is determined by the core diameter, interface surface energy, and materials viscosities (34). The diameter of the produced particles may thus be continuously tuned, for fixed core and cladding materials and processing conditions, by changing the diameter of the fiber, which is readily achieved by adjusting the drawing parameters. This unique feature appropriated from the process of fiber drawing allows for particle size tuning over several orders of magnitude in linear dimension (35, 44–46). After breakup, we extract the COP particles by selective dissolution of the PSU cladding [using dimethylacetamide (DMAC)]. Note that the dissolved cladding polymer may be recycled from the solution and reused as a cladding for a new fiber. In Fig. 1D, we show scanning electron micrographs of representative examples of particles with diameters extending three orders of magnitude from ∼50 μm down to ∼50 nm. In general, this process yields a particle size distribution having ∼10% SD normalized with respect to the mean particle size (details are provided in ref. 35).
Fig. 1.

In-fiber emulsification as a route to producing spherical polymer particles. (A) Preform-to-fiber approach. A centimeter-scale preform (Upper Left) is drawn into a 1-mm-diameter fiber. The Roman numerals identify the three fabrication stages outlined in the main text. (B) Optical transmission micrograph of the fiber cross-section consisting of a 20-μm-diameter COP core inside a PSU cladding. (C) Side views of a section of a fiber undergoing thermal treatment at 321 °C showing the temporal evolution of the PRI starting from the intact cylindrical COP core until it breaks up into a necklace of particles embedded in the PSU cladding. (D) Size tunability of COP particles, released from the PSU cladding using DMAC, demonstrated in scanning electron micrographs. [Scale bar: D, successively reduced by a factor of 5 (Upper to Lower)]. Particle diameters are ∼53 μm, 10.6 μm, 2.2 μm, 270 nm, and 61 nm. Details of the fabrication process are provided in Supporting Information.
The scalability of particle production may be enhanced by incorporating a large number of identical cores into the preform. Two examples are shown in Fig. 2 to highlight this unique feature: a 14-core fiber (each with a 20-μm diameter; Fig. 2 A–D) and a 1,000-core fiber (each with a 500-nm diameter; Fig. 2 E–G), with both having a 1-mm o.d. Thermal treatment of either fiber leads to the simultaneous breakup of all the cores via the PRI into a 3D arrangement of particles held stationary in the cladding matrix. The limit on the particle production rate is thus set by the fiber-drawing speed, the fiber o.d., and the fiber-filling ratio (percentage of the fiber transverse cross-section occupied by the cores). Using currently available fiber fabrication technology, our process may yield on the order of a few kilograms of structured polymeric particles per day independent of the particle size or structure.
Fig. 2.

Scalable in-fiber emulsification. Microscale (A–D) and nanoscale (E–G) in-fiber emulsification. (A) Optical transmission micrograph of the cross-section of a 14-core fiber with 20-μm-diameter COP cores in a PSU cladding. (Inset) Individual core cross-section. (B) Scanning electron micrograph of the fiber side view showing the 14 intact COP cores after dissolving the PSU cladding. The cores coalesce after removing the fiber from the solvent. (C) Optical transmission micrograph of the fiber side view after thermally inducing the PRI, resulting in the cores breaking up into spherical particles held stationary in the cladding matrix. (D) Optical transmission micrograph of the released COP particles after dissolving the PSU cladding. (E and F) Scanning electron micrographs of 1,000 intact 500-nm-diameter COP cores emerging from the fiber after dissolving the PSU cladding matrix. (G) Scanning electron micrograph of the released COP particles. (Inset) Scanning electron micrograph of a single particle. (Scale bar: 500 nm.)
Our approach relies on placing the desired polymer in the core within a sacrificial thermoplastic polymer cladding chosen such that the pair of polymers may be thermally codrawn (32, 33). A unique aspect of this physical process is that it depends on the rheology of the materials and not on their chemistry, as long as they are immiscible. This was confirmed by producing particles from a wide range of polymers, including polystyrene (PS), polycarbonate (PC), acrylonitrile butadiene styrene (ABS), polyetherimide (PEI), and polyethersulfone (PES) (Supporting Information). These polymers have very different biochemical, mechanical, and optical characteristics, and some of them (e.g., COP and ABS) have yet to be produced in particle form. For example, ABS at room temperature is a rigid polymer, but its characteristics at room temperature do not come into play during breakup under thermal treatment. Although PS is extensively used in biomedicine due to its low density (allowing for suspension of PS particles in aqueous solutions) and its chemical inertness, COP maintains these characteristics with the additional benefits of its excellent protein adsorption characteristics (47) and resistance to many more chemicals than PS, such as acids, alkalis, and most organic polar solvents (e.g., acetone, methanol, isopropyl alcohol) (48). Furthermore, the in-fiber process allows for combining inorganic glasses and polymers in a single-particle geometry (35).
Thus, although the particle formation process itself is independent of the chemistry of the core and cladding materials, extracting the particles by selective dissolution of the cladding, on the other hand, does indeed depend on the chemistry of the fiber materials, which places restrictions on the potential polymeric core/cladding pairings. This constraint is lifted by separating the core and cladding polymers with a thermally compatible inorganic buffer layer added in the preform, in which case the core and cladding may even be the same polymer. We realize this design here using PES in both the core and the cladding separated by a layer of the thermally compatible chalcogenide glass, As2Se3 (49) (Fig. 3 B and C and Supporting Information). The PRI yields particles having a core (polymer)/shell (glass) structure that are released by dissolving the polymer cladding (using DMAC), and the polymer particles are extracted by subsequent removal of the shell (using NaOH in deionized water; Fig. 3D).
Fig. 3.

Inorganic sacrificial buffer layer to facilitate particle extraction. (A) Schematic of the fiber structure. (B and C) Optical reflection micrographs of the fiber cross-section at two stages in the fabrication process (Supporting Information). The core consists of a PES (P) cylinder surrounded by a layer of an inorganic glass, As2Se3 (G). The core is embedded in a PES cladding matrix. (D) Time-lapse transmission optical micrographs of core (P)/shell (G) particles placed in NaOH solution. The glass shell is dissolved, leaving behind the polymer cores. (E) Theoretically predicted values for the figure-of-merit 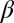 based upon calculated breakup wavelengths
based upon calculated breakup wavelengths 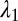 and
and 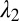 assuming equal-amplitude perturbations (Supporting Information). Black continuous lines are contours of fixed
assuming equal-amplitude perturbations (Supporting Information). Black continuous lines are contours of fixed 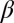 at the values of
at the values of 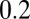 ,
, 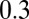 , and
, and  . The dashed red line corresponds to the experimental values of
. The dashed red line corresponds to the experimental values of 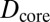 and
and  . D1 and D2 are the experimental values for Dcore and Dshell, respectively.
. D1 and D2 are the experimental values for Dcore and Dshell, respectively.
Because the PRI develops at the two cylindrical interfaces, the polymer-core/glass-buffer inner interface and the glass-buffer/polymer-cladding outer interface (Fig. 3A), the buffer thickness must be chosen to ensure the formation of the core/shell structure. The instability wavelengths at these interfaces (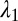 and
and 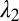 , respectively) will generally be different. To assess whether the chosen physical and geometric parameters yield a core/shell structure, we adopt a figure-of-merit
, respectively) will generally be different. To assess whether the chosen physical and geometric parameters yield a core/shell structure, we adopt a figure-of-merit  , where
, where 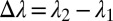 and
and 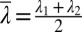 . Small values of
. Small values of 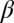 (i.e.,
(i.e.,  ) indicate that the breakup produces the target core/shell geometry, whereas large values of
) indicate that the breakup produces the target core/shell geometry, whereas large values of 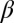 indicate that multiple polymer cores may form within the glass shell. Using a recently developed theoretical model based on linear stability analysis of the PRI in multilayer cylindrical structures (50–52), we calculate
indicate that multiple polymer cores may form within the glass shell. Using a recently developed theoretical model based on linear stability analysis of the PRI in multilayer cylindrical structures (50–52), we calculate 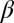 for different ratios of core and shell diameters (
for different ratios of core and shell diameters (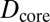 and
and  , respectively) and viscosities (
, respectively) and viscosities (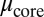 and
and 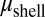 , respectively) assuming that the average initial perturbation amplitudes at the two interfaces are equal (Fig. 3E). This model reveals a wide range of parameters that yield the target structure (taken to occur when
, respectively) assuming that the average initial perturbation amplitudes at the two interfaces are equal (Fig. 3E). This model reveals a wide range of parameters that yield the target structure (taken to occur when 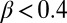 ), including our case of
), including our case of 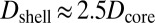 . These results may be generalized to systems comprising three different fluids (50), corresponding to multiple emulsions, a feature that we exploit below for encapsulation.
. These results may be generalized to systems comprising three different fluids (50), corresponding to multiple emulsions, a feature that we exploit below for encapsulation.
A further degree of freedom that may be accessed by the in-fiber approach is the particle internal structure, which follows that of the constructed macroscopic preform. Along these lines, we have produced polymer core/shell particles (COP core and PC shell) and bicompartmentalized polymer Janus particles (PES and PEI hemispheres) (Supporting Information). Applying our general fabrication strategy using different preform structures but the same biocompatible polymer (COP), we produce two distinct particle structures tuned to two different classes of applications. The first class consists of solid particles that we use for demonstrating surface binding of protein and fluorescent antibodies (FAs), a necessary step for biosensing; furthermore, we verify controllable protein coating of the COP particles while maintaining biological specificity. Using the same fabrication procedure applied to a hollow-core fiber design, a second class of particles is developed for encapsulation of selected materials inside a COP shell. We demonstrate this feature using collagen as a model biological material, motivated by recent work demonstrating the utility of collagen nanoparticles in skin tissue engineering (53), bone regeneration (54), and treatment of bacterial infection (55).
We first demonstrate that the surfaces of the polymer particles produced by the fluid instability-based process are amenable to protein binding without pretreatment, and are thus suitable for biosensing applications. By coating solid COP microparticles, removed from the fiber PSU cladding, with antibodies conjugated with fluorophores (FAs) through adsorption, we directly monitor optically the presence of bound proteins using two readily distinguishable FAs: yellow-fluorescing FA1 [Cy3-conjugated anti-rabbit antibodies produced in goat (gAr); Fig. 4A] and red-fluorescing FA2 [Alexa Fluor-647–conjugated anti-mouse antibodies produced in goat (gAm); Fig. 4B] (details are provided in Supporting Information). We next verified that the particle-bound proteins retain their native conformation through optical detection of specific protein–protein interactions. We coated the particles with mouse serum proteins and monitored the fluorescence emitted by two FAs incubated with the particles, one that recognizes mouse serum proteins (FA2, gAm antibodies; Fig. 4D) compared with one that does not (FA1, gAr antibodies; Fig. 4C). Lack of fluorescence from FA1 indicates that mouse serum proteins remain uniformly bound to the particles in their native conformation, thereby preventing FA1 from binding directly to the particles. Moreover, protein binding may be promoted through adding a positive charge to the hydrophobic polymer particle surface by coating it with poly-l-lysine, a positively charged synthetic amino acid chain. This enhancement was confirmed by detecting stronger fluorescence from bound antibodies (compare the panels in Fig. 4 A and E) and also by observing Förster resonant energy transfer (FRET) between two tandem layers of particle-bound FAs (56, 57) from FA1 to FA3 (Alexa Fluor-647–conjugated anti-goat antibodies produced in donkey). Antibodies FA3 recognize FA1 and fluorescence from FA1 overlaps with the FA3 excitation band, thereby allowing for FRET transfer (Fig. 4E). We further verified that protein binding is independent of particle diameter down to 500-nm diameters by incubating 500-nm-diameter COP particles (Figs. 2G and 4F) with FA4 (Alexa Fluor-488–conjugated gAm antibodies).
Fig. 4.

Quantitative assessment of preferential protein coating to polymer particles. (A and B) COP particles coated with FA1 (gAr) or FA2 (gAm) yield yellow or red fluorescence when excited at 561 nm or 647 nm, respectively. Here, 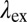 is the excitation wavelength,
is the excitation wavelength, 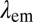 is the emission wavelength, and green and red arrows correspond to the excitation with
is the emission wavelength, and green and red arrows correspond to the excitation with 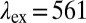 nm and
nm and 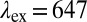 nm, respectively. (C and D) COP particles coated with mouse serum (MS) yield red fluorescence with MS-specific FA2 (gAm) and none with nonspecific FA1 (gAr). (E) COP particles after treatment with poly-l-lysine are coated with FA1 and then FA3 [anti-goat antibodies produced in donkey (dAg), which recognize FA1]. Both antibodies independently produce fluorescence upon excitation. When FA1 is excited (at
nm, respectively. (C and D) COP particles coated with mouse serum (MS) yield red fluorescence with MS-specific FA2 (gAm) and none with nonspecific FA1 (gAr). (E) COP particles after treatment with poly-l-lysine are coated with FA1 and then FA3 [anti-goat antibodies produced in donkey (dAg), which recognize FA1]. Both antibodies independently produce fluorescence upon excitation. When FA1 is excited (at 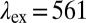 nm) and the red fluorescence of FA3 at
nm) and the red fluorescence of FA3 at 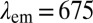 nm is monitored, we observe evidence for FRET transfer from FA1 to FA3. (F, i) Differential interference contrast (DIC) micrograph of a COP nanoparticle (diameter of ∼500 nm; Fig. 2G). FL, fluorescence. (F, ii) Native autofluorescence of the COP particle under short-wavelength (405 nm) excitation. (F, iii) Fluorescence micrograph of the COP particle after coating with FA4. (F, iv) Combined DIC and fluorescence image of the FA4-coated COP nanoparticle. (Scale bars: 500 nm.)
nm is monitored, we observe evidence for FRET transfer from FA1 to FA3. (F, i) Differential interference contrast (DIC) micrograph of a COP nanoparticle (diameter of ∼500 nm; Fig. 2G). FL, fluorescence. (F, ii) Native autofluorescence of the COP particle under short-wavelength (405 nm) excitation. (F, iii) Fluorescence micrograph of the COP particle after coating with FA4. (F, iv) Combined DIC and fluorescence image of the FA4-coated COP nanoparticle. (Scale bars: 500 nm.)
In addition to binding proteins uniformly to the polymer particles, we obtained clear evidence for quantitative control over the relative composition of the immobilized proteins using different mixtures of cofilin and BSA as examples. We confirmed that the relative composition of the particle-bound proteins matches that of the mixtures used in solution, first by dissociation via boiling in a denaturing buffer and quantification of the recovered proteins by electrophoresis in SDS polyacrylamide gels stained to identify BSA and cofilin. Second, we monitored the amount of particle-bound cofilin directly by incubation with specific FAs and measuring the fluorescence (Supporting Information). These results thus confirm that the particles can be coated with a controlled amount of a protein that can be retrieved for subsequent characterization and quantification.
Finally, we demonstrate a unique feature of our polymer particle fabrication strategy that highlights its versatility with respect to structural design. By modifying the macroscopic preform structure, we produce a unique particle geometry that enables us to establish microencapsulation of biological materials inside a polymeric shell of the same polymer used in the solid particles used above in the protein-binding experiments. We use collagen as the encapsulant, which may be useful for cosmetics and dental applications (58, 59) and serves as a model for other potential biological materials or drugs. Like many biological materials, collagen is a globular solution lacking uniform fluid consistency, and is thus incompatible with fiber drawing and traditional microfluidics-based multiple-emulsion approaches. We obviate this limitation by injecting the desired encapsulant inside a drawn hollow fiber in which a COP layer lines the PSU cladding (Fig. 5A and Supporting Information). We inject collagen into the 50-μm-diameter hollow core (Fig. 5C) and thermally induce the PRI, which results in the core breaking up into a necklace of collagen-filled COP microcapsules (Fig. 5D) held stationary in the PSU cladding, which are then released using DMAC (Fig. 5E). We confirm that the microcapsules contain protein by dissolving the COP shell (Fig. 5F) and then using a Coomassie Brilliant Blue dye Bradford protein-binding assay (60) with the recovered cargo (Supporting Information).
Fig. 5.

Microencapsulation of biological materials in polymer shells. (A) Schematic of the steps for producing collagen-filled COP microcapsules. (Inset) Hollow-fiber core lined with COP after dissolving the PSU cladding at the fiber tip. Optical transmission micrograph of the side view of the fiber before (B) and after (C) being filled with collagen (slightly doped with a dye for visualization). (D) Encapsulated collagen particles (COP shell) held in the PSU cladding. (E) Collagen/COP microcapsules released from the cladding. (Scale bars: B–E, 200 μm.) (F) Collagen recovered after dissolving the COP shell. (G) A confocal fluorescence micrograph of a collagen-filled microparticle with surface-bound FAs. (H) An overlay of a bright-field confocal micrograph on the fluorescence image from G. (Scale bars: 20 μm.) P1, COP; P2, PSU.
The two modalities discussed thus far, surface functionalization and volume encapsulation, are combined in Fig. 5 G and H, where we demonstrate surface binding of FA4 to collagen-filled COP microcapsules. In comparison to current encapsulation approaches that usually target biodegradable polymeric shells (61, 62), our approach enables encapsulation with a wider range of polymers, in addition to access to a broad range of sizes.
Discussion
We anticipate that by embedding multiple hollow cores within a polymer shell and filling each with different, and potentially incompatible, biological or chemical agents, the PRI-driven breakup will produce a polymeric particle that encapsulates these agents in isolated enclaves such that the encapsulants do not come into contact at any point during the process. Consequently, each particle may be considered a multicompartment microreactor that produces localized chemical interactions using prescribed ratios of reactants upon delivery of the cargo.
Such size-, material-, and structure-tunable polymeric particles fabricated using the scalable, in-fiber, fluid instability-mediated process are thus compatible with protein binding and biosensing through protein–protein interactions. We expect that this methodology will have broad applications in developing diagnostic tools and cell-specific targeting of biomaterials. In addition, optical, magnetic, and potentially plasmonic functionalities may be embedded within each particle through impregnation of the fiber core at the preform level with appropriate dopants. An important goal is to extend our methodology to polymers that may be processed at lower temperatures to reduce damage or modification to encapsulated or impregnated biological materials, thereby paving the way to therapeutic applications in drug delivery. Combining our results on the precise structuring of multimaterial particles using this in-fiber approach (35) with the biomedical capabilities described here paves the way for the digital design of a unique class of nanoengineered theranostic tools (63), where the chemical, hydroscopic, optical, and biomedical properties are all tuned simultaneously within each structured particle through judicious design of the macroscopic preform from which the fiber is drawn.
Materials and Methods
All the steps and details of the fiber fabrication, particle production, and extraction are provided in Supporting Information. Additionally, we give the details of surface functionalization and fluorescence measurements. Further data on quantifying protein attachment to the particle surfaces is provided.
Supplementary Material
Acknowledgments
This work was supported by the Air Force Office of Scientific Research under Contract FA-9550-12-1-0148; National Institutes of Health Shared Instrument Grant S10RR027142 (to R.C.); and, in part, by the Materials Research Science and Engineering Program of the US National Science Foundation under Award DMR-0819762.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1310214110/-/DCSupplemental.
References
- 1.Derveaux S, et al. Synergism between particle-based multiplexing and microfluidics technologies may bring diagnostics closer to the patient. Anal Bioanal Chem. 2008;391(7):2453–2467. doi: 10.1007/s00216-008-2062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J, et al. Motion-based DNA detection using catalytic nanomotors. Nat Commun. 2010 doi: 10.1038/ncomms1035. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, et al. Synthesis, biodistribution, and microsingle photon emission computed tomography (SPECT) imaging study of technetium-99m labeled PEGylated dendrimer poly(amidoamine) (PAMAM)-folic acid conjugates. J Med Chem. 2010;53(8):3262–3272. doi: 10.1021/jm901910j. [DOI] [PubMed] [Google Scholar]
- 4.Bhavsar MD, Amiji MM. Polymeric nano- and microparticle technologies for oral gene delivery. Expert Opin Drug Deliv. 2007;4(3):197–213. doi: 10.1517/17425247.4.3.197. [DOI] [PubMed] [Google Scholar]
- 5.Champion JA, Katare YK, Mitragotri S. Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. J Control Release. 2007;121(1-2):3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh R, Lillard JW., Jr Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ElKaoutit M, Naggar AH, Naranjo-Rodríguez I, de Cisneros JL. Graphite grains studded with silver nanoparticles: description and application in promoting direct biocatalysis between heme protein and the resulting carbon paste electrode. Colloids Surf B Biointerfaces. 2012;92:42–49. doi: 10.1016/j.colsurfb.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 8.Bai M-Y, et al. A facile and general method for the encapsulation of different types of imaging contrast agents within micrometer-sized polymer beads. Adv Funct Mater. 2012;22(4):764–770. doi: 10.1002/adfm.201102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng Y, et al. Long-circulating polymeric nanoparticles bearing a combinatorial coating of PEG and water-soluble chitosan. Biomaterials. 2009;30(12):2340–2348. doi: 10.1016/j.biomaterials.2008.12.070. [DOI] [PubMed] [Google Scholar]
- 10.Tsai MJ, et al. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. International Journal of Pharmaceutics. 2012;423(2):461–470. doi: 10.1016/j.ijpharm.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26(1):57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Farokhzad OC, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;103(16):6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, et al. Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J Control Release. 2008;128(3):209–216. doi: 10.1016/j.jconrel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Greenhalgh K, Turos E. In vivo studies of polyacrylate nanoparticle emulsions for topical and systemic applications. Nanomedicine. 2009;5(1):46–54. doi: 10.1016/j.nano.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Hu CM, Fang RH, Copp J, Luk BT, Zhang L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotechnol. 2013;8(5):336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109(28):11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vollath D. Nanomaterials: An Introduction to Synthesis, Properties and Application. Weinheim, Germany: Wiley–VCH; 2008. [Google Scholar]
- 18.Sjöblom J. Encyclopedic Handbook of Emulsion Technology. New York: Dekker; 2001. [Google Scholar]
- 19.Kelly JY, DeSimone JM. Shape-specific, monodisperse nano-molding of protein particles. J Am Chem Soc. 2008;130(16):5438–5439. doi: 10.1021/ja8014428. [DOI] [PubMed] [Google Scholar]
- 20.Gañán-Calvo AM, Gordillo JM. Perfectly monodisperse microbubbling by capillary flow focusing. Phys Rev Lett. 2001;87(27 Pt 1):274501. doi: 10.1103/PhysRevLett.87.274501. [DOI] [PubMed] [Google Scholar]
- 21.Thorsen T, Roberts RW, Arnold FH, Quake SR. Dynamic pattern formation in a vesicle-generating microfluidic device. Phys Rev Lett. 2001;86(18):4163–4166. doi: 10.1103/PhysRevLett.86.4163. [DOI] [PubMed] [Google Scholar]
- 22.Nisisako T, Torii T, Higuchi T. Droplet formation in a microchannel network. Lab Chip. 2002;2(1):24–26. doi: 10.1039/b108740c. [DOI] [PubMed] [Google Scholar]
- 23.Anna SL, Bontoux N, Stone HA. Formation of dispersions using “flow focusing” in microchannels. Appl Phys Lett. 2003;82(3):364–366. [Google Scholar]
- 24.Cramer C, Fischer P, Windhab EJ. Drop formation in a coflowing ambient fluid. Chem Eng Sci. 2004;59(15):3045–3058. [Google Scholar]
- 25.Utada AS, et al. Monodisperse double emulsions generated from a microcapillary device. Science. 2005;308(5721):537–541. doi: 10.1126/science.1109164. [DOI] [PubMed] [Google Scholar]
- 26.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9(5):1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 27.Wong C, et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci USA. 2011;108(6):2426–2431. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doiron AL, Clark B, Rinker KD. Endothelial nanoparticle binding kinetics are matrix and size dependent. Biotechnol Bioeng. 2011;108(12):2988–2998. doi: 10.1002/bit.23253. [DOI] [PubMed] [Google Scholar]
- 29.Plateau JAF. Statique Experimentale et Theorique des Liquides Soumis aux Seules Forces Moleculaires. Vol 2. Paris: Gauthier Villars; 1873. [Google Scholar]
- 30.Rayleigh L. On the capillary phenomena of jets. Proc R Soc Lond. 1879;29:71–97. [Google Scholar]
- 31.Eggers J, Villermaux E. Physics of liquid jets. Rep Prog Phys. 2008;71(3):036601. [Google Scholar]
- 32.Abouraddy AF, et al. Towards multimaterial multifunctional fibres that see, hear, sense and communicate. Nat Mater. 2007;6(5):336–347. doi: 10.1038/nmat1889. [DOI] [PubMed] [Google Scholar]
- 33.Tao G, Stolyarov AM, Abouraddy AF. Multimaterial fibers. International Journal of Applied Glass Science. 2012;3(4):349–368. [Google Scholar]
- 34.Shabahang S, Kaufman JJ, Deng DS, Abouraddy AF. Observation of the Plateau-Rayleigh capillary instability in multi-material optical fibers. Appl Phys Lett. 2011;99(16):161909. [Google Scholar]
- 35.Kaufman JJ, et al. Structured spheres generated by an in-fibre fluid instability. Nature. 2012;487(7408):463–467. doi: 10.1038/nature11215. [DOI] [PubMed] [Google Scholar]
- 36.Agemy L, et al. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci USA. 2011;108(42):17450–17455. doi: 10.1073/pnas.1114518108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H, et al. Antibody conjugated magnetic iron oxide nanoparticles for cancer cell separation in fresh whole blood. Biomaterials. 2011;32(36):9758–9765. doi: 10.1016/j.biomaterials.2011.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu JN, et al. Simultaneous nuclear imaging and intranuclear drug delivery by nuclear-targeted multifunctional upconversion nanoprobes. Biomaterials. 2012;33(29):7282–7290. doi: 10.1016/j.biomaterials.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 39.Souto EB, Müller RH. Cosmetic features and applications of lipid nanoparticles (SLN, NLC) Int J Cosmet Sci. 2008;30(3):157–165. doi: 10.1111/j.1468-2494.2008.00433.x. [DOI] [PubMed] [Google Scholar]
- 40. Gong Y, Cabodi M, Porter TM (2009) Measurement of the attenuation coefficient for monodisperse populations of ultrasound contrast agents. Conf Proc IEEE Eng Med Biol Soc, 10.1109/IEMBS.2009.5333441. [DOI] [PubMed]
- 41.You J, et al. Effective photothermal chemotherapy using doxorubicin-loaded gold nanospheres that target EphB4 receptors in tumors. Cancer Res. 2012;72(18):4777–4786. doi: 10.1158/0008-5472.CAN-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunes PS, Ohlsson PD, Ordeig O, Kutter JP. Cyclic olefin polymers: Emerging materials for lab-on-a-chip applications. Microfluid Nanofluid. 2010;9(2-3):145–161. [Google Scholar]
- 43.Tomotika S. On the instability of a cylindrical thread of a viscous liquid surrounded by another viscous fluid. Proc R Soc Lond A Math Phys Sci. 1935;150(870):322–337. [Google Scholar]
- 44.Kaufman JJ, et al. Thermal drawing of high-density macroscopic arrays of well-ordered sub-5-nm-diameter nanowires. Nano Lett. 2011;11(11):4768–4773. doi: 10.1021/nl202583g. [DOI] [PubMed] [Google Scholar]
- 45.Deng DS, et al. In-fiber semiconductor filament arrays. Nano Lett. 2008;8(12):4265–4269. doi: 10.1021/nl801979w. [DOI] [PubMed] [Google Scholar]
- 46.Deng DS, et al. Processing and properties of centimeter-long, in-fiber, crystalline-selenium filaments. Appl Phys Lett. 2010;96(2):023102. [Google Scholar]
- 47. Kai J, Sohn YS, Ahn CH (2003) Proceedings of uTAS 2003–Seventh International Conference on Miniaturized Chemical and Biochemical Analysis Systems, eds Northrup MA, Jensen KF, Harrison DJ (Transducers Research Foundation, Squaw Valley, CA), pp 1101–1104.
- 48.Jena RK, Yue CY. Cyclic olefin copolymer based microfluidic devices for biochip applications: Ultraviolet surface grafting using 2-methacryloyloxyethyl phosphorylcholine. Biomicrofluidics. 2012;6(1):12822–1282212. doi: 10.1063/1.3682098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao G, Shabahang S, Banaei E-H, Kaufman JJ, Abouraddy AF. Multimaterial preform coextrusion for robust chalcogenide optical fibers and tapers. Opt Lett. 2012;37(13):2751–2753. doi: 10.1364/OL.37.002751. [DOI] [PubMed] [Google Scholar]
- 50.Liang X, Deng DS, Nave J-C, Johnson SG. Linear stability analysis of capillary instabilities for concentric cylindrical shells. J Fluid Mech. 2011;683:235–262. [Google Scholar]
- 51.Deng DS, Nave J-C, Liang X, Johnson SG, Fink Y. Exploration of in-fiber nanostructures from capillary instability. Opt Express. 2011;19(17):16273–16290. doi: 10.1364/OE.19.016273. [DOI] [PubMed] [Google Scholar]
- 52. Liang X (2013) Modeling of fluids and waves with analytics and numerics. PhD thesis (Massachusetts Institute of Technology, Cambridge, MA)
- 53.Chen KY, et al. Asymmetric chitosan membrane containing collagen I nanospheres for skin tissue engineering. Biomacromolecules. 2009;10(6):1642–1649. doi: 10.1021/bm900238b. [DOI] [PubMed] [Google Scholar]
- 54.Sakai K, Hashimoto Y, Baba S, Nishiura A, Matsumoto N. Effects on bone regeneration when collagen model polypeptides are combined with various sizes of alpha-tricalcium phosphate particles. Dent Mater J. 2011;30(6):913–922. doi: 10.4012/dmj.2011-126. [DOI] [PubMed] [Google Scholar]
- 55.Alarcon EI, et al. The biocompatibility and antibacterial properties of collagen-stabilized, photochemically prepared silver nanoparticles. Biomaterials. 2012;33(19):4947–4956. doi: 10.1016/j.biomaterials.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 56.Lippincott-Schwartz J, Snapp E, Kenworthy AK. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2(6):444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 57.Volkhard H. Principles of Computational Cell Biology. Weinheim, Germany: Wiley–VCH; 2008. [Google Scholar]
- 58.Helary C, et al. Concentrated collagen hydrogels as dermal substitutes. Biomaterials. 2010;31(3):481–490. doi: 10.1016/j.biomaterials.2009.09.073. [DOI] [PubMed] [Google Scholar]
- 59.Thoma DS, Sancho-Puchades M, Ettlin DA, Hämmerle CH, Jung RE. Impact of a collagen matrix on early healing, aesthetics and patient morbidity in oral mucosal wounds—A randomized study in humans. J Clin Periodontol. 2012;39(2):157–165. doi: 10.1111/j.1600-051X.2011.01823.x. [DOI] [PubMed] [Google Scholar]
- 60.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72(1-2):248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 61.Imoto T, Kida T, Matsusaki M, Akashi M. Preparation and unique pH-responsive properties of novel biodegradable nanocapsules composed of poly(gamma-glutamic acid) and chitosan as weak polyelectrolytes. Macromol Biosci. 2010;10(3):271–277. doi: 10.1002/mabi.200900272. [DOI] [PubMed] [Google Scholar]
- 62.Hudson D, Margaritis A. Biopolymer nanoparticle production for controlled release of biopharmaceuticals. Crit Rev Biotechnol. 2013 doi: 10.3109/07388551.2012.743503. [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, et al. PSMA-targeted theranostic nanoplex for prostate cancer therapy. ACS Nano. 2012;6(9):7752–7762. doi: 10.1021/nn301725w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.