

Targeted genome engineering has been instrumental for the study of biological processes, and it holds great promise for the treatment of disease. Historically, gene targeting by homologous recombination has been the preferred method to modify specific genes in mouse and human cells (Fig. 1). However, this approach is hampered by low efficiency, the requirement for drug selection to detect targeted cells, and the limited number of cell types and organisms amenable to the method. To overcome these limitations, technologies based on sequence-specific zinc finger (ZF) and transcription activator-like effector (TALE) proteins have been developed. These proteins can be engineered to theoretically recognize any DNA sequence in the genome. When fused to a nuclease domain and assembled in pairs flanking a target site of interest, ZF and TALE nucleases (ZFNs and TALENs) will introduce double-strand breaks (DSBs) on DNA. DSBs serve as substrates for nonhomologous end joining (NHEJ) or homology-directed repair (HDR), which in turn facilitate the engineering of targeted mutations, repair of endogenous mutations, or introduction of transgenic DNA elements. The clustered, regularly interspaced, short palindromic repeat (CRISPR)-CRISPR-associated (Cas) system represents the latest addition to this arsenal of tools for site-specific genome engineering (see below). Although each of these three gene modification approaches has advantages and disadvantages (Fig. 1), the pace and ease with which the CRISPR-Cas systems have been adapted to modify genes in different cell types and organisms suggests that it may very well become the new method of choice for genome engineering. In PNAS, Hou et al. introduce a unique variant of the Cas9 enzyme (1), which provides additional flexibility and specificity to the CRISPR system and to genome-modifying tools in general.
Fig. 1.
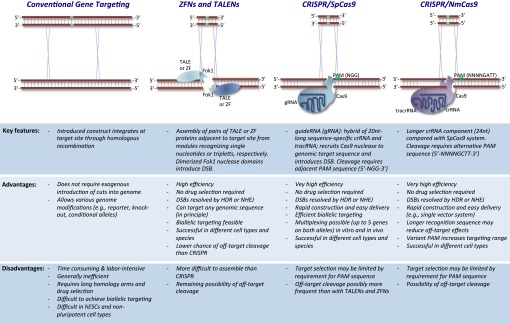
Different approaches to manipulate the mammalian genome. Shown are currently available methods to modify the genome in a site-specific manner with a list of key features, advantages, and disadvantages of each approach. Illustration of homology-directed repair (HDR) to generate a knock-in allele. See text for details and abbreviations.
CRISPR and Cas9 were initially identified as components of the bacterial immune response to bacteriophages (2). In this system, a short CRISPR RNA component (crRNA) recognizes a complementary stretch of nucleotides (the protospacer) within foreign DNA, thus conferring sequence specificity (Fig. 1). In addition, a transactivating CRISPR RNA, dubbed tracrRNA, is required to form ribonucleoprotein complexes with the Cas9 nuclease, generating site-specific DSBs (3). By combining these two RNA components of CRISPR into a single chimeric molecule termed “guide RNA” (gRNA) and expressing it alongside Cas9 protein, several groups have demonstrated the adaptability of this system to modify endogenous loci in eukaryotic cells. DSBs generated by CRISPR are preferentially repaired by NHEJ, which is an error-prone process and thus useful to introduce insertions and deletions into mammalian cells (4, 5). In addition, DSBs are resolved at lower frequency by HDR and therefore can be used to modify endogenous loci (e.g., replacement of point mutations, insertion of knock-in alleles) (4, 5). Notably, this approach requires much shorter homology arms compared with traditional gene targeting systems. Although ZFNs and TALENs enable similar genome modifications, the CRISPR-Cas system offers a few advantages. First and foremost, the CRISPR-Cas system is based on simple gRNA/DNA hybrids to confer sequence specificity, which enables rapid design and delivery of targeting constructs. In contrast, ZFNs and TALENs require modular assembly of pairs of proteins that recognize areas flanking a target site, a process that is more tedious and time-consuming. Another advantage of the CRISPR-Cas system is its amenability to multiplexing, allowing for the simultaneous generation of up to five mutations from a single transfection event (6). This ability is expected to facilitate epistatic gene studies and should allow for dissection of phenotypes that are normally masked by compensatory mechanisms. Furthermore, multiplexing may enable the study of complex genetic diseases in a Petri dish using human embryonic stem cells (hESCs) or patient-derived induced pluripotent stem cells (iPSCs), as has recently been accomplished for monogenic diseases with TALENs (7).
Although the CRISPR/Cas system undoubtedly holds great potential for genome editing, there are a number of important limitations to consider. Target site cleavage by the CRISPR-Cas system requires a so-called protospacer adjacent motif (PAM) immediately downstream of the protospacer element to which the gRNA binds, restricting targeting range (Fig. 1). Interestingly, the PAM sequence varies in size and nucleotide composition in different bacterial strains from which Cas9 proteins have been isolated. The two previously characterized Cas9 proteins from Streptococcus pyogenes (SpCas) and Streptococcus thermophiles (StCas) require PAMs of 5′-NGG-3′ and 5′-NNAGAAW-3′, respectively (2). While these sites are found quite frequently in the genome, as often as every 8 bp (4), specific genomic regions may be difficult to target with these sequence constraints. Another major concern of CRISPR-Cas technology is the potential for off-target effects. This issue is particularly relevant with respect to potential therapeutic applications of this technology. Two recent in-depth studies report that up to five mismatches in the protospacer and a 5′-NAG-3′ variation within the PAM sequence are tolerated by S. pyogenes CRISPR-Cas9 without impairing the ability to cleave DNA (8, 9). This implies that dozens of nontarget loci may be cut by CRISPR-Cas9 when aiming to cleave individual genes in vivo. Indeed, a recent report documented extensive cleavage within the genome when examining a number of predicted off-targets sites (8). Different approaches have been proposed to mitigate off-target effects including titration of gRNA and Cas9 levels, rational design of gRNAs based on bioinformatic predictions (9), as well as the use of pairs of modified Cas9 enzymes that introduce single strand nicks, as opposed to DSBs, on opposite strands of a target site (akin to ZNF and TALEN design) (10). A final possibility to increase specificity and decrease off-target cleavage is the isolation of alternative CRISPR-Cas systems with more stringent interactions between gRNA, target sequence, and PAM from other strains of archaea or bacteria (2).
In PNAS, Hou et al. present the identification and analysis of a unique Cas9 nuclease isolated from Neisseria meningitidis (NmCas9). Unlike the SpCas9 and StCas9 enzymes, NmCas9 binds a 24nt protospacer sequence on its target DNA, conferring additional specificity over the previous 20nt protospacer (Fig. 1). Moreover, NmCas9 differs in its PAM, requiring a 5′-NNNNGATT-3′ or 5-NNNNGCTT-3′ sequence to achieve efficient target DNA cleavage. This extended PAM sequence should enhance specificity and will increase the number of loci within the genome that are amenable to targeting by CRISPR-Cas. Critically, NmCas9, like SpCas9, is capable of achieving efficient cleavage of target DNA sequences both in vitro and in vivo. To document the utility of this unique system for generating mutations in hESCs, the authors generated a puromycin-selectable episomal vector containing crRNA, tracrRNA, and Cas9 on a single construct. This modification will afford increased efficacy over alternative genome editing systems that require two or more plasmids. Indeed, the authors generated an Oct4-GFP knock-in reporter in hESCs and iPSCs at efficiencies of around 60%, which is higher than comparable efforts using TALENs. This report further demonstrates the feasibility of inserting large genomic fragments into cells by HDR using CRISPR-Cas technology, indicating that it should now be possible to generate similar reporter alleles for other genes of interest. Additional single gene and multigene reporter cell lines (derived through multiplexing) will be invaluable tools for tracking cell fate and isolating rare cell populations in different biological contexts.
Of interest, CRISPR-Cas technology is not limited to introducing mutations or reporters into cells, but can, in principle, be used to repair disease-associated mutations in patients. In addition, recent reports showed that transcriptional activators or repressors can be directed to defined genomic elements to reactivate or silence gene expression using CRISPR-Cas technology (10, 11). Together, these examples document the remarkable versatility and amenability of the CRISPR-Cas system and its potential to revolutionize the fields of reverse genetics and epigenetics. The unique Cas9 protein, described by Hou et al., will certainly add to this powerful set of tools and may in fact solve some of the current limitations of CRISPR technology in basic biology and cell therapy.
Footnotes
The authors declare no conflict of interest.
See companion article on page 15644.
References
- 1.Hou Z, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 3.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013a;12(2):238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells [published online ahead of print June 23, 2013] Nat Biotechnol. 2013 doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases [published online ahead of print July 21, 2013] Nat Biotechnol. 2013 doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering [published online ahead of print August 1, 2013] Nat Biotechnol. 2013b doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert LA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]