

Abstract
In November 1973, my colleagues A. C. Y. Chang, H. W. Boyer, R. B. Helling, and I reported in PNAS that individual genes can be cloned and isolated by enzymatically cleaving DNA molecules into fragments, linking the fragments to an autonomously replicating plasmid, and introducing the resulting recombinant DNA molecules into bacteria. A few months later, Chang and I reported that genes from unrelated bacterial species can be combined and propagated using the same approach and that interspecies recombinant DNA molecules can produce a biologically functional protein in a foreign host. Soon afterward, Boyer’s laboratory and mine published our collaborative discovery that even genes from animal cells can be cloned in bacteria. These three PNAS papers quickly led to the use of DNA cloning methods in multiple areas of the biological and chemical sciences. They also resulted in a highly public controversy about the potential hazards of laboratory manipulation of genetic material, a decision by Stanford University and the University of California to seek patents on the technology that Boyer and I had invented, and the application of DNA cloning methods for commercial purposes. In the 40 years that have passed since publication of our findings, use of DNA cloning has produced insights about the workings of genes and cells in health and disease and has altered the nature of the biotechnology and biopharmaceutical industries. Here, I provide a personal perspective of the events that led to, and followed, our report of DNA cloning.
Keywords: restriction enzyme, pSC101, EcoRI, genetic engineering, gene cloning
In a PNAS paper entitled “Construction of Biologically Functional Bacterial Plasmids In Vitro,” my colleagues A. C. Y. Chang, H. W. Boyer, R. B. Helling, and I reported in November 1973 that individual genes can be cloned and isolated by enzymatically fragmenting DNA molecules, linking the pooled fragments to autonomously replicating circular bacterial genetic elements known as plasmids, and introducing the resulting recombinant DNA molecules into bacteria (1). Boyer and I were young faculty at the University of California, San Francisco (UCSF) and Stanford, respectively. Annie Chang was a Research Technician in my laboratory and Bob Helling was a University of Michigan professor on sabbatical leave in Boyer's laboratory. A few months later, Chang and I reported that genes from totally unrelated bacterial species can be combined and propagated using the same approach (2) and that interspecies recombinant DNA molecules can produce a biologically functional protein in a foreign host. Soon afterward, Boyer’s laboratory and mine published collaborative experiments demonstrating that genes from eukaryotic cells can be cloned in bacteria (3).
Bacterial viruses and plasmids had been shown to pick up DNA from the chromosomes of their hosts (4); hybrid viruses from animal cells also had been reported (5, 6). However, it had long been known that only closely related species can interbreed and produce viable offspring, and hybrids displaying heritable characteristics of very different species exist only in mythology; thus, there was uncertainty about whether so-called “natural barriers created during evolution” (7, 8) would prevent propagation of genes across different biological domains. Stringent host range limitations to virus propagation had been observed, and, in some instances, impediments to survival of foreign DNA had been found even among subgroups of the same species (9). Supporting the notion that DNA was unlikely to survive in cells of an unrelated species was the finding that individual biological species maintain characteristic ratios of A+T to G+C base pairs (10, 11). Our discovery that DNA can be transplanted to, and propagated in, a different species, and even in a different biological kingdom, by attaching it to a vector indigenous to the recipient led to the realization that natural barriers to DNA survival are not so constraining after all, and that “genetic engineering”—at least at the cellular level—is possible (8). It also provided a protocol that enabled such engineering to be done by virtually any laboratory having modest genetic and biochemical capabilities.
Our DNA cloning experiments resulted from the pursuit of fundamental biological questions rather than goals that most observers might regard as practical or “translational.” I was investigating mechanisms underlying the ability of plasmids to acquire genes conferring antibiotic resistance and to exist separately from bacterial chromosomes; Herb Boyer was studying enzymes that restrict and destroy foreign DNA. The PNAS publications resulting from these pursuits generated considerable scientific excitement—and work aimed at repeating and extending the findings was undertaken almost immediately by other researchers. The papers also prompted a highly public controversy about potential hazards of “genetic tinkering,” a decision by Stanford University and the University of California to seek patents on the technology that Boyer and I had invented, and efforts by entrepreneurs and industry to implement DNA cloning methods for commercial purposes. In the 40 years that have now passed since publication of these PNAS papers, use of DNA cloning methods has produced important insights about the workings of genes and cells in health and disease and has profoundly altered the biotechnology and pharmaceutical industries. I provide here a personal perspective of these events.
Plasmids and Antibiotic Resistance
After the development of antimicrobial agents in the 1940s, the notion was prevalent that these drugs would end infectious diseases caused by bacteria. Of course that did not happen, and the reason was the occurrence of antibiotic resistance. Investigations carried out primarily in laboratories in Japan and the United Kingdom in the early 1960s showed that antibiotic resistance in bacteria commonly is associated with the acquisition of genes—often multiple genes—capable of destroying antibiotics or otherwise interfering with their actions. The resistance properties commonly did not map genetically to the bacterial chromosomes, suggesting that the genes encoding resistance were located on separate elements (some had called them episomes) analogous to the fertility factor (F-factor) discovered earlier (12). Like F-factors, resistance factors (R-factors) were capable of being transferred between bacteria by cell-to-cell contact (13, 14). In 1952, Joshua Lederberg had given the name “plasmids” to such extrachromosomal genetic elements (15). The antibiotic-inactivating genes carried by resistance plasmids provide a biological advantage to host bacteria in populations exposed to antimicrobial drugs, and, in barely a decade after the introduction of antibiotics to treat human infections, R-plasmid–mediated multidrug resistance had become a major medical problem as well as a scientific enigma.
The transfer of resistance properties between bacteria was found by centrifugation analysis to be sometimes associated with the acquisition of heterogeneous DNA bands (16, 17). However, the molecular nature of this DNA was controversial. Particularly uncertain was whether the resistance and transfer components of R-plasmids were carried by the same DNA molecule or were located on separate molecules that can interact transiently during interbacterial transfer (18, 19). Importantly, nothing was known about the genetic recombination mechanisms that had enabled the accumulation of multiple resistance genes on the same genetic element. Before moving to Stanford, I prepared National Institutes of Health (NIH) and National Science Foundation (NSF) research proposals aimed at addressing these questions. As a postdoctoral fellow in the laboratory of Jerard Hurwitz at the Albert Einstein College of Medicine, I had found that genes on different segments of DNA of bacteriophage λ were controlled temporally at the level of transcription during the production of virus particles (20), and I thought that some of the approaches I used in those experiments could be applied to the study of R-plasmids. I had been trained as both a physician and scientist and believed that an understanding of resistance plasmids was both medically and scientifically important. Bacteriophage λ was the most extensively and competitively investigated bacteriophage of that era, but the role of R-plasmids in antibiotic resistance was being studied in only a small number of microbiology laboratories; I liked the prospect of working in what was still a quiet backwater of scientific research.
Research in the burgeoning field of molecular biology during the 1960s focused on bacteriophages for an important reason: a bacterial cell infected by a virus generates thousands of identical copies—clones—of a single infecting genome during the normal viral life cycle. Thus, phenotypic effects can be correlated with the results of biochemical analyses. I realized that elucidation of how resistance genes function and how R-plasmids evolve required a way to clone individual plasmid DNA molecules and to isolate the resistance genes. Genetic mapping of R-plasmid properties had led to the prediction that bacterial plasmids exist as DNA circles (19, 21, 22), and I proposed to use circularity to isolate intact resistance plasmid DNA. If I could obtain R-plasmid circles—I reasoned—I could apply DNA fragmentation approaches I had used to study λ gene expression, together with ultracentrifugation analysis and DNA hybridization methods, to assess changes in circle size associated with the gain or loss of resistance phenotypes or transferability. Stanley Falkow, whose published investigations at Walter Reed Medical Center and Georgetown University had been instrumental in attracting me to plasmid biology, agreed to provide bacterial strains and plasmids for my initial experiments. However, Falkow also told me that he planned to stop working on plasmids to pursue other scientific interests, and the decision of an established expert to abandon a field that I had just decided to enter was disconcerting! However, there was also good news: in October 1967, Donald Helinski and his colleagues at the University of California, San Diego published the first molecular evidence for circular DNA forms of a transmissible plasmid (23), supporting the notion that R-factors, too, would be DNA circles.
I arrived at Stanford in March 1968, and, the following year, Christine Miller, who was then a newly hired laboratory technician, and I reported the purification of intact circular DNA of the large antibiotic resistance plasmid R1 (24). Several months later, I was invited to Caltech by Jerome Vinograd, a pioneer in the study of circular DNA of animal cells, to give a seminar talk about our plasmid results. Discussions with Norman Davidson and his graduate student Phillip Sharp during that visit initiated a collaboration between our laboratories aimed at using electron microscope-based heteroduplex analysis, which had been developed earlier in Davidson’s laboratory (25) and also at the University of Wisconsin (26), to compare regions of sequence similarity on different plasmids: annealing of homologous DNAs results in smooth thickened DNA segments that are distinguishable visually from thinner, more kinky regions of single-stranded DNA. We expected that such experiments would provide information about the structural relationships between resistance genes that had been picked up by plasmids during their meandering through bacterial populations. The results of these experiments and also of separate investigations from Davidson’s laboratory (27, 28) showed remarkable sequence conservation among large segments of different R-plasmids and, importantly, provided direct physical evidence that plasmid sequences associated with interbacterial DNA transfer had become linked covalently to resistance genes to form large circles of R-plasmid DNA. Sharp’s electron microscopy also detected a phenomenon that we didn’t yet understand the significance of: short inverted repeats of DNA sequences that bracket regions containing certain resistance genes—presaging by a couple of years the discovery of insertion sequence (IS) elements and antibiotic resistance transposons, which enter plasmids by illegitimate (i.e., nonhomologous) recombination and lead to rearrangements of plasmid structure (for reviews, see refs. 29 and 30).
Bacterial Transformation by Plasmid DNA
The methods for heteroduplex analysis that I learned from Sharp during our collaboration enabled me to use electron microscopy in my plasmid studies at Stanford when he and Davidson later moved on to other scientific interests. However, still missing from a growing collection of tools available to investigate the molecular biology of plasmids was a method for reintroducing plasmid DNA molecules into bacteria. Genetic transformation using naked DNA had been shown for pneumococcus, Bacillus species, and certain other bacteria, but not for Escherichia coli or other microbes that carry the R-plasmids I was studying. Quite fortuitously, a critical discovery by Morton Mandel and Akiko Higa reported in the October 1970 issue of the Journal of Molecular Biology (31) pointed the way toward such a method.
Mandel and Higa found that E. coli cells treated with calcium ions can take up DNA of the temperate bacteriophages λ and P22 and that such cells can release infective virus. The procedure is robust, and Peter Lobban, a graduate student working with A. Dale Kaiser in the Stanford Department of Biochemistry, had begun using it to introduce phage P22 DNA into Salmonella typhimurium, a close relative of E. coli. Mandel and Higa had reported an attempt to genetically transform E. coli to express an antibiotic resistance gene present on the bacterial chromosome and had met with failure. However, plasmids, like phages, are extrachromosomal elements; if R-plasmid DNA could be taken up by E. coli at even a low frequency, and if the antibiotic resistance genes carried by circular R-plasmid replicons were expressed in these cells, colonies of bacteria that acquire plasmids could be selected using culture media containing appropriate antibiotics. Such selection might enable cloning of single molecules of plasmid DNA. I asked Leslie Hsu, a first-year medical student who had come to my laboratory for research training, to undertake such experiments.
By late 1971, Leslie, with the help of Annie Chang, had shown that bacterial clones containing autonomously replicating plasmid molecules can be obtained using a modification of the Mandel and Higa procedure. Later work by others (32) indicated that the failure of chromosomal DNA to transform calcium chloride-treated E. coli in Mandel and Higa’s earlier experiments had resulted from exonucleolytic digestion of fragmented chromosomal DNA by a bacterial enzyme. Luckily, the circularity of R-plasmid DNA molecules had avoided this pitfall. The resulting E. coli colonies each contained a circular DNA species having the resistance, fertility, and sedimentation properties of the parental genetic element. Publication of our paper reporting these findings in August 1972 (33) interested plasmid researchers but, so far as I could determine, was hardly noticed by others. There was scant awareness in the phage-oriented world of 1972 molecular biology of the implications of being able to clone plasmid DNA molecules, and our report did nothing to alter the backwater nature of the field of plasmid biology. That was fine with me: I was a junior scientist whose laboratory included just a few students and postdocs, plus two research assistants. My primary academic appointment at Stanford was then in the Department of Medicine, and my clinical teaching responsibilities affected the time I had available for laboratory research; the quiet reception that our paper received allowed me to proceed with less pressure to undertake the experiments I had long been planning.
In May 1972, Annie and I began to break apart molecules of the large multidrug resistance plasmids R6 and R6-5 using the mechanical shearing procedure I had used 6 years earlier to separate and study the two halves of the bacteriophage λ genome (20). The fragmented plasmid DNA was introduced into calcium chloride-treated bacteria, and transformants were screened for cells that acquired individual resistance determinants. I knew from the heteroduplex experiments carried out with Sharp and Davidson that R6 and R6-5 contained repeats of some DNA sequences and hoped that ordinary genetic recombination between these homologous segments would lead to circularization of shear-generated fragments in calcium chloride-treated cells—and perhaps even the formation of novel plasmids lacking some of the original DNA resistance determinants. If such recombinants occurred even infrequently, antibiotic selection might identify colonies of bacteria acquiring them. We did in fact identify bacteria containing a small plasmid that expressed only tetracycline resistance and that we thought had been derived from R-5 (34). However, our later investigations (35) indicated that this was a natural plasmid that had contaminated the DNA preparations; we had underestimated the power of antibiotic selection to identify rare bacterial cells transformed by resistance plasmids. Whereas our efforts in mid-1972 to clone resistance genes from plasmids used largely the same strategy that Boyer and I and our colleagues used successfully less than a year later, they lacked a key ingredient of the later experiments: an enzyme that cuts each plasmid DNA molecule identically and produces complementary sequences at the ends of DNA fragments it generates—i.e., the restriction endonuclease EcoRI.
Restriction Endonucleases
The ability of bacteria to restrict the growth of phage that had been propagated on other strains had been known since the late 1930s, but work aimed at understanding the mechanism underlying this phenomenon didn’t begin for another 20 years. Much of that work was carried out by the Swiss microbiologist and geneticist Werner Arber and his student Daisy Dussoix, who showed that the DNA of restricted phage is enzymatically degraded (9). In 1970, Hamilton Smith and his colleagues at Johns Hopkins University reported that a restriction enzyme they named HindII—a protein isolated from the bacterial pathogen, Haemophilus influenzae—recognizes particular nucleotide sequences in DNA and cuts duplex DNA site-specifically at these sequences (36). The following year, Karen Danna and Daniel Nathans found that the HindII endonuclease cleaves DNA of the mammalian tumor virus SV40 into 11 fragments that can be separated by acrylamide gel electrophoresis, demonstrating the utility of restriction endonucleases for DNA analysis (37). Arber, Nathans, and Smith received the 1978 Nobel Prize in Physiology or Medicine for these accomplishments.
Herb Boyer was studying restriction endonucleases in his laboratory at UCSF. Collaborating with him was Assistant Professor in Residence Daisy Dussoix (then Roulland-Dussoix), who as a student had participated with Arber in early investigations of the restriction/modification phenomenon. Some plasmids had been found to encode restriction enzymes (38, 39), and the Ph.D. thesis project of Robert Yoshimori, a graduate student working with Boyer and Roulland-Dussoix, was to identify new restriction enzymes that might be encoded by E. coli plasmids. He found two such enzymes (40), and Boyer and his colleagues proceeded to purify them and investigate their properties. One of the enzymes was EcoRI (E. coli Restriction endonuclease I). Like HindII, EcoRI cleaved DNA site-specifically, and Boyer set out to determine the nucleotide sequence attacked by the enzyme. Boyer also provided a sample of EcoRI to Norman Salzman at the NIH, whose laboratory found that the oncogenic virus SV40 is cleaved once by this endonuclease and used the cleavage site to map the origin and direction of SV40 DNA replication (41). John Morrow, a Stanford graduate student of Paul Berg, who also received EcoRI from Boyer for testing on SV40 DNA, made the same observation (42). Additional experiments with EcoRI samples that Boyer had given to others yielded an unexpected dividend: evidence that EcoRI, unlike HindII, cleaves the DNA sequence it recognizes asymmetrically, generating single-strand extensions that contain nucleotides complementary to those present at the ends of other EcoRI-generated fragments.
Hydrogen bonding between dA and dT deoxynucleotides and between dGs and dCs had been known for a decade to be able to hold DNA strands together. Alfred Hershey and his colleagues at the Cold Spring Harbor Laboratory had reported in 1963 that bacteriophage λ DNA contains complementary single-strand segments at its ends, enabling linear DNA that had been packaged in a viral particle to become circular and insert into the bacterial chromosome (43). Cohesive ends on λ DNA molecules were used as substrate by Martin Gellert (44) and others (45) to isolate an enzyme, DNA ligase, that covalently joins λ DNA segments held together by complementary ends. Complementary ends were thus well recognized as a device for joining together DNA molecules (46). Attribution of credit for who first made the observation that cleavage of duplex DNA by EcoRI generates fragments that have complementary cohesive termini has been controversial, but what is shown by the published record is that three separate papers simultaneously reporting this finding appeared in the November 1972 issue of PNAS: the papers were authored by Janet E. Mertz and Ronald W. Davis of the Stanford Department of Biochemistry (47), by Vittorio Sgaramella of the Stanford Department of Genetics (48), and by Boyer and his coworkers Howard Goodman and Joe Hedgpeth (49) at UCSF.
The Hawaii Meeting and the Initial DNA Cloning Experiments
In early 1972, I agreed to join Tsutomu Watanabe, a Keio University microbiologist who was a pioneer in studies of bacterial antibiotic resistance, and Donald Helinski in organizing a United States–Japan conference on plasmids later that year. A few weeks before the meeting, which was held in mid-November at the University of Hawaii in Honolulu, Don contacted me to suggest that Herb Boyer, whose work on the plasmid-encoded EcoRI enzyme he had just learned about, be added to the list of speakers. I telephoned our invitation to Herb and thus began a scientific interaction that less than 6 months later resulted in the cloning of antibiotic resistance genes from plasmids.
The actual collaboration began during a long walk near Honolulu’s Waikiki Beach in search of a sandwich shop to have a late evening snack. Boyer and I were joined by Stanley Falkow, who recently had moved his laboratory to the University of Washington, Charles Brinton, a microbiologist from the University of Pittsburgh, and Charles’s wife, Ginger. During that walk, Herb and I discussed recent results from our laboratories. I described our experiments showing that E. coli could be transformed genetically with naked plasmid DNA, and our plasmid DNA shearing experiments, which had not yet been published, and Herb described the similarly unpublished sequencing data that he, Joe Hedgpeth, and Howard Goodman had obtained for the EcoRI cleavage site. As Herb and I talked, I realized that EcoRI was the missing ingredient needed for molecular analysis of antibiotic resistance plasmids. Large plasmids would be cut specifically and reproducibly by the enzyme, and this method of cleavage would surely be better than the haphazard mechanical shearing methods I had been using for fragmentation of plasmid DNA circles. Because EcoRI recognizes a six base pair sequence, cleavage sites on duplex DNA would be on average about 4,100 base pairs apart, and each of the DNA fragments produced would likely contain only a few genes. The number of fragments would be few enough to separate them by centrifugation, enabling their use in DNA–DNA hybridization experiments. Because of the asymmetry of cleavage of the EcoRI recognition sequence, the ends of the multiple plasmid DNA fragments generated by EcoRI would be complementary—and under the right conditions individual plasmid DNA fragments in the mixture could join to each other in different combinations. If cleavage by EcoRI left the replication function of the plasmid intact, the region encoding this function might join randomly to different combinations of antibiotic resistance genes in the fragment mixture, forming DNA circles that could be sealed using DNA ligase, as had been shown for the complementary extensions at the ends of λ DNA (44, 45). And the plasmid DNA transformation procedure would enable us to select for, and hopefully to separately clone, specific resistance genes using appropriate combinations of antibiotics on culture plates.
Herb initially didn’t appear to be especially interested in studying R-plasmid DNA structure and offered simply to provide the enzyme that he generously had given to others. However, he was excited about the use of autonomously replicating R-plasmids to clone EcoRI-generated DNA fragments. By the time we encountered a small delicatessen having an enticing window sign that read, “Shalom,” in place of the ubiquitous “Aloha,” we had decided to proceed collaboratively and agreed on the basic design of the project that our laboratories would jointly carry out. We would target the R6-5 plasmid, which Sharp, Davidson, and I had learned much about from heteroduplex analysis, and which Chang and I had been shearing using a mechanical stirring device and metal blades, in our initial experiments. A few minutes later, over warm corned beef sandwiches and cold beer (Fig. 1), Herb and I sketched out an experimental plan on napkins taken from the dispenser at our table.
Fig. 1.

Cartoon by D. Adair in the Honolulu Advertiser newspaper, September 26, 1988 accompanying an article reporting demolition of the Waikiki beach delicatessen where the initial DNA cloning experiments were planned. The persons depicted clockwise are presumed to be H. Boyer (12 o’clock), S. Cohen, G. Brinton, C. Brinton, and S. Falkow. Reprinted by permission.
Our strategy was straightforward (Fig. 2), but there was no assurance that it would work. Yes, we knew that we could genetically transform E. coli with plasmid DNA and use antibiotic resistance genes to identify cells that acquire plasmids, and we expected from the nucleotide sequence at the EcoRI cleavage site that the restriction enzyme would cut the DNA of our large plasmids reproducibly into multiple fragments. We knew from published earlier results that DNAs having complementary ends would link together by base pairing: Khorana and his colleagues had joined together double-stranded fragments of synthetic DNAs in vitro by chemically adding complementary nucleotides to them one at a time (50)—demonstrating that such recombination is independent of the sequence of the duplex segments being joined. Jensen et al. (51) had used the strategy of bringing natural DNAs together by enzymatically adding stretches of complementary nucleotides to their termini, and Peter Lobban in Dale Kaiser’s laboratory and David Jackson in Paul Berg’s laboratory showed that disparate fragments from either the same genome or different genomes that were held together by enzymatically installed complementary single-strand segments can be ligated to create covalently bonded junctions (52, 53). Berg later commented: “it doesn’t take a genius to figure out that if you can create artificial ends that are complementary to each other, the two DNA molecules will come together” (54). Moreover, results obtained by Mertz and Davis (47) and by Sgaramella (48) and published during the month of the Hawaii meeting showed that the four nucleotide single-strand extensions generated by EcoRI are sufficient in length to enable DNA fragments to be spliced together in vitro.
Fig. 2.

Schematic diagram of the strategy used for construction of biologically functional plasmids (1). R6-5 plasmid DNA fragments generated by cleavage using the EcoRI endonuclease were allowed to associate randomly in vitro and were then covalently joined by DNA ligase. DNA in the resulting mixture was introduced into calcium chloride-treated E. coli, and bacterial colonies expressing individual antibiotic resistance phenotypes encoded by R6-5 were selected on media containing antibiotics. Plasmid constructs isolated from these E. coli clones contained DNA fragments carrying specific resistance genes.
However, notwithstanding our expectation that we would be able to biochemically join the complementary ends of EcoRI-generated fragments of plasmid DNA, there were important biological unknowns in the experiments that Boyer and I planned. Would cleavage of R6-5 with EcoRI disrupt regions needed for plasmid DNA replication or expression of antibiotic resistance? And would recombinant DNA molecules created in the laboratory be reproduced and transcribed in bacterial cells? DNA junctions formed during legitimate genetic recombination in cells are generated by a process that has resulted from billions of years of evolution; would the random joining of DNAs by artificial means create anomalous chromatin conformations that prevent propagation of the molecules? These multiple issues led Falkow, who together with Charles Brinton had participated in the discussion and who envisioned the possibility of isolating an enteric bacterial toxin gene he had been studying using the procedure that Boyer and I had just sketched out, to remark, “If it works, let me know” (55). A senior Stanford colleague whom I spoke with after my return to Palo Alto was considerably less sanguine, proffering the opinion that nothing interpretable was likely to come from the “messy” experimental design.
We began the experiments shortly after the new year. They went more smoothly than we could have hoped, and by March 1973 we had demonstrated the feasibility of the DNA cloning approach that Boyer and I had outlined a few months earlier on delicatessen napkins. During a visit to the Cold Spring Harbor Laboratory that winter to give a seminar talk, Herb learned about the still unpublished agarose gel electrophoresis/DNA staining method that Phillip Sharp, Bill Sugden, and Joseph Sambrook had developed to separate and visualize fragments of DNA generated by restriction enzymes (56); this advance offered a hugely important addition to the centrifugation and heteroduplex methods we were using to analyze plasmids. In the collaboration, Herb’s laboratory purified the restriction endonuclease we used and characterized plasmid DNA in ethidium bromide-stained agarose gels. My laboratory isolated and purified plasmid DNA, did bacterial transformations and selection, and characterized the products by heteroduplex analysis and ultracentrifugation in gradients. Data were analyzed at both places, and results were discussed between laboratories almost daily. I’d arrive in the laboratory early in the morning to look at the culture plates when colonies produced by cells plated late the previous evening were still tiny. I often wished that the bacteria would grow faster so that we could obtain results sooner. Annie lived in San Francisco and carried materials between Stanford and UCSF. We’d hurry to isolate plasmid DNA so that she could carry some of it to Herb’s laboratory for gel analysis the next day. It was an extraordinarily exciting time for all of us.
By introducing a mixture of ligated EcoRI-generated R6-5 DNA fragments into E. coli, we recovered a plasmid that expressed kanamycin resistance but not the other resistance genes of R6-5. This replicon included only three of the DNA fragments characteristic of the parental plasmid (Fig. 3, Top, lanes a and b). Further analysis indicated that one of these fragments encoded functions and sites necessary for autonomous DNA replication but contained no detectable resistance gene; a second fragment lacking the capability for autonomous replication, but carrying a kanamycin resistance gene, had been attached in the fragment mixture to the replication region, and, during bacterial transformation and selection of kanamycin resistant cells, the gene had been cloned. We were absolutely elated but knew that robust DNA cloning would require a plasmid vector that includes both a selectable gene and replication capabilities on a single EcoRI-generated fragment. We found that the tetracycline resistance plasmid that Chang and I had isolated during our mechanical shearing experiments—a replicon we named pSC101 (Fig. 3, Top, lane c and Fig. 3, Middle)—had exactly these properties. Using pSC101 as a vector, we were able to identify the specific R6-5 DNA fragment that carries the kanamycin resistance gene (Fig. 3, Bottom).
Fig. 3.
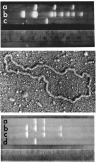
DNA analysis in the initial DNA cloning experiments. (Top) Agarose-gel electrophoresis of (lane a) the pSC102 plasmid containing three of the multiple EcoRI-generated fragments of R6-5 DNA (lane b). Lane c shows that EcoRI cleavage of the pSC101 vector produces a single DNA fragment of the expected size. (Middle) Electron photomicrograph of pSC101, the first plasmid used successfully as a vector for DNA cloning. (Bottom) Agarose gel electrophoresis showing cloning of the kanamycin resistance gene of R6-5: (lane d) EcoRI-cleaved DNA of the pSC101 plasmid vector, (lane c) EcoRI-generated fragments of a novel plasmid (pSC102) that had been constructed from R6-5 (see Top) and that expresses the kanamycin resistance determinant of the parental R6-5 replicon, (lane b) mixture of the DNAs shown in lanes c and d, and (lane a) EcoRI-generated fragments of a novel plasmid (pSC105) expressing both the tetracycline resistance gene of the pSC101 vector and the kanamycin resistance gene, which had been cloned from pSC102 by attaching it to pSC101. Top and Bottom are from ref. 1.
By early May, we had shown that our cloning results were reproducible, and we met to decide on the figures for the manuscript we would be preparing. I outlined the paper’s format in a notebook that sometimes has been referred to as my “laboratory notebook” (57) but which was used for jotting down ideas and future plans rather than for recording experimental data. The paper (1) was completed in early June and, after being modified to address small points raised by reviewers, was communicated to PNAS by Academy member Norman Davidson.
Herb and I had recognized that small antibiotic resistance plasmids such as pSC101 might enable the cloning of eukaryotic cell genes in E. coli and included this statement in our paper’s Discussion section. However, which eukaryotic DNA should be used to test the notion? We couldn’t specifically select bacteria containing cloned eukaryotic DNA, and only a few cellular genes of eukaryotes had been purified and characterized well enough to identify them unambiguously in bacterial isolates. Besides, there were other experiments that each of us wanted to do, and our priorities were different. Herb was eager to use a rapidly expanding collection of restriction enzymes to construct high copy-number vectors that offer increased flexibility of cloning sites. The most pressing issue for me was to learn whether DNA hybrids containing very different components derived from unrelated species could be propagated and cloned. Our laboratories exchanged experimental tools and set out separately to address our different priorities.
Testing of “Interspecies Barriers”
Design of specific experiments that I believed could provide an initial test of the hypothesized barriers to interspecies gene transfer began after completion of the Cohen, Chang, Boyer, and Helling manuscript. Richard Novick of the Public Health Research Institute in New York and his colleagues had described an 18-kb plasmid named pI258 (58) that replicates autonomously in Staphyloccus aureus, but not in E. coli. pI258 had been shown to carry a β-lactamase gene encoding resistance to penicillins, and such resistance might be used to select E. coli transformants carrying hybrid plasmids expressing β-lactamase. Whether DNAs known to be highly disparate in nucleotide composition (11) and taken from microbes as different as the Gram-positive coccus S. aureus and the Gram-negative rod-shaped E. coli could be propagated as part of the same replicon and whether the staphylococcal gene would be expressed in the new host was questionable. However, if these events occurred, the density gradient analysis methods that Miller and I had used earlier (24) would aid in establishing the origin of DNA segments that differ in A+T/G+C ratio. E. coli cells resistant to both penicillin/ampicillin and tetracycline were already highly prevalent, so combining pI258 and pSC101 DNAs would not produce a novel resistance combination.
The experiments themselves were not complicated and the results were conclusive. We cleaved pI258 DNA and pSC101 DNA using the EcoRI enzyme, characterized, and then combined, the DNA fragments, introduced the ligated mixture into calcium chloride-treated E. coli, and selected bacterial colonies that expressed both the ampicillin resistance of pI258 and the tetracycline resistance encoded by pSC101 (Fig. 4, Upper). Buoyant density ultracentrifugation (Fig. 4, Lower), agarose gel electrophoresis, and electron microscope heteroduplex analysis showed that the E. coli colonies contained plasmids that included DNA sequences from the two species (2).
Fig. 4.

Cloning of S. aureus plasmid DNA in E. coli. (Upper) Schematic diagram of strategy used for testing the viability of interspecies DNA hybrids (2). DNA of the pI258 plasmid, which carries a β lactamase gene encoding resistance to penicillins in S. aureus was cleaved by EcoRI endonuclease and mixed with similarly cleaved DNA of the pSC101 vector encoding tetracycline resistance. After ligation, the mixture was introduced into E. coli cells, and colonies that expressed both resistance phenotypes were identified. (Lower) Centrifugation analysis in isopycnic density gradient of plasmid DNA (pSC112) isolated from an E. coli clone expressing both resistances and showing DNA species that band at buoyant densities characteristic of E. coli (ρ = 1.710) and S. aureus (ρ = 1.68–1.69) DNAs and reflect the distinctly different A+T/G+C nucleotide ratios of these unrelated bacterial species. Lower is from ref. 2.
Gordon Conference Discussions About Biohazard Concerns and Cloning of Eukaryotic DNA in Bacteria
Sometime in late May or early June 1973, Boyer received an invitation to give an informal talk at the Gordon Research Conference on Nucleic Acids scheduled for mid-June. He described his laboratory’s work at a session on “Bacterial Enzymes in the Analysis of DNA” and, near the end of his presentation, reported results from our collaborative experiments, which had not yet been accepted for publication. His presentation prompted Bill Sugden, one of the inventors of agarose gel electrophoresis attending the Gordon Conference to comment, “well, now we can put together any DNAs we want to” (59). The following morning on the last day of the meeting, a special session was called by cochairs Maxine Singer and Dieter Söll to discuss the implications of the data that Boyer had shown. Seven months earlier, the ability of EcoRI to generate cohesive DNA ends that could be used for joining DNAs biochemically had been reported; however, now presented with evidence that EcoRI-generated, ordinarily nonreplicating DNA fragments can actually be propagated in bacteria by attaching them to plasmid DNA—and that hybrid DNAs created in this way are biologically functional—the attendees were concerned that hybrids “with biological activity of unpredictable nature may eventually be created” (60). They voted to communicate this concern to the presidents of the National Academy of Sciences (NAS) and its Institute of Medicine, suggesting that a study committee be established to consider the issue and to “recommend specific actions or guidelines.”
A discussion at the same Gordon Conference between Boyer and John Morrow, who had completed his Ph.D. thesis project in Paul Berg’s laboratory at Stanford but had not yet moved to a postdoctoral position with Donald Brown at the Carnegie Institution of Washington laboratory in Baltimore, MD, led to a second collaboration between Boyer’s laboratory and mine. Brown had purified and characterized the ribosomal genes of the African frog, Xenopus laevis, and Morrow had found that this DNA was cleaved by the EcoRI enzyme preparation that Boyer had provided for analysis of SV40 viral DNA. Morrow and Boyer discussed trying to clone EcoRI-generated fragments of frog ribosomal DNA using the approach that Boyer's lab and mine had employed to clone plasmid DNA fragments. Brown agreed to allow the DNA he had given to Morrow to be used for the attempt. However, how to identify cloned ribosomal RNA? When Herb returned to UCSF, he phoned me to discuss this question and to invite my participation in the proposed project. We agreed that multiple parameters would be needed to show unambiguously that DNA from another biological kingdom was being propagated in bacteria. Although there were no phenotypic properties that would enable bacterial colonies that acquired plasmids carrying ribosomal DNA inserts to be selected, ribosomal genes, which were known to be extraordinarily rich in G+C base pairs (61), could be distinguished from E. coli DNA by buoyant density differences during isopycnic centrifugation, as well as by fragment size during agarose gel analysis and by electron microscopy heteroduplex analysis. I thought that it might be necessary to screen a large number of individual bacterial colonies to find recombinant plasmids, but it was worth a try.
Morrow and Chang carried out the bulk of the experiments, which were done largely in my laboratory at Stanford during the late summer and early fall of 1973—still several months before the November publication of results from the initial collaboration with Boyer. As data began to accumulate to support the conclusion that eukaryotic DNA can actually be propagated in bacteria, our examination of grids prepared for electron microscope heteroduplex analysis removed any remaining uncertainty (Fig. 5): heteroduplexes formed between the ribosomal RNA taken from frogs and recombinant plasmids isolated from E. coli revealed homology at two locations that were spaced at the distance that Brown’s laboratory had shown to be the spacing between sequence repeats present on ribosomal RNA genes (3). And additional DNA/RNA hybridization experiments indicated that transcripts capable of interacting with ribosomal RNA genes of the frog were produced in E. coli.
Fig. 5.
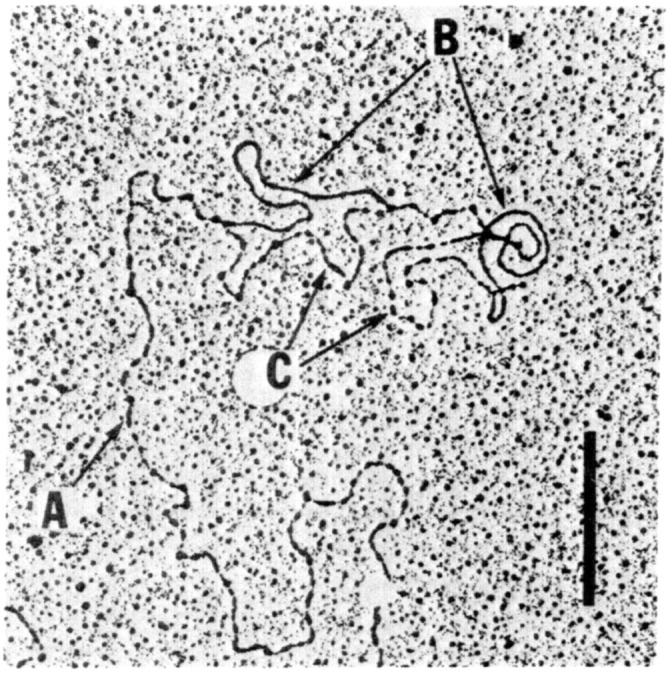
Electric photomicrograph of heteroduplex showing homology between DNA isolated from X. laevis oocytes and plasmid DNA isolated from bacteria and containing fragments of ribosomal RNA genes that had been cloned by attaching the eukaryotic cell DNA to the pSC101 vector. (A) Single strand of X. laevis rDNA. (B) Double-stranded regions of homology between X. laevis rDNA and plasmid DNA isolated from E. coli. (C), Single-strand DNA regions corresponding in length to the pSC101 vector, which shares no homology with X. laevis rDNA. (Scale bar: 1 μm.) Figure is from ref. 3.
The news that eukaryotic DNA can be cloned and amplified in bacteria spread immediately in the scientific community, and requests for the pSC01 plasmid began to arrive at my laboratory. The first to receive pSC101 was David Hogness, a distinguished Stanford Department of Biochemistry professor who had been attempting unsuccessfully to clone Drosophila melanogaster DNA by using λdv, a nonlytic phage variant that Kenichi Matsubara and Dale Kaiser had shown can replicate autonomously in E. coli (62). Attaching the Drosophila DNA to the pSC101 plasmid enabled Hogness and his coworkers to confirm in late 1974 the cloning of eukaryotic cell genes in bacteria and to use DNA cloning to map sequences in Drosophila chromosomes (63). Concurrently, other laboratories focused on constructing new bacteriophage λ variants able to produce viable molecular hybrids (64, 65), and, in November 1974, Ron Davis and his coworkers at Stanford reported that such hybrids can be propagated as plaque-forming phage (66).
Biohazard Speculations Mount
In response to the vote of attendees at the nucleic acids Gordon Conference and the consequent letter from Singer and Söll, NAS President Phillip Handler chose Paul Berg to form a committee to evaluate possible biohazardous consequences of constructing hybrid DNAs. Berg had thought deeply about this issue and was a perfect choice: using complementary ends installed onto DNA and further manipulations that Berg and his postdoctoral fellows David Jackson and Robert Symons credited to Dale Kaiser’s graduate student Peter Lobban, Berg’s laboratory had biochemically attached DNA of the SV40 tumor virus to a version of the bacteriophage λdv replicon that includes the galactose (gal) operon of E. coli (52). Berg later received the 1980 Nobel Prize in Chemistry in recognition of “his fundamental studies of the biochemistry of nucleic acids, with particular regard to recombinant DNA.” Berg and his graduate student Janet Mertz planned to introduce these SV40-λdvgal hybrid DNA molecules into mammalian cells to determine whether the bacterial gene would function there (54, 67), and he and Mertz have written that they also wished to propagate the SV40-λdvgal hybrid DNA molecules in E. coli (67,68). However, at a Cold Spring Harbor Laboratory summer course in 1971 where Mertz described her proposed experiments, biologist Robert Pollack raised biohazard concerns about the possibility of creating oncogenic E. coli by such experiments, and Berg was persuaded to forego attempts at cloning the biochemically spliced SV40-λdvgal DNA molecules in either eukaryotic cells or bacteria (54, 68). Ironically with regard to Pollack’s scenario, Mertz’s 1975 PhD dissertation (67) stated that “scientific problems have been encountered during attempts to use λdvgal as a vector for replicating other DNA molecules” and that “the plasmid is unstable and readily lost from its E. coli host” (67). It was later learned that insertion of foreign DNA into the λdvgal site that the Berg team had used for construction of hybrid DNA molecules (52, 54) disrupts a gene essential for λdvgal replication in bacteria (54, 66, 69), possibly explaining the lack of success of the DNA cloning attempts reported in Mertz’s dissertation (67). But Pollack’s concerns and Berg’s decision had importantly raised awareness about possible biohazardous consequences of creating novel DNA combinations (54).
The committee that Berg formed to address the biohazard concerns of the Gordon Conference participants consisted mostly of experts on oncogenic viruses, and it initially focused on issues related to the introduction of mammalian cell virus sequences into bacteria. However, during discussions by the group, its focus expanded to address the possible construction of novel resistance-gene combinations, and Herb Boyer, David Hogness, Ron Davis, and I were invited to participate in the formulation of the committee’s final recommendations. These recommendations, which were published concurrently in July 1974 in PNAS, Science, and Nature as a letter entitled, “Potential Biohazards of Recombinant DNA Molecules,” proposed a voluntary moratorium on the introduction of resistance genes into bacterial species that do not already express that type of resistance and on the linkage of animal virus genes to plasmids (70–72).
An article by New York Times journalist Victor McElheny, who learned about our DNA cloning experiments from Berg et al. committee member David Baltimore, appeared in the Times a few weeks before release of the committee’s moratorium proposal (73). In this article, which was headlined, “Animal Gene Shifted to Bacteria; Aid Seen to Medicine and Farm,” McElheny and the scientists he interviewed spoke optimistically about the potential benefits of DNA cloning, which was by then increasingly referred to as “recombinant DNA technology.” However, a press conference arranged by the NAS to announce the moratorium proposed by the Berg et al. letter resulted in an abrupt shift of public focus to biohazard issues. The notion that prompted the shift: “if the researchers themselves are concerned, then the dangers must be truly horrific.” The unprecedented effort of scientists to restrict their own research in order to guard against hazards that were not known to exist was so novel that this effort was widely interpreted as implying that danger was likely. A more extensive personal perspective on the Berg et al. letter, the Asilomar Conference that it led to, and the post-Asilomar period of interaction between scientists and legislators is provided in the oral history I recorded with University of California Berkeley historian Sally Smith Hughes for the Bancroft Library in the mid-1990s (74).
Stanford/UCSF DNA Cloning Patents
McElheny’s upbeat article in May 1974 was read by Niels Reimers, whose job as Director of the Office of Technology Licensing at Stanford was to help fund the university’s academic programs by promoting the licensing of inventions made at the university. The day after the article appeared, I received a telephone call from Reimers indicating that he wanted to discuss patenting the technology that Boyer and I had invented. My first reaction was quite negative. Could findings of basic research funded by the public be patented, and should they be? I told him that our work depended on years of fundamental research on plasmid biology by many laboratories and on properties of DNA, DNA ligase, and restriction enzymes that had been discovered by others. And would a patent adversely affect advancement of the science? Reimers pointed out that prior knowledge is a pillar for every invention and that a well-honed legal process determines whether a particular advance is novel and patentable, as well as the validity of the inventorship claimed in the application. He explained that only commercial entities would pay royalties, that a patent would not impede noncommercial use of DNA cloning methods, and that funds received by Stanford and UCSF would aid research programs at these institutions. I discussed Reimer’s proposal with Herb, and together we agreed to let our universities proceed with applications for patents that eventually had 461 licensees before their expiration in 1997. Reimers’ oral history is a source of further information about the events that led to these patents (75).
Converting Promise into Reality
As experiments using DNA cloning procedures proceeded without adverse incident in laboratories around the world, biohazard fears dissipated (74). And multiple scientific advances in these laboratories began to turn the early promise of DNA cloning for producing fundamental knowledge and practical applications into reality. Better strategies for detecting cloned genes and their products were devised (76, 77), and methods were soon developed for cloning enzymatically generated DNA sequences complementary to mRNA (78). The strategies we had used for cloning DNA in E. coli were shown to be applicable to multiple eukaryotic and prokaryotic hosts. Immunologically reactive (79–81) and then biologically functional (82) eukaryotic proteins were produced in bacteria. Specialized vectors were developed by academic and industrial laboratories to express human proteins such as insulin and growth hormone in E. coli, to produce vaccines containing antigens expressed from cloned genes, and to identify genetic regulatory signals using reporter genes. Together, these advances provided a foundation for the creation of biotechnology companies. Herb Boyer and entrepreneur Robert A. Swanson founded an enterprise that became preeminent among these companies: Genentech. Efficient DNA sequencing methods invented by Allan Maxam and Walter Gilbert (83) and by Frederick Sanger and his colleagues (84) dramatically facilitated analysis of cloned DNA, and, together with the invention of the PCR by Kary Mullis (85), information that DNA sequencing yielded about the structure and function of cloned genes (86) led to the birth of the field of genomics. The number and breadth of the scientific discoveries that have occurred during a four-decade time frame seem unprecedented, and the consequent growth of knowledge in biology and chemistry has been almost logarithmic. However, although this article has been retrospective, in reality, the accelerated scientific journey that has resulted from the ability to clone DNA has only begun.
Acknowledgments
I thank Herb Boyer, Annie Chang, and Bob Helling for their partnership in the scientific collaboration that resulted in successful DNA cloning, and both Annie Chang and Christine Miller for the other contributions they made to my laboratory and to science over decades. I also thank the many scientists whose prior research made possible the experiments I’ve described, especially plasmid biologists/microbiologists Stanley Falkow, Donald Helinski, and Richard Novick; biochemists Martin Gellert, Robert Lehman, Jerard Hurwitz, and Phillip Sharp; geneticists/microbiologists Dale Kaiser, Peter Lobban, Ronald Davis, and Vittorio Sgaramella; and biologist James D. Watson. The contributions and support of now-deceased friends and colleagues, especially Joshua Lederberg, Norton Zinder, Jerome Vinograd, and Norman Davidson, have also been of major importance to me, both scientifically and personally. I apologize to those whose relevant contributions have not been specifically mentioned. For more extensive information about my perspective of the events I’ve related here, I refer readers to my Bancroft Library oral history (74), and for the perspectives of others, to the Bancroft Library oral histories of Herbert W. Boyer (87), Paul Berg (54), and Niels Reimers (75).
Footnotes
The author declares no conflict of interest.
This article is a PNAS Direct Submission.
This article was invited in recognition of the 40th anniversary of the November 1973 PNAS paper by S. N. Cohen, A. C. Y. Chang, H. W. Boyer, and R. B. Helling reporting a method for constructing and cloning biologically functional DNA molecules (1).
References
- 1.Cohen SN, Chang ACY, Boyer HW, Helling RB. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci USA. 1973;70(11):3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang ACY, Cohen SN. Genome construction between bacterial species in vitro: Replication and expression of Staphylococcus plasmid genes in Escherichia coli. Proc Natl Acad Sci USA. 1974;71(4):1030–1034. doi: 10.1073/pnas.71.4.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrow JF, et al. Replication and transcription of eukaryotic DNA in Escherichia coli. Proc Natl Acad Sci USA. 1974;71(5):1743–1747. doi: 10.1073/pnas.71.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinder ND, Lederberg J. Genetic exchange in Salmonella. J Bacteriol. 1952;64(5):679–699. doi: 10.1128/jb.64.5.679-699.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis AM, Jr, Prigge KO, Rowe WP. Studies of adenovirus-SV40 hybrid viruses. IV. An adenovirus type 2 strain carrying the infectious SV40 genome. Proc Natl Acad Sci USA. 1966;55(3):526–531. doi: 10.1073/pnas.55.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowe WP, Pugh WE. Studies of adenovirus-SV40 hybrid viruses. V. Evidence for linkage between adenovirus and SV40 genetic materials. Proc Natl Acad Sci USA. 1966;55(5):1126–1132. doi: 10.1073/pnas.55.5.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chargaff E (1977) Research with Recombinant DNA: An Academy Forum March 7–9, 1977 (National Academy of Sciences, Washington, DC), pp 56–57.
- 8.Sinsheimer RL. Recombinant DNA. Annu Rev Biochem. 1977;46:415–438. doi: 10.1146/annurev.bi.46.070177.002215. [DOI] [PubMed] [Google Scholar]
- 9.Arber W. Host-controlled modification of bacteriophage. Annu Rev Microbiol. 1965;19:365–378. doi: 10.1146/annurev.mi.19.100165.002053. [DOI] [PubMed] [Google Scholar]
- 10.Falkow S. Nucleic acids, genetic exchange and bacterial speciation. Am J Med. 1965;39(5):753–765. doi: 10.1016/0002-9343(65)90095-1. [DOI] [PubMed] [Google Scholar]
- 11.Marmur J, Falkow S, Mandel M. New approaches to bacterial taxonomy. Annu Rev Microbiol. 1963;17:329–372. doi: 10.1146/annurev.mi.17.100163.001553. [DOI] [PubMed] [Google Scholar]
- 12.Lederberg J, Cavalli LL, Lederberg EM. Sex compatibility in Escherichia coli. Genetics. 1952;37(6):720–730. doi: 10.1093/genetics/37.6.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada K, Kameda M, Suzuki M, Mitsuhashi S. Drug resistance of enteric bacteria. 3. Acquisition of transferability of nontransmissible R(Tc) factor in cooperation with F factor and formation of Fr(Tc) J Bacteriol. 1964;88:1257–1265. doi: 10.1128/jb.88.5.1257-1265.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe T, Ogata C, Sato S. Episome-mediated transfer of drug resistance in Enterobacteriaceae. 8. Six-drug-resistance R factor. J Bacteriol. 1964;88:922–928. doi: 10.1128/jb.88.4.922-928.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lederberg J. Cell genetics and hereditary symbiosis. Physiol Rev. 1952;32(4):403–430. doi: 10.1152/physrev.1952.32.4.403. [DOI] [PubMed] [Google Scholar]
- 16.Falkow S, Citarella RV, Wohlhieter JA. The molecular nature of R-factors. J Mol Biol. 1966;17:102–116. doi: 10.1016/s0022-2836(66)80097-9. [DOI] [PubMed] [Google Scholar]
- 17.Rownd R, Nakaya R, Nakamura A. Molecular nature of the drug-resistance factors of the Enterobacteriaceae. J Mol Biol. 1966;17(2):376–393. doi: 10.1016/s0022-2836(66)80149-3. [DOI] [PubMed] [Google Scholar]
- 18.Anderson ES, Lewis MJ. Characterization of a transfer factor associated with drug resistance in Salmonella typhimurium. Nature. 1965;208(5013):843–849. doi: 10.1038/208843a0. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe T, Fukasawa T. Episome-mediated transfer of drug resistance in Enterobacteriaceae. I. Transfer of resistance factors by conjugation. J Bacteriol. 1961;81:669–678. doi: 10.1128/jb.81.5.669-678.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen SN, Maitra U, Hurwitz J. Role of DNA in RNA synthesis. XI. Selective transcription of gamma DNA segments in vitro by RNA polymerase of Escherichia coli. J Mol Biol. 1967;26(1):19–38. doi: 10.1016/0022-2836(67)90258-6. [DOI] [PubMed] [Google Scholar]
- 21.Campbell AM. Episomes. Adv Genet. 1963;11:101–145. [Google Scholar]
- 22.Jacob F, Brenner S, Cuzin F. On the regulation of DNA replication in bacteria. Cold Spring Harb Symp Quant Biol. 1963;28:329–348. [Google Scholar]
- 23.Hickson FT, Roth TF, Helinski DR. Circular DNA forms of a bacterial sex factor. Proc Natl Acad Sci USA. 1967;58(4):1731–1738. doi: 10.1073/pnas.58.4.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen SN, Miller CA. Multiple molecular species of circular R-factor DNA isolated from Escherichia coli. Nature. 1969;224(5226):1273–1277. doi: 10.1038/2241273a0. [DOI] [PubMed] [Google Scholar]
- 25.Davis RW, Davidson N. Electron-microscopic visualization of deletion mutations. Proc Natl Acad Sci USA. 1968;60(1):243–250. doi: 10.1073/pnas.60.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westmoreland BC, Szybalski W, Ris H. Mapping of deletions and substitutions in heteroduplex DNA molecules of bacteriophage lambda by electron microscopy. Science. 1969;163(3873):1343–1348. doi: 10.1126/science.163.3873.1343. [DOI] [PubMed] [Google Scholar]
- 27.Sharp PA, Cohen SN, Davidson N. Electron microscope heteroduplex studies of sequence relations among plasmids of Escherichia coli. II. Structure of drug resistance (R) factors and F factors. J Mol Biol. 1973;75(2):235–255. doi: 10.1016/0022-2836(73)90018-1. [DOI] [PubMed] [Google Scholar]
- 28.Sharp PA, Hsu MT, Otsubo E, Davidson N. Electron microscope heteroduplex studies of sequence relations among plasmids of Escherichia coli. I. Structure of F-prime factors. J Mol Biol. 1972;71(2):471–497. doi: 10.1016/0022-2836(72)90363-4. [DOI] [PubMed] [Google Scholar]
- 29.Cohen SN. Transposable genetic elements and plasmid evolution. Nature. 1976;263(5580):731–738. doi: 10.1038/263731a0. [DOI] [PubMed] [Google Scholar]
- 30.Starlinger P, Saedler H. IS-elements in microorganisms. Curr Top Microbiol Immunol. 1976;75:111–152. doi: 10.1007/978-3-642-66530-1_4. [DOI] [PubMed] [Google Scholar]
- 31.Mandel M, Higa A. Calcium-dependent bacteriophage DNA infection. J Mol Biol. 1970;53(1):159–162. doi: 10.1016/0022-2836(70)90051-3. [DOI] [PubMed] [Google Scholar]
- 32.Cosloy SD, Oishi M. Genetic transformation in Escherichia coli K12. Proc Natl Acad Sci USA. 1973;70(1):84–87. doi: 10.1073/pnas.70.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen SN, Chang ACY, Hsu L. Nonchromosomal antibiotic resistance in bacteria: Genetic transformation of Escherichia coli by R-factor DNA. Proc Natl Acad Sci USA. 1972;69(8):2110–2114. doi: 10.1073/pnas.69.8.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen SN, Chang ACY. Recircularization and autonomous replication of a sheared R-factor DNA segment in Escherichia coli transformants. Proc Natl Acad Sci USA. 1973;70(5):1293–1297. doi: 10.1073/pnas.70.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen SN, Chang AC. Revised interpretation of the origin of the pSC101 plasmid. J Bacteriol. 1977;132(2):734–737. doi: 10.1128/jb.132.2.734-737.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith HO, Wilcox KW. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J Mol Biol. 1970;51(2):379–391. doi: 10.1016/0022-2836(70)90149-x. [DOI] [PubMed] [Google Scholar]
- 37.Danna K, Nathans D. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc Natl Acad Sci USA. 1971;68(12):2913–2917. doi: 10.1073/pnas.68.12.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takano T, Watanabe T, Fukasawa T. Specific inactivation of infectious lambda DNA by sonicates of restrictive bacteria with R factors. Biochem Biophys Res Commun. 1966;25(2):192–198. doi: 10.1016/0006-291x(66)90579-1. [DOI] [PubMed] [Google Scholar]
- 39.Takano T, Watanabe T, Fukasawa T. Mechanism of host-controlled restriction of bacteriophage lambda by R factors in Escherichia coli K12. Virology. 1968;34(2):290–302. doi: 10.1016/0042-6822(68)90239-0. [DOI] [PubMed] [Google Scholar]
- 40.Yoshimori R, Roulland-Dussoix D, Boyer HW. R factor-controlled restriction and modification of deoxyribonucleic acid: Restriction mutants. J Bacteriol. 1972;112(3):1275–1279. doi: 10.1128/jb.112.3.1275-1279.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fareed GC, Garon GF, Salzman NP. Origin and direction of simian virus 40 deoxyribonucleic acid replication. J Virol. 1972;10(3):484–491. doi: 10.1128/jvi.10.3.484-491.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrow JF, Berg P. Cleavage of Simian virus 40 DNA at a unique site by a bacterial restriction enzyme. Proc Natl Acad Sci USA. 1972;69(11):3365–3369. doi: 10.1073/pnas.69.11.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hershey AD, Burgi E, Ingraham L. Cohesion of DNA molecules isolated from phage lambda. Proc Natl Acad Sci USA. 1963;49(5):748–755. doi: 10.1073/pnas.49.5.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gellert M. Formation of covalent circles of lambda DNA by E. coli extracts. Proc Natl Acad Sci USA. 1967;57(1):148–155. doi: 10.1073/pnas.57.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gefter ML, Becker A, Hurwitz J. The enzymatic repair of DNA. I. Formation of circular lambda-DNA. Proc Natl Acad Sci USA. 1967;58(1):240–247. doi: 10.1073/pnas.58.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaiser AD, Wu R. Structure and function of DNA cohesive ends. Cold Spring Harb Symp Quant Biol. 1968;33:729–734. doi: 10.1101/sqb.1968.033.01.083. [DOI] [PubMed] [Google Scholar]
- 47.Mertz JE, Davis RW. Cleavage of DNA by R 1 restriction endonuclease generates cohesive ends. Proc Natl Acad Sci USA. 1972;69(11):3370–3374. doi: 10.1073/pnas.69.11.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sgaramella V. Enzymatic oligomerization of bacteriophage P22 DNA and of linear Simian virus 40 DNA. Proc Natl Acad Sci USA. 1972;69(11):3389–3393. doi: 10.1073/pnas.69.11.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedgpeth J, Goodman HM, Boyer HW. DNA nucleotide sequence restricted by the RI endonuclease. Proc Natl Acad Sci USA. 1972;69(11):3448–3452. doi: 10.1073/pnas.69.11.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khorana HG. Polynucleotide synthesis and the genetic code. Harvey Lect. 1966-1967;62:79–105. [PubMed] [Google Scholar]
- 51.Jensen RH, Wodzinski RJ, Rogoff MH. Enzymatic addition of cohesive ends to T7 DNA. Biochem Biophys Res Commun. 1971;43(2):384–392. doi: 10.1016/0006-291x(71)90765-0. [DOI] [PubMed] [Google Scholar]
- 52.Jackson DA, Symons RH, Berg P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad Sci USA. 1972;69(10):2904–2909. doi: 10.1073/pnas.69.10.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lobban PE, Kaiser AD. Enzymatic end-to-end joining of DNA molecules. J Mol Biol. 1973;78(3):453–471. doi: 10.1016/0022-2836(73)90468-3. [DOI] [PubMed] [Google Scholar]
- 54. Berg P (2000) A Stanford Professor's Career in Biochemistry, Science Politics, and the Biotechnology Industry (typescript of an oral history conducted in 1997 by Sally Smith Hughes) (Regional Oral History Office, The Bancroft Library, University of California, Berkeley, CA)
- 55.Falkow S. Oral history with Charles Weiner, MIT Oral History Program Recorded on May 29, 1976 and Feb 26, 1977. Cambridge, MA: MIT Institute Archives; 1977. [Google Scholar]
- 56.Sharp PA, Sugden B, Sambrook J. Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose—ethidium bromide electrophoresis. Biochemistry. 1973;12(16):3055–3063. doi: 10.1021/bi00740a018. [DOI] [PubMed] [Google Scholar]
- 57. Sharrer T (October 1, 2006) Recombinant DNA: The first report. The Scientist, p100.
- 58.Peyru G, Wexler LF, Novick RP. Naturally occurring penicillinase plasmids in Staphylococcus aureus. J Bacteriol. 1969;98(1):215–221. doi: 10.1128/jb.98.1.215-221.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lear J. Recombinant DNA: The Untold Story. New York: Crown Publishers; 1978. [Google Scholar]
- 60.Singer M, Soll D. Guidelines for DNA hybrid molecules. Science. 1973;181(4105):1114. doi: 10.1126/science.181.4105.1114. [DOI] [PubMed] [Google Scholar]
- 61.Davison PF. Isopycnic centrifugation for the isolation of DNA strands coding for ribosomal RNA. Science. 1966;152(3721):509–512. doi: 10.1126/science.152.3721.509. [DOI] [PubMed] [Google Scholar]
- 62.Matsubara K, Kaiser AD. Lambda dv: An autonomously replicating DNA fragment. Cold Spring Harb Symp Quant Biol. 1968;33:769–775. doi: 10.1101/sqb.1968.033.01.088. [DOI] [PubMed] [Google Scholar]
- 63.Wensink PC, Finnegan DJ, Donelson JE, Hogness DS. A system for mapping DNA sequences in the chromosomes of Drosophila melanogaster. Cell. 1974;3(4):315–325. doi: 10.1016/0092-8674(74)90045-2. [DOI] [PubMed] [Google Scholar]
- 64.Hershfield V, Boyer HW, Yanofsky C, Lovett MA, Helinski DR. Plasmid ColEl as a molecular vehicle for cloning and amplification of DNA. Proc Natl Acad Sci USA. 1974;71(9):3455–3459. doi: 10.1073/pnas.71.9.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murray NE, Murray K. Manipulation of restriction targets in phage lambda to form receptor chromosomes for DNA fragments. Nature. 1974;251(5475):476–481. doi: 10.1038/251476a0. [DOI] [PubMed] [Google Scholar]
- 66.Thomas M, Cameron JR, Davis RW. Viable molecular hybrids of bacteriophage lambda and eukaryotic DNA. Proc Natl Acad Sci USA. 1974;71(11):4579–4583. doi: 10.1073/pnas.71.11.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mertz JE (1975) Deletion mutants of simian virus 40. PhD dissertation (Stanford University, Stanford, CA)
- 68.Berg P, Mertz JE. Personal reflections on the origins and emergence of recombinant DNA technology. Genetics. 2010;184(1):9–17. doi: 10.1534/genetics.109.112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helling RB, Goodman HM, Boyer HW. Analysis of endonuclease R-EcoRI fragments of DNA from lambdoid bacteriophages and other viruses by agarose-gel electrophoresis. J Virol. 1974;14(5):1235–1244. doi: 10.1128/jvi.14.5.1235-1244.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anonymous Potential biohazards of recombinant DNA molecules. Proc Natl Acad Sci USA. 1974;71(7):2593–2594. doi: 10.1073/pnas.71.7.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anonymous NAS ban on plasmid engineering. Nature. 1974;250:175. [Google Scholar]
- 72.Berg P, et al. Letter: Potential biohazards of recombinant DNA molecules. Science. 1974;185(4148):303. [PubMed] [Google Scholar]
- 73. McElheny VK (May 20, 1974) Animal gene shifted to bacteria; aid seen to medicine and farm. NY Times, p 61.
- 74. Cohen SN (2009) Science, Biotechnology, and Recombinant DNA: A Personal History (typescript of an oral history conducted in 1995 by Sally Smith Hughes) (Regional Oral History Office, The Bancroft Library, University of California, Berkeley, CA)
- 75. Reimers N (1998) Stanford's Office of Technology Licensing and the Cohen/Boyer Cloning Patents (typescript of an oral history conducted in 1997 by Sally Smith Hughes) (Regional Oral History Office, The Bancroft Library, University of California, Berkeley, CA), p 85.
- 76.Grunstein M, Hogness DS. Colony hybridization: A method for the isolation of cloned DNAs that contain a specific gene. Proc Natl Acad Sci USA. 1975;72(10):3961–3965. doi: 10.1073/pnas.72.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kedes LH, Chang ACY, Houseman D, Cohen SN. Isolation of histone genes from unfractionated sea urchin DNA by subculture cloning in E. coli. Nature. 1975;255(5509):533–538. doi: 10.1038/255533a0. [DOI] [PubMed] [Google Scholar]
- 78.Efstratiadis A, Kafatos FC, Maxam AM, Maniatis T. Enzymatic in vitro synthesis of globin genes. Cell. 1976;7(2):279–288. doi: 10.1016/0092-8674(76)90027-1. [DOI] [PubMed] [Google Scholar]
- 79.Goeddel DV, et al. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc Natl Acad Sci USA. 1979;76(1):106–110. doi: 10.1073/pnas.76.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Itakura K, et al. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science. 1977;198(4321):1056–1063. doi: 10.1126/science.412251. [DOI] [PubMed] [Google Scholar]
- 81.Villa-Komaroff L, et al. A bacterial clone synthesizing proinsulin. Proc Natl Acad Sci USA. 1978;75(8):3727–3731. doi: 10.1073/pnas.75.8.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang ACY, et al. Phenotypic expression in E. coli of a DNA sequence coding for mouse dihydrofolate reductase. Nature. 1978;275(5681):617–624. doi: 10.1038/275617a0. [DOI] [PubMed] [Google Scholar]
- 83.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA. 1977;74(2):560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saiki RK, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 86.Nakanishi S, et al. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278(5703):423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 87. Boyer HW (2001) Recombinant DNA Research at UCSF and Commercial Application at Genentech (typescript of an oral history conducted in 1994 by Sally Smith Hughes) (Regional Oral History Office, The Bancroft Library, University of California, Berkeley, CA)