

Abstract
Artificial membrane systems allow researchers to study the structure and function of membrane proteins in a matrix that approximates their natural environment and to integrate these proteins in ex vivo devices such as electronic biosensors, thin-film protein arrays, or biofuel cells. Given that most membrane proteins have vectorial functions, both functional studies and applications require effective control over protein orientation within a lipid bilayer. In this work, we explored the role of the bilayer surface charge in determining transmembrane protein orientation and functionality during formation of proteoliposomes. We reconstituted a model vectorial ion pump, proteorhodopsin, in liposomes of opposite charges and varying charge densities and determined the resultant protein orientation. Antibody-binding assay and proteolysis of proteoliposomes showed physical evidence of preferential orientation, and functional assays verified the vectorial nature of ion transport in this system. Our results indicate that the manipulation of lipid composition can indeed control orientation of an asymmetrically charged membrane protein, proteorhodopsin, in liposomes.
Introduction
Living systems use membrane proteins to regulate transport and signaling across different cellular compartments. The amphiphilic properties of membrane proteins, which contribute to their stability and ability to partition into membranes, also make it difficult for researchers to investigate their structure and functionality outside of living systems. Fortunately, reconstitution of membrane proteins into proteoliposomes—lipid vesicles that contain membrane proteins embedded in the lipid bilayer—has allowed extensive biochemical manipulation and transport characterization in these systems, and today reconstitution studies have become a standard approach for working with membrane proteins. For example, researchers have used vesicles containing antigenic proteins and lipids as a target for toxin (1,2) and hormone binding (3). Proteoliposomes can fuse with cellular membranes and organelles to incorporate nonnative proteins and thus rescue ion exchange in cells deficient in an appropriate receptor (4). Proteoliposome systems are also useful for immobilization of membrane proteins on solid surfaces (5), which are becoming increasingly relevant for biotechnology applications such as biosensors, biofuel cells, and protein arrays. Membrane proteins embedded in lipid bilayers on a solid support also provide an ideal model system for studying cell signaling and ligand–receptor interactions (6–8) and for developing bioelectronics devices (9). The most common approach for the formation of such supported lipid bilayers involves proteoliposome rupture and fusion onto the substrate (10).
The vectorial nature of many membrane proteins often creates a technical problem for fundamental studies and biotechnology applications. Once membrane proteins are solubilized, their orientation becomes randomized. If this random orientation persists on membrane reconstitution, it could lead to reduced or even completely undetectable signature of protein activity. In most cases, the orientation of protein in a liposomal membrane is random (11); in others, is it either exclusively inside-out or outside-out relative to the native orientation (12) and cannot be tailored to a specific application. Yet, researchers need robust approaches to control the protein orientation to maximize the signal detectable in a specific application or to be able to work with the proteins that can only be activated from one side of the membrane. For formation of supported lipid bilayers, a substrate surface functionalized with a monolayer of nitrilotriacetic acid (13,14) can be used to recruit hexahistidine-tagged proteins in a directed orientation; yet, this technique is limiting because it requires protein labeling and irreversible (and often undesirable) modification of substrate surface. The major means of orienting membrane proteins in supported bilayers is to fuse vesicles containing preoriented proteins onto the solid support; researchers can then benefit from an ability to form uniformly oriented and vectorially functional proteoliposome systems.
Researchers have created preoriented proteoliposome systems by using large silicate bead particles attached to one end of the protein to enforce membrane insertion via the opposite end (15), by forming giant unilamellar vesicles using the water-in-oil droplet transfer method (16), or by using blockers after liposome formation to inactivate half of the randomly oriented protein population (17). Although these methods are viable for certain systems, they often dilute the sample with multiple washing steps, lead to the formation of large cell-sized vesicles, or waste half of the protein amount in the sample.
In this study, we show that we can use composition and surface charge of the lipid bilayer to create an oriented population of the membrane protein. In particular, we found that surface charge of the bilayer can direct preferential orientation of an asymmetrically charged protein during the liposome formation. We used proteolytic and antibody-labeling assays to demonstrate asymmetric insertion of a light-driven proton pump into vesicles of opposite charge and showed that this insertion leads to a preferential proton transport in a direction consistent with the insertion orientation.
Materials and Methods
Plasmids and strains
The SAR proteorhodopsin (pR) coding sequence with C-terminal herpes simplex virus (HSV) and hexahistidine tags in the pET27b(+) vector (18) was a kind gift of Dr. Ernst Bamberg (Max Plank Institute of Molecular Physiology, Dortmund, Germany). It was transformed and expressed in the Escherichia coli strain BL21(DE3).
Expression and purification of pR
E. coli cultures were inoculated in 1-L Luria Bertani broth containing 50 μg/mL kanamycin and grown at 37°C with 275 rpm shaking. When the A578 nm reached ∼0.9 OD, gene expression was induced by addition of isopropylthiogalactoside to 1 mM, and 20 μM all-trans retinal (Sigma-Aldrich, St. Louis, MO) was supplied as cofactor. Cultures were grown for an additional 12–16 h at 37°C with 275 rpm shaking. Cells were harvested by centrifugation at 16,000 g, and the resulting cell pellet was pink-orange in color because of pR expression. The cells were washed and resuspended in 10 mM HEPES buffer pH 7.6 and lysed at 1.5–2.0 kbar for 30 min on ice using an EmulsiFlex-C3 cell homogenizer (Avestin, Ottawa, ON, Canada). The crude membrane fraction was pelleted by centrifugation of the cell lysate at 100,000 rcf for 2 h at 4°C. The membrane fraction was then solubilized with 2% (w/v) Triton X-100 overnight at 4°C, and insoluble debris were pelleted by centrifugation at 10,000 g for 1 h at 4°C. The resulting supernatant was incubated with nitrilotriacetic acid agarose resin (Qiagen, Valencia, CA) for ∼45 min at 4°C with stirring to allow binding of the pR to the resin. After being packed into a gravity flow column, the resin was rinsed thoroughly with at least 10 column volumes of wash buffer containing 50 mM TRIS, 100 mM NaCl, 4 M urea, 40 mM imidazole, 0.5 mM dithiothreitol, and 0.1% (w/v) n-dodecyl-β-D-maltoside (β-DDM), pH 7.6. The protein was eluted in 50 mM Tris, 100 mM NaCl, 300 mM imidazole, 1 mM dithiothreitol, pH 7.6, and exchanged into 10 mM HEPES, 0.05% (w/v) β-DDM, pH 7.6 using spin dialysis membranes (40K MWCO). Purity of the resulting pR was analyzed by SDS-PAGE, and concentration was calculated using the absorbance value at 520 nm and an extinction coefficient of 45,000 M–1 cm–1 (19); pure fractions were stored at 4°C for up to 2 months. The typical protein yields were in the range of 1–2 mg of pR per liter of culture.
Preparation of liposomes
The lipids used in this study were 18:1 (Δ9-Cis) 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 16:0-18:1 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 18:1 (Δ9-Cis) 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG), 16:0–18:1 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), and 18:1 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) lipid (Avanti Polar Lipids, Alabaster, AL).
Unilamellar liposomes were prepared using the extrusion technique. Briefly, lipid solutions in chloroform were mixed to give the desired stoichiometric compositions, dried to a lipid film on a glass vial using a steady stream of N2, hydrated with 10 mM HEPES buffer pH 6.2 to a final concentration of 3 mg/mL lipid, and extruded 20 times through 200-nm polycarbonate membranes (Avanti Polar Lipids). The resulting vesicles were stored at 4°C and used within a week. To form negatively or positively charged liposomes, we used POPC:POPG (80:20 mol %) and DOPC:DOTAP (80:20 mol %), respectively. For charge-neutral liposomes, we used 100% DOPC lipid. To prepare 5 (6) carboxyfluorescein (5(6)CF) dye-entrapped liposomes, the dried lipid film was hydrated with 0.05 mM 5(6)CF in 10 mM HEPES pH 6.2. Excess unencapsulated dye was removed using size-exclusion chromatography with a column containing Sepharose CL-6B (Sigma-Aldrich).
Oriented reconstitution of pR in charged proteoliposomes
Detergent-mediated reconstitution (20) of pR into these liposomes was performed at a protein:lipid molar ratio of 1:500. We started with 480 μL of 200-nm extruded liposome solution in 10 mM HEPES pH 6.2 ([lipid] = 3 mg/mL) and added 95 μL of 0.05% β-DDM in 10 mM HEPES buffer pH 6.2 to allow the detergent to induce permeability in liposomes. After incubating this mixture for 45–60 min, we added 8.1 μL of pR–micelle (10.8 mg/mL) solution in 10 mM HEPES buffer, 0.05% β-DDM. The final detergent concentration in this solution is 0.0088%, just above the critical micelle concentration. The lipid–pR–detergent complex was incubated at room temperature for 1 h. To ensure complete removal of the detergent, we used polystyrene biobeads SM2 (Bio-Rad, Hercules, CA), which were thoroughly washed using the Holloway method (21). Specifically, the biobeads were washed with 100% methanol two times, washed with deionized water three to four times, and then equilibrated in 10 mM HEPES buffer. Biobeads (160 mg/mL) were added to the lipid–pR–detergent mixture and gently stirred at room temperature in the dark for 6–8 h, during which time the solution was transferred into a fresh batch of biobeads each hour. Following this, the complex was further incubated with biobeads overnight at 4°C with gentle stirring. To form proteoliposomes in pH 5 and pH 9 environments, we used 10 mM MES and 10 mM TRIS buffers, respectively, to hydrate dried lipid films and to prepare 0.05% β-DDM solutions. We monitored removal of β-DDM and changes in the particle size over time using ultraviolet-visible spectroscopy (NanoDrop 2000, Thermo Scientific, Pittsburgh, PA) and dynamic light scattering measurements (Zetasizer Nano Range, Malvern, Worcestershire, UK) after each biobead exchange. For experiments requiring disrupted proteoliposomes, we added 10% (w/v) aqueous solution of Triton X-100 detergent (Thermo Scientific, Logan, UT) and mixed the solution by pipetting. To form proteoliposomes containing bacteriorhodopsin (Sigma-Aldrich), we followed similar procedures as described above.
Proteolytic digestion of pR-containing proteoliposomes
To assay orientation of pR in proteoliposomes, we added proteinase K (Thermo Scientific, Pittsburgh, PA) to the proteoliposomes to a final concentration of 2.5 mg/mL (specific activity 2.5 units/mg). These samples were incubated for 2 h at 37°C. To stop the reaction, we added the protease inhibitor phenylmethanesulfonylfluoride in absolute ethanol to a concentration of 10 mM and cooled the reaction on ice for 30 min. The reaction products were loaded onto 4–20% Tris-HCl Criterion Precast Gels (Bio-Rad).
Zeta potential measurements
Zeta potential measurements were taken using a dip cell configuration on a Malvern Zetasizer Nano instrument at 25°C. In all cases, the final lipid concentration was 2.5 mg/ml in 10 mM HEPES buffer pH 6.2.
Determination of pR-bound HSV antibodies using fluorimetry and ultraviolet-visible spectroscopy
For this experiment, we used a fluorescein-conjugated HSV Tag Goat anti-HSV polyclonal antibody (FITC-α-HSV, LifeSpan Biosciences, Seattle, WA). Proteoliposomes were made from DOPC, POPC-POPG, and DOPC-DOTAP lipid combinations with a pR:lipid mole ratio of 1:500 using methods described previously. These samples were incubated with FITC-α-HSV (1:1 pR:FITC-α-HSV molar ratio) for 2 h at room temperature in the dark, and excess unbound antibody was removed using size-exclusion chromatography. A gravity-flow, size exclusion column packed with a CL-6B Sepharose agarose (Sigma-Aldrich) and equilibrated with 10 mM HEPES buffer pH 7.6 was used for this purpose. Elution fractions were collected in a 96-well plate format and an absorbance spectrum was taken for each well using a SpectraMax Plate Reader. The fractions showing both light-scatter from the proteoliposomes and a pR absorbance peak at 520 nm were pooled into a single sample. The fluorescein emission from bound FITC-α-HSV was measured for each proteoliposome sample using a Jobin–Yvon fluorimeter (ex. λ = 494 nm, em. λ = 516 nm). Following the fluorescence emission measurement, the proteoliposome samples were measured by ultraviolet-visible spectroscopy to determine the mole fraction of protein to antibody in each of the samples. Because the scattering from intact proteoliposomes obscures the pR absorption, we first used a 10% (w/v) aqueous solution of Triton X-100 to break apart the liposomes and incorporate the protein into detergent micelles. FITC-α-HSV and pR concentrations were then determined using absorbance values at 494 nm and 520 nm, respectively, and extinction coefficients of 70,000 M–1 cm–1 and 45,000 M–1 cm–1, respectively (19).
Fluorescence measurements for determination of proton pumping
We prepared 0.05 mM 5(6)CF-entrapped proteoliposomes of varying lipid composition using methods described above. The proteoliposome solutions were diluted to 0.6 mg/mL lipid concentration in 10 mM HEPES buffer pH 6.8, allowed to equilibrate in the dark for 1 h, and then were placed in the fluorimeter measuring chamber. To induce proton pumping by pR, we illuminated the solutions with 530-nm light using a band-pass filter (Newport, Irvine, CA) at 50-s intervals by using a flexible light guide directed above the cuvette. The shutter was closed during the external illumination periods to protect the fluorimeter detector. After each illumination period, 5(6)CF fluorescence emission was measured at 514 nm ± (4) nm (slit width). The observed fluorescence changes were converted to pH using a calibration curve that we obtained by measuring fluorescence intensity of 0.05 mM 5(6)CF in a series of buffer solutions of known pH.
Results and Discussion
Recent simulation studies have shown that nonspecific and long-distance electrostatic interactions can facilitate protein recruitment to charged surfaces such as biomembranes (22). To determine whether the surface charge influences the direction of protein recruitment in an ex vivo environment into artificial membranes, we inserted a model membrane protein into bilayers formed by lipids with differently charged polar headgroups and determined the resulting protein orientation (Fig. 1 A). Our experiments used a light-driven proton pump: pR. pR has seven transmembrane α-helices that enclose a retinal chromophore. On light absorption and under conditions of neutral pH, the protein undergoes a series of conformational shifts that translocate one proton across the membrane from the C-terminal face of pR to its N-terminal face (23). Interestingly, insertion into the lipid bilayer is required for pR to be functional. The structure of proteorhodopsin also gives it an inherent asymmetric charge density (24); the C-terminal has an overall positive charge because of an abundance of positively charged residues, and the N-terminal contains a number of negatively charged residues. To investigate whether this charge asymmetry could produce preferential orientation during insertion into the bilayer, we compared proteoliposomes created by pR insertion into cationic, anionic, and charge–neutral liposomes (Fig. 1 B). Furthermore, we varied several reconstitution parameters such as pH, presence of low concentration of divalent cations, and lipid tail saturation level to determine whether they influence pR insertion or orientation. We expressed pR in E. coli and purified it via nitrilotriacetic acid affinity chromatography; purified pR exhibited the absorption spectrum maximum at 530 nm and pH-dependent absorbance shift that is characteristic for a protein that is bound to its retinal cofactor and properly folded (see Fig. S1 in the Supporting Material). pR reconstitution produces ca. 200 nm proteoliposomes that display the characteristic pR absorption peak at 530 nm, indicating that structurally intact pR is incorporated into the vesicle’s lipid bilayer.
Figure 1.
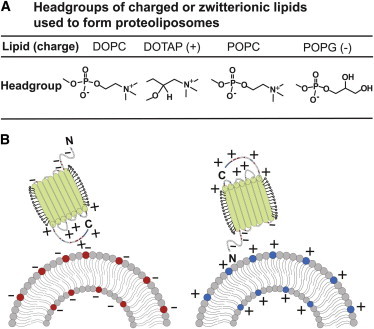
Liposome surfaces and protein orientation during insertion. (A) Headgroups of phospholipids used in this study. (B) Schematic showing an asymmetrically charged membrane protein approaching charged surface of a proteoliposome with electrostatic interactions inducing a preferential protein orientation.
Limited proteolysis
Similar to the approaches of Gerber (25) and Kalmbach (26), we used a proteolytic digestion assay with a nonspecific serine protease, proteinase K (ProtK), to determine the orientation of reconstituted pR in liposomes. When a membrane protein is incorporated into lipid vesicles, the membrane shields the core of the protein and only the hydrophilic residues that form loop regions of a protein remain exposed to the solution. Indeed, a one-dimensional topological model of pR based on hydrophobicity analysis (27) shows that the membrane shields all but the N-terminal, C-terminal, and loop regions; thus, properly folded and embedded pR has only a few sites vulnerable to proteolysis. Critically, these cleavage sites are located asymmetrically in the protein structure; as a result, ProtK-induced cleavage on different sides of the membrane produces a distinct set of protein fragments (Fig. 2 A), which we could then distinguish by SDS-PAGE analysis of the digestion products. pR-containing samples that have not been exposed to ProtK show a single band at ∼24 kDa, independent of whether the protein is in micelles or proteoliposomes prior to SDS-PAGE analysis (Fig. 2 B, lanes 6–9). In contrast, the proteolytic digest results shown in Fig. 2 B reveal distinct patterns depending on the charge state of the lipid in the proteoliposomes.
Figure 2.
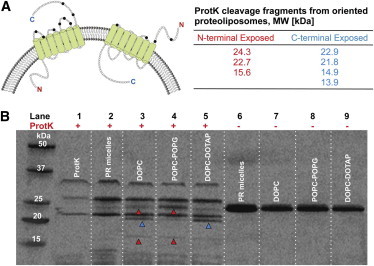
Limited proteolysis with proteinase K (ProtK) shows that pR orientation in proteoliposomes depends on the lipid composition. (A) Expected sizes of proteolytic fragments for ProtK digest of the pR protein when the N-terminal (red numbers) or C-terminal (blue numbers) is exposed to bulk solution. Note that the 24.3 kDa fragment is exclusive to N-exposed pR orientation. The schematic on the top left shows ProtK cleavage sites (black circles). The C-exposed orientation is more vulnerable to cleavage with a greater number of unprotected serine residues. (B) SDS-PAGE gel analysis of the ProtK digestion products. Lane 1 shows bands specific to ProtK enzyme only; lane 2 shows near complete digestion of pR–micelle complexes; lanes 3–5 show digest patterns from 100% zwitterionic, 20% negative, and 20% positive liposomes containing reconstituted pR. Lanes 6–9 show undigested samples corresponding to lanes 2–5, respectively. Red and blue arrows indicate N-exposed and C-exposed fragment sizes, respectively.
ProtK (Fig. 2 B, lane 1) shows three distinct bands at 23, 27, and 34 kDa. This pattern is likely due to aggregation and self-cleavage as ProtK itself has 44 serine residues. We tested pR digestion in the presence of Triton X-100, which should solubilize the protein. Digestion of a pR–micelle complex alone mostly reduces it to fragments smaller than 14 kDa (Fig. 2 B, lane 2). This result suggests that after 2 h of digestion, ProtK most likely cleaves each serine site on the solubilized pR, resulting in smaller sized fragments. The gel also shows some signs of incomplete digestion (∼5% pR remains undigested with some intact protein close to 23–24 kDa), which we can attribute to the stabilizing effect of Triton X-100 detergent monomers on pR native folding (26). This stabilizing effect of Triton X-100 on pR was disrupted by further solubilization in 0.1% SDS solution (Fig. S2). SDS disrupts the protein folding, allowing the protease equal access to all the serine residues, leading to near complete digestion by ProtK (Fig. S2).
We found the digestion products of proteoliposomes (Fig. 2 B, lanes 3–5) to be distinct from the products of pR–micelles digestion. Digestion of zwitterionic DOPC proteoliposomes (Fig. 2 B, lane 3) produced a mixture of digestion products (molecular weights of 24.3 kDa, 21.8 kDa, 15.6 kDa, and smaller fragments around 8 kDa) that correspond to fragments that originated from both C- and N-terminal protein fragments. These results suggest that a charge-neutral surface does not induce significant vectorial pR orientation. Digestion fragments from proteoliposomes made from negatively charged POPC-POPG lipid complex (Fig. 2 B, lane 4) produce the major cleavage product (highest band density) at 24.3 kDa. This size product clearly indicates that ProtK is cutting the protein near the N-terminal. There are also bands present at ∼22 and 15.6 kDa, both also consistent with a predominant N-exposed pR orientation.
Digest fragments of proteoliposomes made with positively charged DOPC-DOTAP lipid complex (Fig. 2 B, lane 5) show a rather different picture. Here we see a strong band at 21 kDa or 22 kDa and a rather weak band at 24.3 kDa; these results strongly indicate a predominantly C-terminal exposed orientation of the pR protein. The molecular weights for the digestion fragments (see table in Fig. 2 A) that were calculated based on protein cleavage sites agree closely with estimated molecular weights based on the Rf values from the gel.
We also used a similar proteolysis assay to investigate the effect of varying solution pH on pR reconstitution and orientation. A previous study by Liang et al. (24) has calculated pH-dependent effective charges associated with pR extramembrane domains at different pH levels. Those calculations show that at pH 5, the N-terminal has an effective net charge of −2 and the C-terminal has an effective net charge of +4; at pH 6.2, the N-terminal charge still remains at −2 and the C-terminal charge is +1.8. Although this pH change does not reverse the charge asymmetry, it does cause a significant shift in charge density. In this case, we expect the proteolysis of proteoliposomes formed in pH 5 or pH 6.2 buffers to produce similar digest fragments, but the incorporation efficiency should be reduced. Indeed, the resultant digest fragments at pH 5 reconstitution (Fig. 3 A) show that DOPC-DOTAP liposomes had a dominant fragment at ∼22 kDa, POPC-POPG liposomes have a dominant fragment at ∼24 kDa, and DOPC liposomes have a mixed population of fragments. This result is still consistent with a C-exposed pR orientation expected for the positive DOPC-DOTAP liposomes and N-exposed orientation for the negative POPC-POPG liposomes. However, densitometry calculations (not shown) reveal that total protein incorporation efficiency in a pH 5 environment is markedly less compared with incorporation at pH 6.2. Previous studies have shown that pR becomes unstable at lower pH values and causes precipitation (28). At pH 9, however, the C-terminal effective charge becomes −3.8 and the N-terminal remains unchanged at −2. In this instance, both terminals are negatively charged, with the C-terminal becoming the more negative of the two protein ends. The cleavage products for pH 9 reconstitution (Fig. 3 B) show a different pattern of fragments from those at pH 5 and 6.2. Here, for the first time, we see a dense 21 kDa band in the POPC-POPG liposomes. In addition, the DOPC-DOTAP sample now shows a marked increase in the density of the 24 kDa band. Also, all three liposome groups show smaller fragments between 13 and 18 kDa. We calculated band densities of the 24 and 21–22 kDa fragments using densitometry (Table 1). A ratio close to 1 suggests no preferential orientation, whereas a ratio > 1 or < 1 is indicative of an N-exposed or C-exposed orientation, respectively. Significantly, DOPC-DOTAP liposomes show predominant C-exposed orientation at pH 6.2 and an N-exposed orientation at pH 9, whereas negatively charged POPC-POPG liposomes show both C- and N-exposed populations at pH 9. These data indicate that by varying the effective protein terminal charge, we are able to tune the fraction of C- or N-exposed liposomal orientation.
Figure 3.
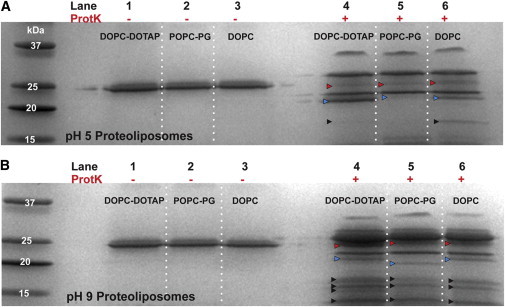
Proteolysis of proteolipsomes in different pH environments shows that pR orientation can be tuned by changing charge density profiles. (A) and (B) SDS-PAGE gel analysis of ProtK digestion products in pH 5 and pH 9. In both gels, lanes 1–3 show undigested samples and lanes 4–6 show digest patterns from 20% positive, 20% negative, and 100% zwitterionic liposomes reconstituted with pR. Red and blue arrows indicate fragments that correlate with N- or C-exposed orientation. Black arrows point to smaller fragments that can be attributed to both orientations.
Table 1.
Ratio of 24 kDa:21–22 kDa fragment density from SDS-PAGE analysis at varying reconstitution pH levels
| pH | Liposome composition (charge) |
||
|---|---|---|---|
| DOPC | POPC-POPG (−) | DOPC-DOTAP (+) | |
| 5 | 1.1 | 1.3 | 0.6 |
| 6.2 | 1.6 | 2.7 | 0.5 |
| 9 | 1.1 | 0.9 | 1.7 |
The lipids that we used for these experiments differ not only by their headgroup charge, but also by the saturation levels of the hydrocarbon chain. To test for the possibility of the hydrocarbon saturation levels influencing the protein insertion orientation, we compared the results of the digest of the proteoliposomes made with POPC-POPG and DOPC-DOPG mixtures, and POPC, and DOPC proteoliposomes under the same proteolysis conditions. Both of these comparisons showed that lipids with the similar headgroup charge but different hydrocarbon chain saturation levels exhibit identical digest patterns (Fig. S3). This result is an indication that protein insertion is directed by the polar headgroup charges and not by lipid tail saturation.
We can also manipulate the bilayer surface charge by adding divalent ions, such as Ca2+. We found that a relatively low concentration of CaCl2, 0.5 mM, can affect protein incorporation yield. Densitometry on the digest fragments produced after the proteolysis experiment showed that the negatively charged liposomes, POPC-PG and DOPC-PG, show decreased incorporation of pR, whereas positively charged liposomes, DOPC and DOTAP, show an increase in pR incorporation, as evidenced by increased the density of the 22 kDa band (Fig. S4). It is likely that Ca2+ binding to negatively charged liposomes reduced the negative surface charge, thereby reducing the driving force for protein incorporation. The origins of the increased binding in positively charged liposomes are harder to decipher, but it is possible that calcium ions bind to the zwitterionic DOPC component of positively charged liposomes and increase surface potential. Note that other factors, such as changes in the conformation and physical properties of membrane lipids caused by calcium ions, could also influence the protein incorporation efficiency (29).
Surface potential measurements
Proteolysis experiments suggest that negatively charged liposomes assume an N-exposed protein orientation, which should further increase the negative charge on the liposome surface because the N-terminal domain has an overall negative charge. In the same manner, positively charged liposomes should promote a C-exposed protein orientation, creating a more positive potential on the liposome surface. Thus, we expect that the overall surface charge will be amplified after the protein insertion. We have tested this hypothesis by quantifying the net charge of the liposomes before and after protein insertion with zeta potential measurements (Fig. 4 A). For anionic POPC:POPG (80:20 mol %) liposomes, the initial zeta potential value was −24.63 ± 4.15 mV; after protein reconstitution, the potential decreased even more to −38.3 ± 16.0 mV. This result is consistent with the notion that an N-exposed protein population is dominant in negatively charged vesicles. For the cationic DOPC:DOTAP (80:20 mol %) liposome sample, the initial potential was at the 24.03 ± 7.13 mV value, which increased to 39.0 ± 7.6 mV after protein incorporation. Again, we attribute this increase to a C-exposed protein orientation being dominant in cationic liposomes. In contrast, the zwitterionic DOPC liposome sample showed only small changes in the surface potential (from −1.58 ± 0.31 mV to −3.95 ± 2.56 mV after pR reconstitution). Overall, samples of zwitterionic DOPC proteoliposomes did not produce a clear trend in zeta potential data. Thus, the analysis of the surface charge measurements validates our conclusion that liposome surface charge can direct the protein orientation during proteoliposome formation.
Figure 4.
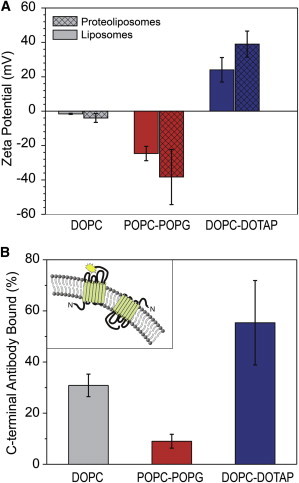
Surface potential and antibody accessibility assays show that pR is predominately N-terminal exposed and C-terminal exposed in negatively charged and positively charged proteoliposomes, respectively. (A) Surface potential values for liposomes (solid bars) and proteoliposomes (patterned bars) composed of different lipids. Charge-induced protein orientation leads to the surface potential increase (decrease) for a positively (negatively) charged membrane. (B) C-terminal epitope accessibility assay of the antibody binding to the HSV epitope tag exposed on the C-terminus of proteorhodopsin. (Inset) Fluorescent label bound to the C-terminus of the protein.
Immunolabeling assay
We can also confirm the preferential protein orientation in the vesicles using an immunolabeling assay. pR amino acid sequence contains an HSV epitope on the C-terminal end of the protein, which can bind an antibody that recognizes this epitope (Fig. 4 B, inset). Because the HSV sequence is proximal to the C-terminal, binding of this antibody could also serve as a convenient way to discriminate between two possible protein orientations. Because the antibody-binding epitope is located on the positively charged end of the molecule, we would expect to see significant binding to positively charged proteoliposomes and reduced binding to the negatively charged proteoliposomes. We exposed proteoliposomes to an excess of fluorescently labeled anti-HSV antibody, isolated the proteoliposome-bound antibodies from those free in solution using size-exclusion chromatography, and then quantified the relative abundance of the antibody from each proteoliposome population using fluorescence measurements. Indeed, the experimental data showed the expected pattern (Fig. 4 B): Positively charged DOPC-DOTAP liposomes showed an almost 6 times higher (52 ± 22%) antibody binding affinity than the negatively charged POPC-POPG vesicles (9 ± 2% binding affinity). Unsurprisingly, uncharged liposomes made from pure DOPC lipid showed the affinity value (31 ± 4%) that fell in between the affinity values obtained for the positively and negatively charged proteoliposomes. We have also verified this analysis by rupturing the proteoliposomes with detergent to reduce scatter and calculating pR and FITC-α-HSV antibody concentration based on the absorbance peaks of this solution (Fig. S5). The conclusions from the absorbance data analysis match the conclusions that we drew from the fluorescence analysis data.
Vectorial proton pumping in proteoliposomes
It is also important to confirm that protein orientation preference during insertion also defines the direction of the proton transport in the proteoliposomes. Green light excitation of the pR reconstituted in phospholipid vesicles induces pH gradient across the lipid membrane with the sign of the pH change indicative of the predominant protein orientation in the vesicles. To assess the proton pumping direction of the protein, we produced proteoliposomes that contain 5(6)CF dye in their lumen. 5(6)CF is a pH-sensitive dye with a low membrane permeability coefficient (30), so we can use it to monitor pH change on the light-induced pR proton pumping. For this particular dye, an increase or decrease in pH causes an increase or decrease in fluorescence emission, respectively. For the orientation of pR protein in our proteoliposomes, we expected the POPC-POPG proteoliposomes to show the increase in the lumen pH on green light excitation and DOPC-DOTAP proteoliposomes to show the opposite trend.
Indeed, exposure of the POPC-POPG proteoliposomes to green light illumination showed a steady increase in 5(6)CF fluorescence emission with each additional light exposure with a change of ∼1.2 pH units after 200 s of illumination (Fig. 5), indicating that vesicle lumen pH was increasing and pR was pumping protons outward. DOPC-DOTAP proteoliposomes showed the opposite trend of steadily decreasing fluorescence with a pH change of ca. 0.85 pH units after a similar illumination period, indicating in that case a net inward pumping of protons. To confirm that these changes in fluorescence result from pR pumping activity, we carried out these experiments on liposomes that do not contain the protein and observed pH change of less than 0.15 pH units. These experiments confirm vectorial proton transport and preferential pR orientation in liposomes of varying charge states.
Figure 5.
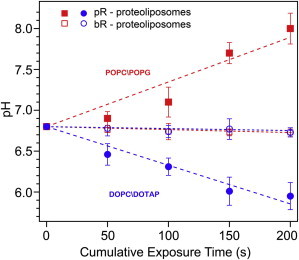
The direction of vectorial proton pumping is dictated by protein orientation in proteoliposomes. Change in pH within the proteoliposomes calculated from changes in fluorescence intensity of the pH-sensitive dye 5(6)CF encapsulated in negatively (POPC-POPG, red lines) and positively (DOPC-DOTAP, blue lines) charged proteoliposomes on a series of successive 50-s 530 nm light exposures. Closed symbols, pR proteoliposomes; open symbols, bacteriorhodopsin proteoliposomes.
As another control, we assayed another photoactivated proton pump, bacteriorhodopsin. Although, bacteriorhodopsin bears ∼24% sequence identity to pR, the crucial difference is that it does not have asymmetrically charged N- and C-terminals in its native form. After following similar procedures for bacteriorhodopsin reconstitution into charged liposomes encapsulating pH-sensitive dye, we measured proton pumping activity (Fig. 5). Significantly, we saw only minimal changes in dye fluorescence (< 2%), which translated to a pH changes less than 0.05 pH units, and there was no clear trend in either positive or negatively charged proteoliposome sample. This observation suggests that the lack of charge asymmetry in the protein led to an overall randomized orientation, which did not allow proton build-up or depletion on either side of the liposome membrane.
Finally, we want to discuss the implications of our findings for a mechanism of protein insertion into the liposomes. It is plausible to assume that initially a detergent-solubilized protein forms a weak complex with the surface of the lipid bilayer in which the orientation of the protein is guided by the interaction of the protein and bilayer surface charges. Significantly, this orientation remains conserved during the subsequent step of detergent removal. Our results indicate that electrostatic attraction between oppositely charged protein regions in pR and liposome surface not only plays an important role in guiding the protein to the bilayer surface, but also helps to insert the protein into the bilayer membrane. As the result, the opposite end of the protein (bearing the same charge as the bilayer in case of pR) is displayed on the outer bilayer surface. pR has several loop structures and C-/N-terminals on opposite ends of the protein, which lends itself to multiple types of assays as we have demonstrated in this work. pR also does not have prominent extramembrane structural features such a large hydrophilic domain as seen in ATPase (31) or α-hemolysin (32), or a conical shape factor as seen in KcsA protein (33). Such structural attributes can help to guide protein insertion into membranes in an oriented manner without the need for a strong external driving force. The overall rectangular shape of pR does not naturally lend itself to a preferential orientation in an ex vivo environment and could enhance the role that membrane charge plays in pR reconstitution. Interestingly, while this article was in review, Vitrac et al. (34) reported that membrane charge also plays a defining role in controlling orientation and membrane topology on another protein, lactose permease. Potentially, even proteins without charge asymmetry can be modified to include charged residues at either terminal end to become suitable for this type of recruitment method.
Conclusion
In this work, we investigated the role of the lipid bilayer surface charge on the recruitment and orientation of proteorhodopsin in a detergent-mediated reconstitution procedure. Several different assay results give strong and consistent evidence of preferential orientation, and a functional assay confirms that such preferential insertion can induce vectorial proton transport. This method of achieving oriented reconstitution can be useful for a number of applications that require control of direction of membrane protein functionality, both in vesicular systems and in supported lipid bilayer systems. These results also could help researchers understand the fundamentals of the membrane protein insertion into lipid bilayer membrane in in vivo and in vitro systems. However, future studies of reconstitution procedures with other large-membrane proteins with asymmetrically charged terminal ends and two hydrophilic domains separated by a membrane-spanning hydrophobic domain are likely to result in the formulation of a general set of principles that would serve as a guide for oriented reconstitution.
Acknowledgments
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering. Parts of this work were performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. R.T. acknowledges support from the LSP program at Lawrence Livermore National Laboratory.
Supporting Material
References
- 1.Eytan G.D. Use of liposomes for reconstitution of biological functions. Biochim. Biophys. Acta. 1982;694:185–202. doi: 10.1016/0304-4157(82)90024-7. [DOI] [PubMed] [Google Scholar]
- 2.Fishman P.H., Moss J., Alving C.R. Liposomes as model membranes for ligand-receptor interactions: studies with choleragen and glycolipids. Biochemistry. 1979;18:2562–2567. doi: 10.1021/bi00579a020. [DOI] [PubMed] [Google Scholar]
- 3.Aloj S.M., Kohn L.D., Meldolesi M.F. The binding of thyrotropin to liposomes containing gangliosides. Biochem. Biophys. Res. Commun. 1977;74:1053–1059. doi: 10.1016/0006-291x(77)91624-2. [DOI] [PubMed] [Google Scholar]
- 4.Volsky D.J., Cabantchik Z.I., Loyter A. Implantation of the isolated human erythrocyte anion channel into plasma membranes of Friend erythroleukemic cells by use of Sendai virus envelopes. Proc. Natl. Acad. Sci. USA. 1979;76:5440–5444. doi: 10.1073/pnas.76.11.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castellana E.T., Cremer P.S. Solid supported lipid bilayers: from biophysical studies to sensor design. Surf. Sci. Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plant A.L., Brigham-Burke M., O’Shannessy D.J. Phospholipid/alkanethiol bilayers for cell-surface receptor studies by surface plasmon resonance. Anal. Biochem. 1995;226:342–348. doi: 10.1006/abio.1995.1234. [DOI] [PubMed] [Google Scholar]
- 7.Yang T., Baryshnikova O.K., Cremer P.S. Investigations of bivalent antibody binding on fluid-supported phospholipid membranes: the effect of hapten density. J. Am. Chem. Soc. 2003;125:4779–4784. doi: 10.1021/ja029469f. [DOI] [PubMed] [Google Scholar]
- 8.Yang T., Jung S., Cremer P.S. Fabrication of phospholipid bilayer-coated microchannels for on-chip immunoassays. Anal. Chem. 2001;73:165–169. doi: 10.1021/ac000997o. [DOI] [PubMed] [Google Scholar]
- 9.Huang S.-C.J., Artyukhin A.B., Noy A. Carbon nanotube transistor controlled by a biological ion pump gate. Nano Lett. 2010;10:1812–1816. doi: 10.1021/nl100499x. [DOI] [PubMed] [Google Scholar]
- 10.Hamai C., Yang T., Musser S.M. Effect of average phospholipid curvature on supported bilayer formation on glass by vesicle fusion. Biophys. J. 2006;90:1241–1248. doi: 10.1529/biophysj.105.069435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fung B.K., Hubbell W.L. Organization of rhodopsin in photoreceptor membranes. 1. Proteolysis of bovine rhodopsin in native membranes and the distribution of sulfhydryl groups in the fragments. Biochemistry. 1978;17:4396–4402. doi: 10.1021/bi00614a007. [DOI] [PubMed] [Google Scholar]
- 12.Cuello L.G., Romero J.G., Perozo E. pH-dependent gating in the Streptomyces lividans K+ channel. Biochemistry. 1998;37:3229–3236. doi: 10.1021/bi972997x. [DOI] [PubMed] [Google Scholar]
- 13.Ataka K., Giess F., Heberle J. Oriented attachment and membrane reconstitution of His-tagged cytochrome c oxidase to a gold electrode: in situ monitoring by surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 2004;126:16199–16206. doi: 10.1021/ja045951h. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y.C., Rieben N., Martinez K.L. Specific and reversible immobilization of histidine-tagged proteins on functionalized silicon nanowires. Nanotechnology. 2010;21:245105. doi: 10.1088/0957-4484/21/24/245105. [DOI] [PubMed] [Google Scholar]
- 15.Pfleger N., Wörner A.C., Glaubitz C. Solid-state NMR and functional studies on proteorhodopsin. Biochim. Biophys. Acta. 2009;1787:697–705. doi: 10.1016/j.bbabio.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 16.Yanagisawa M., Iwamoto M., Oiki S. Oriented reconstitution of a membrane protein in a giant unilamellar vesicle: experimental verification with the potassium channel KcsA. J. Am. Chem. Soc. 2011;133:11774–11779. doi: 10.1021/ja2040859. [DOI] [PubMed] [Google Scholar]
- 17.Drachev A.L., Drachev L.A., Khitrina L.V. The action of lanthanum ions and formaldehyde on the proton-pumping function of bacteriorhodopsin. Eur. J. Biochem. 1984;138:349–356. doi: 10.1111/j.1432-1033.1984.tb07922.x. [DOI] [PubMed] [Google Scholar]
- 18.Friedrich T., Geibel S., Bamberg E. Proteorhodopsin is a light-driven proton pump with variable vectoriality. J. Mol. Biol. 2002;321:821–838. doi: 10.1016/s0022-2836(02)00696-4. [DOI] [PubMed] [Google Scholar]
- 19.Lenz M.O., Huber R., Wachtveitl J. First steps of retinal photoisomerization in proteorhodopsin. Biophys. J. 2006;91:255–262. doi: 10.1529/biophysj.105.074690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rigaud J.L., Paternostre M.T., Bluzat A. Mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. 2. Incorporation of the light-driven proton pump bacteriorhodopsin. Biochemistry. 1988;27:2677–2688. doi: 10.1021/bi00408a007. [DOI] [PubMed] [Google Scholar]
- 21.Holloway P.W. A simple procedure for removal of Triton X-100 from protein samples. Anal. Biochem. 1973;53:304–308. doi: 10.1016/0003-2697(73)90436-3. [DOI] [PubMed] [Google Scholar]
- 22.Talasaz A.H., Nemat-Gorgani M., Davis R.W. Prediction of protein orientation upon immobilization on biological and nonbiological surfaces. Proc. Natl. Acad. Sci. USA. 2006;103:14773–14778. doi: 10.1073/pnas.0605841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakatos M., Lanyi J.K., Váró G. The photochemical reaction cycle of proteorhodopsin at low pH. Biophys. J. 2003;84:3252–3256. doi: 10.1016/S0006-3495(03)70049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang H., Whited G., Stucky G.D. The directed cooperative assembly of proteorhodopsin into 2D and 3D polarized arrays. Proc. Natl. Acad. Sci. USA. 2007;104:8212–8217. doi: 10.1073/pnas.0702336104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerber G.E., Gray C.P., Khorana H.G. Orientation of bacteriorhodopsin in Halobacterium halobium as studied by selective proteolysis. Proc. Natl. Acad. Sci. USA. 1977;74:5426–5430. doi: 10.1073/pnas.74.12.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalmbach R., Chizhov I., Engelhard M. Functional cell-free synthesis of a seven helix membrane protein: in situ insertion of bacteriorhodopsin into liposomes. J. Mol. Biol. 2007;371:639–648. doi: 10.1016/j.jmb.2007.05.087. [DOI] [PubMed] [Google Scholar]
- 27.Béjà O., Aravind L., DeLong E.F. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- 28.Bergo V.B., Sineshchekov O.A., Spudich J.L. His-75 in proteorhodopsin, a novel component in light-driven proton translocation by primary pumps. J. Biol. Chem. 2009;284:2836–2843. doi: 10.1074/jbc.M803792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Macdonald P.M., Seelig J. Calcium binding to mixed phosphatidylglycerol-phosphatidylcholine bilayers as studied by deuterium nuclear magnetic resonance. Biochemistry. 1987;26:1231–1240. doi: 10.1021/bi00379a005. [DOI] [PubMed] [Google Scholar]
- 30.Komatsu H., Chong P.L. Low permeability of liposomal membranes composed of bipolar tetraether lipids from thermoacidophilic archaebacterium Sulfolobus acidocaldarius. Biochemistry. 1998;37:107–115. doi: 10.1021/bi972163e. [DOI] [PubMed] [Google Scholar]
- 31.Walker J.E., Saraste M., Gay N.J. The unc operon. Nucleotide sequence, regulation and structure of ATP-synthase. Biochim. Biophys. Acta. 1984;768:164–200. doi: 10.1016/0304-4173(84)90003-x. [DOI] [PubMed] [Google Scholar]
- 32.Song L., Hobaugh M.R., Gouaux J.E. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 33.Hirano M., Onishi Y., Ide T. Role of the KcsA channel cytoplasmic domain in pH-dependent gating. Biophys. J. 2011;101:2157–2162. doi: 10.1016/j.bpj.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitrac H., Bogdanov M., Dowhan W. In vitro reconstitution of lipid-dependent dual topology and postassembly topological switching of a membrane protein. Proc. Natl. Acad. Sci. USA. 2013;110:9338–9343. doi: 10.1073/pnas.1304375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.