

Significance
The TGF-β family encompasses a large number of secreted proteins that regulate embryonic development and adult tissue homeostasis. Growth and differentiation factor (GDF) -associated serum protein-1 (GASP-1) and GASP-2 are related proteins capable of binding and inhibiting two family members, myostatin and GDF-11. Here, we show that mice genetically engineered to lack GASP-1 and/or GASP-2 exhibit muscle and skeletal phenotypes consistent with overactivity of myostatin and/or GDF-11. These studies also reveal the enormous complexity of this regulatory system in vivo and the delicate balance that must be maintained between signaling molecules and their inhibitory proteins for proper levels of signaling to be achieved.
Abstract
Myostatin (MSTN) and growth and differentiation factor-11 (GDF-11) are highly related TGF-β family members that have distinct biological functions. MSTN is expressed primarily in skeletal muscle and acts to limit muscle growth. GDF-11 is expressed more widely and plays multiple roles, including regulating axial skeletal patterning during development. Several MSTN and GDF-11 binding proteins have been identified, including GDF-associated serum protein-1 (GASP-1) and GASP-2, which are capable of inhibiting the activities of these ligands. Here, we show that GASP-1 and GASP-2 act by blocking the initial signaling event (namely, the binding of the ligand to the type II receptor). Moreover, we show that mice lacking Gasp1 and Gasp2 have phenotypes consistent with overactivity of MSTN and GDF-11. Specifically, we show that Gasp2−/− mice have posteriorly directed transformations of the axial skeleton, which contrast with the anteriorly directed transformations seen in Gdf11−/− mice. We also show that both Gasp1−/− and Gasp2−/− mice have reductions in muscle weights, a shift in fiber type from fast glycolytic type IIb fibers to fast oxidative type IIa fibers, and impaired muscle regeneration ability, which are the reverse of what are seen in Mstn−/− mice. All of these findings suggest that both GASP-1 and GASP-2 are important modulators of GDF-11 and MSTN activity in vivo.
The TGF-β superfamily comprises almost 40 signaling proteins that play important regulatory roles both during embryogenesis and in adult tissues. Because these proteins are potent regulators of cell growth and differentiation, there has been extensive focus on elucidating the molecular mechanisms by which these proteins signal and their activities are regulated, with the long-term goal of developing strategies to modulate their activity levels for a variety of different clinical indications.
Myostatin (MSTN) and growth and differentiation factor-11 (GDF-11) are closely related TGF-β family members that share 89% amino acid sequence identity within the mature C-terminal region (1, 2). Although the activities of these molecules are indistinguishable in in vitro assays, their different tissue expression patterns confer distinct biological functions. MSTN is expressed predominantly in skeletal muscle and functions as a negative regulator of muscle growth (1). As a result, there is considerable interest in developing MSTN inhibitors to improve muscle growth in various clinical settings, including muscular dystrophy, age-related sarcopenia, and cancer cachexia. However, GDF-11 is expressed in a wide range of tissues and has been shown to play important roles in the development of the olfactory system (3), retina (4), and pancreas (5, 6) as well as in anterior–posterior patterning of the axial skeleton (7). In adult mice, GDF-11 circulates in the blood (8), and declining levels of circulating GDF-11 have been implicated in the etiology of age-related cardiac hypertrophy (9).
The regulation of MSTN and GDF-11 seems to be complex, because multiple proteins have been identified that are capable of binding these ligands and inhibiting their activities (8, 10). One of these binding proteins is GDF-associated serum protein-1 (GASP-1), which was isolated as an MSTN-associated protein from mouse and human serum by affinity purification using an anti-MSTN antibody (11). GASP-1, also known as WFIKKNRP or WFIKKN2, contains many conserved domains associated with protease-inhibitory proteins, including a whey acidic protein domain, a follistatin/Kazal domain, an Ig domain, two tandem Kunitz domains, and a netrin domain (11, 12). GASP-1 is closely related to GASP-2, also known as WFIKKN or WFIKKN1, which has the same overall domain structure and shares 54% amino acid sequence identity and 69% sequence similarity with GASP-1 (12, 13). Both GASP-1 and GASP-2 have been shown to be capable of blocking MSTN and GDF-11 activity in vitro (11, 14, 15), and overexpression of GASP-1 in mice has also been shown to cause increased muscle growth, consistent with inhibition of MSTN activity (16, 17). What roles, if any, GASP-1 and GASP-2 normally play in regulating the activity of MSTN and GDF-11 in vivo are not known. Here, we present data showing that both GASP-1 and GASP-2 act by blocking the initial binding of the ligand to its receptor. We also show that mice carrying targeted mutations in Gasp1 and/or Gasp2 have skeletal muscle and axial patterning phenotypes consistent with altered MSTN and GDF-11 signaling, respectively. Our findings show the important roles that GASP-1 and GASP-2 play in regulating the activities of these ligands.
Results
Both GASP-1 and GASP-2 Inhibit MSTN and GDF-11.
Previous studies have shown that GASP-1 and GASP-2 produced as fusion proteins in either COS1 cells or Drosophila S2 cells are capable of binding MSTN and GDF-11 and inhibiting their activities in vitro (11, 14). To characterize further the biological activities of GASP-1 and GASP-2, we sought to obtain highly purified preparations of these proteins in their native state. For this purpose, we generated CHO cell lines expressing high levels of either murine GASP-1 or murine GASP-2 and then purified GASP-1 and GASP-2 from the conditioned medium of these cells. GASP-1 protein was purified by successive fractionation using butyl Sepharose, HiTrap heparin Sepharose, and wheat germ lectin Sepharose. GASP-2 protein was purified by successive fractionation using butyl Sepharose, HiTrap heparin Sepharose, and HiTrap Q Sepharose. Based on silver stain analysis, these fractionation schemes yielded highly purified preparations of GASP-1 and GASP-2 (Fig. 1A).
Fig. 1.

Both GASP-1 and GASP-2 inhibit MSTN and GDF-11 in vitro. (A) Silver stain analysis of purified GASP-1 and GASP-2. (B and C) Effect of GASP-1 (open boxes) and GASP-2 (closed circles) on luciferase reporter activity induced in A204 cells by (B, Left) 20 ng/mL MSTN, (B, Right) 20 ng/mL GDF-11, or 20 ng/mL Activin A (C, Left) or C3H10T1/2 cells by (C, Center) 0.3 ng/mL TGF-β1 and (C, Right) 50 ng/mL BMP-4. RLU, relative light unit. (D) Effect of GASP-1 on GDF-11 binding to ACVR2B/Fc. Immunoblot analysis of GDF-11 (lane 1), purified GASP-1 (lane 2), or protein A Sepharose-bound proteins (lanes 3–7) was carried out using (Top) anti–GDF-11, (Middle) anti–GASP-1, or (Bottom) anti-Fc antibodies. (E) Effect of GASP-2 on GDF-11 binding to ACVR2B/Fc. Immunoblot analysis was performed as in D, except that purified GASP-2 and anti–GASP-2 antibodies were used in place of purified GASP-1 and anti–GASP-1 antibodies, respectively. Data were shown as mean ± SEM.
To confirm that these purified proteins were biologically active, we tested the ability of these proteins to inhibit MSTN and GDF-11 activity in vitro. Specifically, we tested purified GASP-1 and GASP-2 for their ability to block MSTN- and GDF-11–induced reporter gene activity in A204 rhabdomyosarcoma cells carrying an Smad2/3-responsive pGL3-(CAGA)12-luciferase expression construct (18). As shown in Fig. 1B, both GASP-1 and GASP-2 inhibited MSTN and GDF-11 activity in this reporter assay, with GASP-1 having an IC50 value ∼10-fold lower than the IC50 value of GASP-2 (∼833 pM for GASP-1 vs. ∼7,792 pM for GASP-2). To examine the ligand specificity of GASP-1 and GASP-2, we tested the ability of these proteins to inhibit the activities of other TGF-β superfamily members, specifically activin A, TGF-β1, and BMP-4, using the same A204 reporter cells, C3H10T1/2 cells carrying a p3TP-Lux reporter construct (19), or C3H10T1/2 cells carrying a (BRE)2-Luc reporter construct (20), respectively. Consistent with previous reports (11, 14, 15), neither GASP-1 nor GASP-2 was capable of blocking the activities of these other ligands (Fig. 1C). Hence, our results confirm that both of these proteins have a high degree of specificity for MSTN and GDF-11.
Both GASP-1 and GASP-2 Inhibit GDF-11 Binding to the Type II Receptor.
To understand the mechanism by which GASP-1 and GASP-2 exert their inhibitory effects, we attempted to identify the step in the overall MSTN/GDF-11 signaling pathway that is sensitive to GASP-1 and GASP-2. We started by examining the ability of GASP-1 and GASP-2 to block the first step in the pathway, namely the binding of the ligand to the type II receptor. Because MSTN and GDF-11 share 89% amino acid sequence identity in the mature C-terminal region and seem to use the same signaling components (21), we focused our analysis only on GDF-11, because this protein is more readily available. We examined the effect of GASP-1 and GASP-2 on binding of GDF-11 to its high-affinity receptor, the activin type IIB receptor (ACVR2B), using a purified soluble form of the receptor, in which we fused the ligand binding portion of ACVR2B to an Fc domain (ACVR2B/Fc). We preincubated purified GASP-1 and GASP-2 proteins with the C-terminal dimer of GDF-11, added purified ACVR2B/Fc to the mixture, and finally, isolated the ACVR2B/Fc-bound proteins using protein A Sepharose. Immunoblot analysis of the eluted proteins showed that, as expected, GDF-11 bound readily to ACVR2B/Fc in the absence of either GASP-1 (Fig. 1D) or GASP-2 (Fig. 1E). With increasing concentrations of GASP-1 or GASP-2, however, the amount of GDF-11 binding to ACVR2B/Fc was decreased in a concentration-dependent manner. Hence, GASP-1 and GASP-2 are capable of blocking the first step in the signaling cascade by binding the ligand and inhibiting its ability to engage the type II receptor. While this work was being completed, similar results were reported in the work by Szláma et al. (15) using surface plasmon resonance analysis.
Expression Patterns of Gasp1 and Gasp2.
Based on the ability of GASP-1 and GASP-2 to inhibit MSTN and GDF-11 activity in vitro, the identification of GASP-1 as a protein bound to MSTN in the blood (11), and the demonstration that overexpression of GASP-1 can increase muscle growth in mice (16, 17), we investigated the role that GASP-1 and GASP-2 may normally play in the regulation of MSTN and GDF-11 activity in vivo. We first examined the expression patterns of Gasp1 and Gasp2 to determine whether either of these genes is expressed in tissues that would be consistent with regulation of either MSTN or GDF-11 function. By RNA blot analysis (Fig. 2A), the expression of Gasp1 was detected in many adult tissues, including skeletal muscle, which is the predominant site of Mstn expression (1). In contrast, the expression of Gasp2 seemed to be much more tissue-restricted, and its expression in skeletal muscle was below the level of detection by RNA blot analysis (Fig. 2A).
Fig. 2.
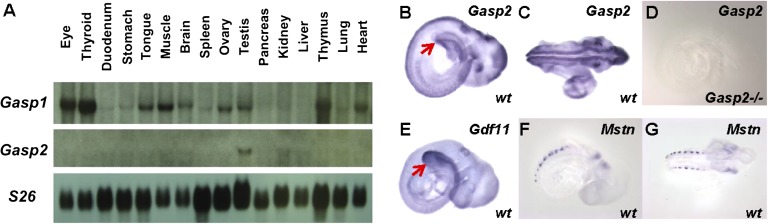
Expression patterns of Gasp1 and Gasp2. (A) RNA blot analysis of adult mouse tissues (20 µg total RNA) with (Top) Gasp1 and (Middle) Gasp2 probes. (Bottom) S26 was used as the loading control. (B–G) Whole-mount in situ hybridization of mouse embryos at 9.5 d postcoitum. Lateral and dorsal views of the same embryo are shown in B and C, respectively. In B, the arrow denotes expression of Gasp2 in the posterior region. (D) Absence of Gasp2 expression in Gasp2−/− embryos. (E) Whole-mount in situ hybridization showing expression of Gdf11 in the posterior region of the embryo (arrow). (F and G) Lateral and dorsal views, respectively, of an embryo hybridized with an Mstn probe showing expression in somites.
We also examined the expression patterns of these genes during embryogenesis. By whole-mount in situ hybridization of early mouse embryos, we were unable to detect any expression of Gasp1. In contrast, Gasp2 expression was readily detected in a variety of sites in day 9.5 postcoitum embryos (Fig. 2 B and C), including the retina, otocyst, and neural tube, consistent with previously reported information on rat embryos (22). One notable difference between our results and the results of the prior study, however, was that we also observed prominent expression of Gasp2 in the posterior region of the embryo (Fig. 2B). To confirm the specificity of this hybridization, we showed that no Gasp2 expression could be detected by whole-mount in situ hybridization analysis of Gasp2−/− embryos (see below) using the same probe (Fig. 2D). This expression pattern was intriguing, because Gdf11 is also known to be expressed in the posterior region of early stage embryos, specifically the primitive streak region and tail bud, and Gdf11 expression in this region is believed to be responsible for the critical role that GDF-11 plays in regulating anterior/posterior axial patterning (7). Although the expression pattern of Gasp2 did not coincide precisely with the expression pattern of Gdf11 (compare Fig. 2B with Fig. 2E), the fact that these two secreted proteins are expressed in the same region raised the possibility that they may functionally interact during development. Also noteworthy was the absence of either Gasp1 or Gasp2 expression in developing somites, which is the primary site of Mstn expression during early embryogenesis (Fig. 2 F and G) (1).
Posteriorly Directed Transformations in Gasp2−/−.
To investigate the functions of GASP-1 and GASP-2 in vivo, we used gene targeting to generate mice carrying loss-of-function mutations in these genes. We generated targeting constructs, in which we flanked exon 2 with LoxP sites (Fig. 3A); because exon 2 contains nearly the entire coding sequence of each gene, it seemed almost certain that deletion of exon 2 by cre-mediated recombination would result in a null allele. For both Gasp1 and Gasp2, mice homozygous for the deletion allele were viable and fertile, and therefore, all subsequent analysis was carried out using these deletion alleles.
Fig. 3.

Role of Gasp2 in axial skeletal patterning. (A) Gene targeting strategy for Gasp1 and Gasp2. NEO, neomycin resistance cassette; TK, thymidine kinase. (B) Vertebral columns of WT, Gasp2−/−, Gasp2−/−;Fst−/−, Gdf11+/−, and Gdf11−/− mice. Note the changes in the number of ribs. *Gasp2−/−;Fst−/− mice also had a reduced number of lumbar vertebrae from six to four, with extensive fusion of lumbar and sacral segments. (C) Cervical and anterior thoracic regions of WT, Gasp2−/−, and Gdf11−/− mice. Note the shift of the position of the anterior tuberculum (arrow) from C6 to C5 in Gasp2−/− mice and from C6 to C7 in Gdf11−/− mice. (D) Vertebrosternal ribs of WT, Gasp2−/−;Fst+/−, Gasp2−/−;Fst−/−, Gdf11+/−, and Gdf11−/− mice. Note changes in the number of attached ribs and additional cervical ribs (arrow). (E) Schematic representation of vertebral columns. Gdf11+/− and Gdf11−/− mice have anteriorly directed homeotic transformations of the axial skeleton. In contrast, mice lacking certain GDF-11 inhibitors (Gasp2−/− and Gasp2−/−;Fst−/−) have posteriorly directed transformations. Cervical (orange), thoracic (purple), and lumbar (sky blue) vertebrae, anterior tuberculi (small blue dots), ectopic ribs from a cervical vertebra (green curved lines), sternums (red curves), and ribs (blue lines) are color coded as indicated. The dashed lines indicate the normal positions of typical vertebral characteristics: 6 for the anterior tuberculum, 14 for the most posterior rib attached to the sternum, 20 for the most posterior thoracic vertebra, and 26 for the most posterior lumbar vertebra.
To investigate possible roles for GASP-1 and GASP-2 in the regulation of GDF-11 activity, we focused on the known role of GDF-11 in regulating axial skeletal patterning. Previous studies have shown that Gdf11−/− mice have dramatic anteriorly directed homeotic transformations of the axial skeleton (i.e., posterior regions take a more anterior fate) (7). If GASP-1 or GASP-2 normally acts to block GDF-11 activity, one prediction is that mice lacking GASP-1 or GASP-2 would have the opposite phenotype, namely posteriorly directed homeotic transformations. We analyzed the axial skeletal patterns of Gasp1−/− and Gasp2−/− mice by staining newborn carcasses for bone and cartilage with Alizarin red and Alcian blue, respectively. Although the axial skeletal patterning of Gasp1−/− mice was similar to the axial skeletal patterning of WT mice, Gasp2−/− mice exhibited posteriorly directed homeotic transformations throughout the axial skeleton with incomplete penetrance (Fig. 3 B and C, Table 1, and Table S1). WT mice most commonly have 7 cervical, 13 thoracic, and 6 lumbar vertebrae. The most frequent abnormality seen in Gasp2−/− mice was a defect in the formation of the 13th rib; almost 90% of homozygous mutants had at least one truncated or absent 13th rib, and over 40% had a complete absence of both ribs, reflecting a transformation of the 13th thoracic segment into a lumbar segment. Posteriorly directed transformations were observed in other regions of the axial skeleton as well, albeit at lower frequencies. The next most common abnormality was the partial or complete loss of one lumbar segment, which was seen in ∼12% of Gasp2−/− mice. In the cervical region, some rarer abnormalities were observed, including a supernumerary rib arising from the seventh cervical (C7) vertebra (cervical rib) and a shift in the position of the anterior tuberculum from C6 to C5 (Fig. 3C). Hence, Gasp2−/− mice exhibited a wide range of defects in axial skeletal patterning, all consistent with posteriorly directed homeotic transformations.
Table 1.
Skeletal analysis of WT, Gasp1−/−, Gasp2−/−, and Fst−/− mice in C57BL/6 background
| Genotype | WT | Gasp1−/− | Gasp2+/− | Gasp2−/− | Gasp2−/−; Fst+/− | Gasp2+/−; Fst−/− | Gasp2−/−; Fst−/− |
| N | 29 | 24 | 65 | 41 | 12 | 6 | 10 |
| Vertebral pattern | |||||||
| C7T13L6 | 28 (96.6) | 24 (100) | 61 (93.8) | 28 (68.3) | — | — | — |
| C7T13L5/6 | — | — | 2 (3.1) | — | 1 (8.3) | — | — |
| C7T13L5 | 1 (3.4) | — | 2 (3.1) | 1 (2.4) | — | 3 (50) | — |
| C7T12L7 | — | — | — | 6 (14.6) | — | — | — |
| C7T12L6/7 | — | — | — | 4 (9.8) | 3 (25) | — | — |
| C7T12L6 | — | — | — | 2 (4.9) | 8 (66.7) | 3 (50) | 9 (90) |
| C7T12L4 | — | — | — | — | — | — | 1 (10) |
| 13th pair of ribs | |||||||
| Intact | 29 (100) | 24 (100) | 63 (96.9) | 12 (29.3) | 1 (8.3) | — | — |
| One anlage | — | — | 2 (3.1) | 5 (12.2) | — | — | — |
| Both anlagen | — | — | — | 3 (7.3) | — | — | — |
| One anlage and one missing | — | — | — | 5 (12.2) | — | 3 (50) | — |
| One missing | — | — | — | 4 (9.8) | — | — | — |
| Both missing | — | — | — | 12 (29.3) | 11 (91.7) | 3 (50) | 10 (100) |
| Attached ribs | |||||||
| 6 | — | — | — | — | — | — | 2 (20) |
| 6.5 | — | — | — | — | — | 1 (16.7) | 6 (60) |
| 7 | 29 (100) | 24 (100) | 65 (100) | 41 (100) | 12 (100) | 5 (83.3) | 2 (20) |
| Cervical ribs | |||||||
| None | 28 (96.6) | 24 (100) | 60 (92.3) | 32 (78) | 3 (25) | 2 (33.3) | — |
| One side | 1 (3.4) | — | 4 (6.2) | 4 (9.8) | 7 (58.3) | 3 (50) | 2 (20) |
| Both sides | — | — | 1 (1.5) | 5 (12.2) | 2 (16.7) | 1 (16.7) | 8 (80) |
The percentage of each genotype is shown in parentheses.
If these posteriorly directed transformations result from lack of inhibition of GDF-11 signaling by GASP-2 during embryogenesis, one prediction is that loss of GASP-2 should have no effect in the complete absence of GDF-11. To test this prediction, we crossed Gasp2 mutant mice to Gdf11 mutant mice and analyzed the skeletal patterns in Gasp2−/−;Gdf11−/− double mutants. Skeletons of Gasp2−/−;Gdf11−/− double mutant mice appeared to be indistinguishable from skeletons of Gdf11 single mutant mice, consistent with the defects seen in Gasp2−/− mice being the result of overactivity of GDF-11 (Table S2).
Functional Redundancy Between Gasp2 and Follistatin in Axial Skeletal Patterning.
We also investigated the possibility that GASP-2 may be functionally redundant with other proteins capable of binding and inhibiting GDF-11. First, we investigated possible genetic interactions between Gasp1 and Gasp2 by generating mice lacking the functions of both genes. As shown in Table S1, the skeletal patterning defects were similar in double Gasp1−/−;Gasp2−/− mutant mice compared with Gasp2−/− single mutants, suggesting that these two genes are not functionally redundant for axial patterning. Second, we examined possible interactions between GASP-2 and follistatin (FST), which is an unrelated protein capable of binding and inhibiting GDF-11 (23). A previous study showed that Fst−/− mice also exhibit posteriorly directed transformations of the axial skeleton, with ∼90% of Fst−/− mice having at least one truncated 13th rib (24) and 30% of Fst−/− mice lacking both 13th ribs, most likely because of overactivity of GDF-11. We generated Gasp2−/−;Fst−/− double mutant mice and found that the complete loss of both genes exacerbated the patterning defects, with 100% of Gasp2−/−;Fst−/− mice lacking both 13th ribs and having at least one cervical rib (Table 1). Moreover, in one of the double mutants, not only was the number of thoracic vertebrae reduced by one but also, the number of lumbar vertebrae was further reduced from six to four, resulting in a C7T12L4 pattern (Fig. 3B). Finally, the number of ribs attached to the sternum (vertebrosternal ribs) was reduced to six from the normal number of seven, presumably reflecting a transformation of T7 to T8 (Fig. 3D). Hence, there seems to be at least some functional redundancy between GASP-2 and FST with respect to axial patterning by GDF-11.
Craniofacial Defects in Gasp1−/−;Gdf11−/− Mutant Mice.
Because Fst−/− and Gdf11−/− mice have been shown to exhibit a range of palatal defects (7, 24) in addition to the axial patterning defects, we analyzed the effect of Gasp1 and Gasp2 mutations in craniofacial bone development as well. As shown in Table S2, mice lacking GASP-2 had normal palates, and loss of GASP-2 also did not seem to increase the frequency of cleft palate in Gdf11−/− mice. In contrast, whereas nearly all Gasp1−/− mice also exhibited normal palate development, loss of GASP-1 had a dramatic effect on the frequency of palatal defects in Gdf11−/− mice, with 15 of 15 Gasp1−/−;Gdf11−/− mice analyzed having cleft palate (Fig. S1 and Table S3). Interestingly, analysis of Gasp1−/−;Fst−/− double mutant mice revealed no effect of loss of GASP-1 on the penetrance of cleft palate in Fst−/− mice.
Effect of Mutations in Gasp1 and Gasp2 on Skeletal Muscle Weight.
Because both Gasp1−/− and Gasp2−/− mice were viable, we were also able to investigate the possible roles of GASP-1 and GASP-2 in regulating MSTN activity. Mice lacking MSTN have been shown to have approximately a doubling of muscle weights as a result of increased numbers of muscle fibers and increased fiber sizes (1). If GASP-1 and GASP-2 normally act to block MSTN, the prediction is that loss of GASP-1 or GASP-2 would have the opposite phenotype, namely a reduction in muscle size. To investigate this possibility, we analyzed wet weights of four muscles (pectoralis, triceps, quadriceps, and gastrocnemius) in both 10-wk- and 8-mo-old mice. Although we did not observe significant differences between WT and mutant mice at 10 wk of age, male mutant mice had small but statistically significant reductions in muscle weights at 8 mo of age compared with age-matched WT mice (Fig. 4A and Table S4). Statistically significant decreases in muscle weights were observed in both mutants, with the effects generally being greater in Gasp1−/− mice (decreases of 5–12%) compared with Gasp2−/− mice (decreases of 6–9%). The effects on the quadriceps were the greatest: 12% decrease in Gasp1−/− mice (P < 0.001) and 8.6% decrease in Gasp2−/− mice (P < 0.001); the effects on the triceps were the lowest: 5.0% decrease in Gasp1−/− mice (P < 0.01) and 7.1% decrease in Gasp2−/− mice (P < 0.001). Although similar trends were also observed in female mice, most of the differences were smaller than in male mice (Table S4).
Fig. 4.

Effect of mutations in Gasp1 and Gasp2 on skeletal muscle. (A) Reductions in muscle mass in Gasp1−/− and Gasp2−/− mice. Numbers represent percent increase or decrease in muscle mass relative to WT mice and were calculated from the data shown in Table S4. Muscles analyzed were pectoralis (red), triceps (gray), quadriceps (blue), and gastrocnemius (green). *P < 0.01 vs. WT; †P < 0.001 vs. WT. (B) Fiber type analysis of the gastrocnemius muscle. Note that muscles of Gasp1−/− and Gasp2−/− mice had an increased ratio of myosin heavy chain IIa (green) to myosin heavy chain IIb (red) positive fibers compared with WT mice, which was most evident in the ventrolateral portion of the muscle. The boxed region is shown at higher magnification next to the image of the entire gastrocnemius muscle. (C) Quantification of the ratio of IIa to IIb fibers. Analysis of 2,000 myofibers in the ventrolateral region of the gastrocnemius was carried out for three mice per group. Note the increase in the percentage of IIa fibers in Gasp1−/− (P = 0.014) and Gasp2−/− (P = 0.017) mice and the decrease in the percentage of IIb fibers in Gasp1−/− (P = 0.010) and Gasp2−/− (P = 0.018) mice compared with WT mice. Data are shown as mean ± SEM.
Fiber Type Shifting to Oxidative Type IIa Fibers in Gasp1−/− and Gasp2−/− Mice.
Loss of MSTN has been shown to result not only in increases in muscle size but also, a shift in the distribution of fiber types to fast glycolytic type IIb fibers (25, 26). To determine whether loss of GASP-1 or GASP-2 has the converse effect, we carried out fiber type analysis of the gastrocnemius muscle of 8-mo-old mutant mice. Although there were no statistically significant differences in the total number of type I fibers (mean ± SEM) among WT (46.3 ± 9.5), Gasp1−/− (35.7 ± 5.8), and Gasp2−/− (22.3 ± 1.3) mice, the ratio of type IIa to type IIb fibers seemed to be increased in both Gasp1−/− and Gasp2−/− mice compared with WT mice. This increase in the percentage of type IIa fibers was most clearly seen in the ventrolateral portion of the gastrocenemius muscle, in which there was a clear spread of the region containing a significant number of type IIa fibers to the outside portion of the muscle (Fig. 4B). In this region, the percentage of IIa fibers increased from 39.9 ± 2.4% in WT mice to 51.3 ± 1.3% and 50.1 ± 1.0% in Gasp1−/− and Gasp2−/− mice, respectively, and the percentage of type IIb fibers decreased correspondingly from 57.8 ± 2.1% in WT mice to 46.9 ± 1.1% and 48.8 ± 1.0% in Gasp1−/− and Gasp2−/− mice, respectively (Fig. 4C). Hence, there seemed to be a shift in fiber type from fast glycolytic type IIb fibers to fast oxidative type IIa fibers in the gastrocnemius muscles of Gasp1−/− and Gasp2−/− mice, which is the reverse of what is seen in Mstn−/− mice.
Impaired Muscle Regeneration in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− Mice After Cardiotoxin-Induced Injury.
Loss of MSTN has been shown to affect not only muscle mass and fiber type distribution but also, the ability of the muscle to regenerate. In particular, loss of MSTN activity has been shown to result in an enhanced regenerative response to injury (27, 28), whereas heterozygous loss of Fst impaired muscle remodeling in response to injury (29). We investigated the possibility that loss of GASP-1 or GASP-2 may affect muscle regeneration by examining the response of the gastrocnemius muscle to cardiotoxin (CTX) -induced injury. We injected CTX into the right gastrocnemius muscles of WT, Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice at the age of 3 wk and analyzed muscle histology at 1, 3, 7, 14, and 28 d after injection. Before CTX injection, muscles from mutant mice were histologically similar to WT muscle. After CTX injection, however, there were clear differences in the response of the muscle to injury in the mutant mice, which could be seen easily on histologic examination (Fig. 5). In WT mice, degenerating fibers were prominent at early stages, and these degenerating fibers were replaced with regenerating myofibers containing centralized nuclei at later stages. Calcified fibers, which could be readily seen as black deposits after von Kossa staining, were present in injured WT muscle at days 7 and 14, but these calcified fibers completely disappeared by 4 wk. Indeed, by 4 wk, the muscle appeared almost completely regenerated, with very little evidence of fibrotic changes. In contrast, this regenerative response was impaired in the mutant mice. As early as 3 d after injury, Gasp2−/− and Gasp1−/−;Gasp2−/− mice exhibited myofiber degeneration that was accompanied by extensive calcifications, and many of these calcified fibers persisted, even at 4 wk after injury. Similarly, although the appearance of calcified fibers was not accelerated in timing in Gasp1−/− mice, these calcified fibers persisted in the mutant muscle. The impairment of regeneration in the mutant mice was also readily seen by examining the extent of fibrotic changes. As shown in Fig. 5, extensive areas of fibrosis could be seen by Masson trichrome staining in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice by 2 wk, and these areas persisted at 4 wk. Hence, although muscles from the mutants were capable of mounting a regenerative response after CTX-induced injury, this response was clearly impaired in the absence of either GASP-1 or GASP-2.
Fig. 5.

Impaired muscle regeneration in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− after CTX injury. Gastrocnemius muscles at various days after CTX injury were analyzed by H&E, von Kossa (to highlight calcified fibers), and Masson trichrome (to highlight areas of fibrosis) staining. Severe myofiber degeneration, accompanied by extensive calcifications, is observed in Gasp2−/− and Gasp1−/−;Gasp2−/− mice as early as 3 d after injury, and extensive areas of fibrosis are seen in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice by 14 d after injury. These abnormalities persisted at 28 d after injury. Please note that the figure is a composite of images taken from representative stained sections prepared from the individual experimental groups as indicated.
Gene Expression Patterns of MSTN Signaling Components During Skeletal Muscle Regeneration.
To begin to understand these histologic changes at a molecular level, we carried out gene expression analysis of the muscles in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice after injury. Specifically, we analyzed the expression patterns of 12 candidate genes using real-time RT-PCR: 8 genes encoding components of the MSTN signaling pathway, namely Mstn, Gdf11, Gasp1, Gasp2, Fst, follistatin-like 3 (Fstl3), Acvr2b, and mammalian tolloid (mTld), and 4 genes known to be involved in skeletal muscle regeneration, namely paired box 7 (Pax7), myogenic differentiation 1 (MyoD), myogenic factor 5 (Myf5), and myogenin (Myog). The mRNA levels of each gene were normalized to 18S rRNA and then compared with levels in the muscles before injury.
Consistent with a key role for MSTN signaling in muscle regeneration, expression levels of many of the components of the MSTN regulatory system changed after CTX-induced injury in WT mice (Fig. 6). Several genes were down-regulated at early time points, including Gasp1, Mstn, and Acvr2b. The expression levels of each of these genes gradually increased during the regenerative process and in the case of Gasp1 and Acvr2b, were even slightly higher at 4 wk compared with time 0. Other components were up-regulated after injury. Perhaps the most dramatic change was seen for Fstl3, which was up-regulated by more than eightfold at day 1 and then quickly restored to almost normal levels by day 7. Fst, mTld, and Gasp2 were also up-regulated early, albeit to a lesser extent than Fstl3, and these increased expression levels persisted through day 14. Expression of Gdf11 was not changed in the early time points (1–7 d) after injury but up-regulated in the later time points, with a peak being reached at 14 d after injury. Finally, consistent with previous studies (27, 28, 30), we observed up-regulation of genes known to be important for skeletal muscle regeneration, specifically Pax7, MyoD, Myf5, and Myog, with peak expression levels being seen at 3 d after injury.
Fig. 6.

Gene expression patterns in WT muscles after CTX-induced injury. Real-time RT-PCR was performed on gastrocnemius muscles of WT mice before injury and at 1, 3, 7, 14, and 28 d after CTX injection (n = 3). Eight genes encoding components of the MSTN signaling pathway, including (A) Gasp1, (B) Gasp2, (C) Fst, (D) Fstl3, (E) Mstn, (F) Gdf11, (G) Acvr2b, and (H) mTld, and four genes known to be involved in skeletal muscle regeneration, including (I) Pax7, (J) MyoD, (K) Myf5, and (L) Myog, were evaluated. The mRNA levels of each gene were normalized to 18S rRNA and then compared with levels in the muscles before injury. Data were shown as mean ± SEM.
Altered Patterns of Gene Expression in Gasp1 and Gasp2 Mutants During Skeletal Muscle Regeneration.
The overall gene expression patterns in Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice after injury were generally similar to the expression patterns of WT mice. As shown in Fig. 7 A–D, expression of Pax7, MyoD, Myf5, and Myog were all induced by CTX injury in mutant mice like in WT mice, with the highest expression levels being observed at day 3. We did, however, observe some small but statistically significant differences at specific time points, and in general, these differences reflected a slight reduction in expression levels in the mutants. For example, the induction of both Pax7 and Myog expressions was slightly blunted at day 3 in mutant compared with WT mice. Similarly, expression levels of all four genes were generally lower at day 14 in the injured mutant muscles.
Fig. 7.

Gene expression patterns in Gasp1 and Gasp2 mutant muscles after CTX-induced injury. Real-time RT-PCR was performed on gastrocnemius muscles of WT (green), Gasp1−/− (yellow), Gasp2−/− (blue), and Gasp1−/−;Gasp2−/− (red) before injury and at 1, 3, 7, 14, and 28 d after CTX injection (n = 3). The mRNA levels of each gene were normalized to 18S rRNA and then compared with levels in the WT muscles before injury. (A) Pax7. (B) MyoD. (C) Myf5. (D) Myog. (E) Mstn. (F) Gdf11. (G) Fst. (H) Fstl3. (I) Acvr2b. (J) mTld. Data were shown as mean ± SEM.
Expression analysis of the eight genes representing components of the MSTN regulatory system in mutant mice is shown in Fig. 7 E–J. Although the expression patterns of some genes, like Mstn and Acvr2b, were very similar in mutant compared with WT mice, we observed some differences in the expression patterns of the other genes. For example, the extent of induction of Gdf11 expression was significantly reduced in all of the mutant samples at day 14, which was the time point at which the highest levels of Gdf11 expression were seen in WT mice. Similarly, the induction of mTld and Fstl3 expressions was also blunted, particularly in Gasp1−/−;Gasp2−/− mice. Finally, in the case of Fst, the time course of induction seemed to be delayed, with a peak being seen at day 7 in Gasp1−/−;Gasp2−/− mice compared with day 1 in WT mice. The general blunting of the magnitude of the gene expression changes in mutant mice is consistent with the impaired regeneration that was evident on histological examination of the muscles.
Discussion
In this study, we have investigated the biological functions of GASP-1 and GASP-2, which are related proteins capable of modulating the activities of certain members of the TGF-β superfamily, namely MSTN and GDF-11. Previous studies have shown that GASP-1 and GASP-2 are capable of binding MSTN and GDF-11 and inhibiting their activities in vitro (11, 14, 15). Although a role for GASP-1 in regulating MSTN activity in vivo was suggested by the finding that these proteins could be detected complexed together in the blood (11), the precise biological functions of GASP-1 and GASP-2 in vivo were not known. Here, we have provided a detailed characterization of the activities of GASP-1 and GASP-2 in vitro using highly purified preparations of native proteins, and we have presented evidence that these molecules act by blocking the ability of the ligand to engage the type II receptor, which is the initial event in the signaling cascade. Most importantly, we have presented genetic data showing that mice carrying targeted mutations in Gasp1 and/or Gasp2 have phenotypes consistent with regulation of MSTN and GDF-11 in vivo.
Based on our genetic studies, it is clear that GASP-2 plays an important role in modulating the activity of GDF-11 during embryogenesis. We showed that Gasp2, like Gdf11, is expressed in the posterior region of midgestation embryos and that mice lacking GASP-2 exhibit posteriorly directed homeotic transformations of axial skeleton. A schematic summary of the axial skeletal patterning effects of targeting Gasp2 is shown in Fig. 3E. For example, mice lacking GASP-2 are characterized by homeotic transformations of the last thoracic vertebra into a lumbar vertebra and the last lumbar vertebra into a sacral vertebra, the presence of supernumerary ribs on cervical segments, and an anterior shift in the position of the anterior tuberculi, which is normally seen at the level of C6. These shifts are the reverse of the shifts that are observed in mice carrying mutations in Gdf11. When GDF-11 levels are slightly reduced (for example, in mice heterozygous for a Gdf11 loss-of-function mutation), one lumbar vertebra undergoes an anteriorly directed homeotic transformation to have a morphology typical of a thoracic vertebra (7). In the complete absence of GDF-11 (i.e., in Gdf11 homozygous mutants), the anteriorly directed homeotic transformations are much more extensive; for example, five lumbar segments are transformed into thoracic segments, eight sacral and caudal segments are transformed into lumbar segments, and there is a posterior shift in the position of the anterior tuberculi (7). Based on the results presented here, it seems clear that GASP-2 plays an important role in axial skeletal patterning, most likely by regulating the activity of GDF-11 in the posterior region of the developing embryo, where segment identity is being specified.
The function of GASP-2 in regulating axial patterning seems to be redundant with the function of FST, which is also known to block GDF-11 activity. In mice lacking both GASP-2 and FST, the posteriorly directed homeotic transformations are more extensive; double mutant mice are characterized, for example, by a reduction in the number of vertebrae with ribs touching the sternum, transformation of additional lumbar segments into sacral segments, and significantly increased penetrance of other abnormalities seen in Gasp2−/− mice. Hence, proper anterior–posterior patterning of the axial skeleton occurs within a narrow range of GDF-11 signaling activity, which must be precisely regulated by balancing levels of GDF-11 with levels of its inhibitory binding proteins, including GASP-2 and FST. Slight deviations in GDF-11 signaling activity either up or down can lead to homeotic transformations that are posteriorly or anteriorly directed, respectively.
An interesting feature of this regulatory system is that axial patterning is remarkably sensitive to altered levels of signaling, even though there is functional redundancy among the regulatory components. As discussed above, it is clear that there is some functional redundancy among the inhibitory binding proteins, GASP-2 and FST, although loss of either one alone leads to a clear patterning phenotype. There also seems to be functional redundancy among the ligands, because complete loss of both GDF-11 and MSTN results in an even more severe patterning phenotype than loss of GDF-11 alone, with the most dramatic manifestations being the presence of 20–21 thoracic segments and an additional pair of forelimbs (31).
In this respect, the range of effects in the number of vertebral segments that can be altered with respect to their positional identity by manipulating GDF-11 activity is remarkable and much wider than the range of effects seen in mice carrying loss-of-function mutations in individual Hox genes, which generally exhibit only mild axial changes (32). The wide range of vertebral patterns that can be generated by targeted manipulation of this pathway in mice is reminiscent of the wide variation in skeletal patterns seen in different species. It is tempting to speculate that naturally occurring genetic changes leading to alterations in the activity levels of this signaling pathway during embryonic development may have been responsible for the generation of new vertebral patterns during the evolution of the vertebrate body plan.
We have also presented data supporting roles for GASP-1 and GASP-2 in the regulation of MSTN activity in vivo. Skeletal muscle analysis of adult mice revealed that both Gasp1 and Gasp2 mutants had small but statistically significant decreases in overall muscle weights as well as a shift in fiber type from fast glycolytic type IIb fibers to fast oxidative type IIa fibers. These phenotypes contrast with the increased muscle weights and the shift in fiber types to type IIb fibers seen in Mstn−/− mice (25, 26), and therefore, they are consistent with overactivity of MSTN in Gasp1 and Gasp2 mutants. Considering the dramatic muscle phenotype resulting from loss of MSTN, however, the effects seen in Gasp1 and Gasp2 mutants were relatively mild. One potential explanation for the mild phenotype may be possible functional redundancy between these proteins and other MSTN binding proteins. In this respect, it will be important to investigate the effect of targeting GASP-1 and GASP-2 combined with other known MSTN inhibitors, such as FST and FSTL-3 (33).
Much more pronounced effects of GASP-1 and GASP-2 loss were seen in the setting of cardiotoxin-induced muscle injury and regeneration. Histologically, muscles of mutant mice exhibited signs of increased damage and impaired regeneration after CTX injection compared with muscles of WT mice. For example, calcified fibers appeared as early as 3 d after injury in Gasp2−/− and Gasp1−/−;Gasp2−/− mice compared with 7 d in WT mice, and unlike in WT muscle, many of the calcified fibers in the mutant muscles persisted, even 4 wk after injury. Moreover, extensive fibrotic changes were observed by 2 wk in both Gasp1−/− and Gasp2−/− single mutant mice as well as Gasp1−/−;Gasp2−/− double mutant mice, and this fibrosis persisted at 4 wk.
At a molecular level, we also detected gene expression changes consistent with an overall blunting of the regenerative response in the mutant mice. We first showed that expression of many components of the MSTN regulatory system are normally either up- or down-regulated after CTX-induced injury, consistent with an important role for this signaling pathway in muscle regeneration. Examination of muscles from CTX-injured mutant mice revealed that these expression patterns were altered in the absence of GASP-1 or GASP-2. For certain components of this regulatory system, like Gdf11, mTld, and Fstl3, the magnitude of changes in expression levels was generally lower in the mutant mice, whereas for Fst, the time course of induction seemed to be delayed. We also observed that the magnitude of induction of Pax7 and the MyoD family of myogenic regulators after CTX-induced injury was reduced in the mutants compared with WT mice.
This impairment in the regenerative response after CTX-induced injury in mice lacking GASP-1 and/or GASP-2 contrasts with the enhanced regenerative response seen in mice lacking MSTN. The mechanisms by which MSTN loss leads to improved muscle healing are not fully understood, but studies have suggested that MSTN is capable of inhibiting myoblast proliferation by arresting cell cycles in the G1 phase (34) and blocking myoblast differentiation by inhibiting MyoD activity (35). A number of studies have suggested that one role for MSTN may be to maintain satellite cells in quiescence (27, 34, 36, 37). MSTN has also been suggested to accelerate fibrotic changes in injured muscle by directly stimulating muscle fibroblasts (38, 39). Hence, in the absence of MSTN, muscle healing may be enhanced through increased activation of satellite cells and reduced activity of muscle fibroblasts (27, 28). Whatever the underlying mechanisms, the contrasting phenotypes seen in mice lacking MSTN vs. mice lacking GASP-1 and/or GASP-2 suggest that the impairment of muscle regeneration seen in Gasp1 and/or Gasp2 mutant mice likely reflects overactivity of the MSTN signaling pathway.
Based on the results presented here, it is clear that both GASP-1 and GASP-2 are important modulators of GDF-11 and MSTN activity in vivo. Because members of the TGF-β superfamily are potent regulators of cell growth and differentiation, there is considerable interest in understanding the molecular mechanisms by which their activities are regulated, with the long-term goal of exploiting their activities for clinical applications. In this regard, there is considerable effort being directed at developing strategies for modulating MSTN activity in vivo to enhance muscle strength, function, and regeneration in patients with muscle loss. Hence, the elucidation of the precise biological roles that binding proteins, like GASP-1 and GASP-2, play in this regulatory system will be important for not only understanding how the activity of this system is modulated in different physiological states but also, developing the best strategies for targeting this regulatory network for clinical applications.
Methods
Functional Assays.
GASP-1 and GASP-2 proteins in their native state were purified as described in SI Methods. MSTN, GDF-11, and activin A activities were measured in A204 rhabdomyosarcoma cells carrying an Smad2/3-responsive pGL3-(CAGA)12-luciferase expression construct (18). TGF-β1 and BMP-4 activities were measured using C3H10T1/2 cells carrying either a p3TP-Lux reporter construct (plasmid 11767; Addgene) (19) or a (BRE)2-Luc reporter construct (provided by P. ten Dijke, Leiden University Medical Center, Leiden, The Netherlands) (20), respectively.
RNA Blot Analysis and Whole-Mount in Situ Hybridization.
Full-length cDNA of Gasp1 was used as a probe for the RNA blot and in situ hybridization. For analysis of Gasp2 expression patterns, we used a probe corresponding to the C-terminal region (452 bp) of exon 2 (chromosome 17: 26,014,635–26,015,086) to avoid repetitive elements. For whole-mount in situ hybridization, embryos were prepared and stained as described (7).
KO Mice.
Gasp1 and Gasp2 KO mice were generated as described in SI Methods. Fst KO mice were obtained from Martin M. Matzuk (Baylor College of Medicine, Houston, TX). Skeletal analysis of newborn mice and fetuses at 18.5 d postcoitum was carried out as described (7). All animal experiments were carried out in accordance with protocols that were approved by the Institutional Animal Care and Use Committees at Johns Hopkins University School of Medicine.
Muscle Weight and Fiber Type Analysis.
For measurement of muscle weights, individual muscles from both sides of 10-wk-old and 8-mo-old mice were dissected, and the average weight was used for each muscle. For muscle fiber type analysis, 10-µm frozen cross-sections were taken from the right gastrocnemius and subjected to immunohistochemistry for three different isoforms of myosin heavy chain. Information on the primary antibodies for myosin heavy chain staining can be found in SI Methods.
CTX Injury.
For skeletal muscle injury experiments, 250 μL CTX (10 µM Naja mossambica mossambica; Sigma-Aldrich) were injected into the right gastrocnemius muscles of WT, Gasp1−/−, Gasp2−/−, and Gasp1−/−;Gasp2−/− mice at the age of 3 wk, and muscles were harvested at 1, 3, 7, 14, and 28 d after induction of injury. A total of 144 mice muscles was evaluated for skeletal muscle regeneration after CTX injury: six mice per each genotype (four different genotype groups) at six different time points. One-half was harvested for histological analysis, and the other one-half was harvested for gene expression analysis.
Supplementary Material
Acknowledgments
We thank Charles Hawkins and Ann Lawler for carrying out the blastocyst injections and embryo transfers, Martin M. Matzuk for the generous gift of Fst KO mice, Min-Sung Kim for assistance with protein purification, Adam Lehar for assistance with sectioning, Suzanne M. Sebald for assistance with CTX injection, and Jeremy Nathans for helpful comments on the manuscript. This work was supported by the National Institutes of Health Grants R01AR059685, R01AR060636, and P01NS0720027 (to S.-J.L.).
Footnotes
Conflict of interest statement: Under a licensing agreement between Pfizer, Inc. and Johns Hopkins University, S.-J.L. is entitled to a share of royalty received by the University on sales of products related to myostatin. The terms of this arrangement are being managed by the University in accordance with its conflict of interest policies.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309907110/-/DCSupplemental.
References
- 1.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 2.Nakashima M, Toyono T, Akamine A, Joyner A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech Dev. 1999;80(2):185–189. doi: 10.1016/s0925-4773(98)00205-6. [DOI] [PubMed] [Google Scholar]
- 3.Wu HH, et al. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37(2):197–207. doi: 10.1016/s0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- 4.Kim J, et al. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308(5730):1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- 5.Harmon EB, et al. GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development. 2004;131(24):6163–6174. doi: 10.1242/dev.01535. [DOI] [PubMed] [Google Scholar]
- 6.Dichmann DS, Yassin H, Serup P. Analysis of pancreatic endocrine development in GDF11-deficient mice. Dev Dyn. 2006;235(11):3016–3025. doi: 10.1002/dvdy.20953. [DOI] [PubMed] [Google Scholar]
- 7.McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22(3):260–264. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- 8.Souza TA, et al. Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol. 2008;22(12):2689–2702. doi: 10.1210/me.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loffredo FS, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153(4):828–839. doi: 10.1016/j.cell.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- 11.Hill JJ, Qiu Y, Hewick RM, Wolfman NM. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: A novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17(6):1144–1154. doi: 10.1210/me.2002-0366. [DOI] [PubMed] [Google Scholar]
- 12.Trexler M, Bányai L, Patthy L. Distinct expression pattern of two related human proteins containing multiple types of protease-inhibitory modules. Biol Chem. 2002;383(1):223–228. doi: 10.1515/BC.2002.023. [DOI] [PubMed] [Google Scholar]
- 13.Trexler M, Bányai L, Patthy L. A human protein containing multiple types of protease-inhibitory modules. Proc Natl Acad Sci USA. 2001;98(7):3705–3709. doi: 10.1073/pnas.061028398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kondás K, Szláma G, Trexler M, Patthy L. Both WFIKKN1 and WFIKKN2 have high affinity for growth and differentiation factors 8 and 11. J Biol Chem. 2008;283(35):23677–23684. doi: 10.1074/jbc.M803025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szláma G, Kondás K, Trexler M, Patthy L. WFIKKN1 and WFIKKN2 bind growth factors TGFβ1, BMP2 and BMP4 but do not inhibit their signalling activity. FEBS J. 2010;277(24):5040–5050. doi: 10.1111/j.1742-4658.2010.07909.x. [DOI] [PubMed] [Google Scholar]
- 16.Haidet AM, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008;105(11):4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monestier O, et al. Ubiquitous Gasp1 overexpression in mice leads mainly to a hypermuscular phenotype. BMC Genomics. 2012;13(1):541. doi: 10.1186/1471-2164-13-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thies RS, et al. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18(4):251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- 19.Wrana JL, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71(6):1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 20.Logeart-Avramoglou D, Bourguignon M, Oudina K, Ten Dijke P, Petite H. An assay for the determination of biologically active bone morphogenetic proteins using cells transfected with an inhibitor of differentiation promoter-luciferase construct. Anal Biochem. 2006;349(1):78–86. doi: 10.1016/j.ab.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23(20):7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida AT, et al. OC29 is preferentially expressed in the presumptive sensory organ region of the otocyst. Dev Dyn. 2004;231(4):766–774. doi: 10.1002/dvdy.20180. [DOI] [PubMed] [Google Scholar]
- 23.Gamer LW, et al. A novel BMP expressed in developing mouse limb, spinal cord, and tail bud is a potent mesoderm inducer in Xenopus embryos. Dev Biol. 1999;208(1):222–232. doi: 10.1006/dbio.1998.9191. [DOI] [PubMed] [Google Scholar]
- 24.Matzuk MM, et al. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374(6520):360–363. doi: 10.1038/374360a0. [DOI] [PubMed] [Google Scholar]
- 25.Girgenrath S, Song K, Whittemore LA. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve. 2005;31(1):34–40. doi: 10.1002/mus.20175. [DOI] [PubMed] [Google Scholar]
- 26.Hennebry A, et al. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009;296(3):C525–C534. doi: 10.1152/ajpcell.00259.2007. [DOI] [PubMed] [Google Scholar]
- 27.McCroskery S, et al. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118(Pt 15):3531–3541. doi: 10.1242/jcs.02482. [DOI] [PubMed] [Google Scholar]
- 28.Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102(7):2519–2524. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SJ, et al. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24(10):1998–2008. doi: 10.1210/me.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao P, Hoffman EP. Embryonic myogenesis pathways in muscle regeneration. Dev Dyn. 2004;229(2):380–392. doi: 10.1002/dvdy.10457. [DOI] [PubMed] [Google Scholar]
- 31.McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev Biol. 2009;9:24. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellik DM. Hox genes and vertebrate axial pattern. Curr Top Dev Biol. 2009;88:257–278. doi: 10.1016/S0070-2153(09)88009-5. [DOI] [PubMed] [Google Scholar]
- 33.Hill JJ, et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277(43):40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- 34.Thomas M, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275(51):40235–40243. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- 35.Langley B, et al. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- 36.McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162(6):1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamada M, et al. High concentrations of HGF inhibit skeletal muscle satellite cell proliferation in vitro by inducing expression of myostatin: A possible mechanism for reestablishing satellite cell quiescence in vivo. Am J Physiol Cell Physiol. 2010;298(3):C465–C476. doi: 10.1152/ajpcell.00449.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, et al. Relationships between transforming growth factor-beta1, myostatin, and decorin: Implications for skeletal muscle fibrosis. J Biol Chem. 2007;282(35):25852–25863. doi: 10.1074/jbc.M704146200. [DOI] [PubMed] [Google Scholar]
- 39.Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283(28):19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.