

Abstract
HIV-1 cell entry is initiated by the interaction of the viral envelope glycoprotein gp120 with CD4, and chemokine coreceptors CXCR4 and CCR5. The molecular recognition of CXCR4 or CCR5 by the HIV-1 gp120 is mediated through the V3 loop, a fragment of gp120. The binding of the V3 loop to CXCR4 or CCR5 determines the cell tropism of HIV-1 and constitutes a key step before HIV-1 cell entry. Thus, elucidating the molecular recognition of CXCR4 by the V3 loop is important for understanding HIV-1 viral infectivity and tropism, and for the design of HIV-1 inhibitors. We employed a comprehensive set of computational tools, predominantly based on free energy calculations and molecular-dynamics simulations, to investigate the molecular recognition of CXCR4 by a dual tropic V3 loop. We report what is, to our knowledge, the first HIV-1 gp120 V3 loop:CXCR4 complex structure. The computationally derived structure reveals an abundance of polar and nonpolar intermolecular interactions contributing to the HIV-1 gp120:CXCR4 binding. Our results are in remarkable agreement with previous experimental findings. Therefore, this work sheds light on the functional role of HIV-1 gp120 V3 loop and CXCR4 residues associated with HIV-1 coreceptor activity.
Introduction
The primary step of human immunodeficiency virus type 1 (HIV-1) cell entry is the interaction of the viral envelope glycoprotein (comprising subunits gp41 and gp120) with the host leukocyte glycoprotein receptor, CD4, and the two chemokine receptors CXCR4/CCR5 on the surface of the host cells (1–5). Specifically, the glycoprotein gp120 interaction with CD4 triggers conformational changes in gp120 that increase the exposure of the third variable region (V3) loop. Subsequently, the protein gp120, via its V3 loop, binds to chemokine receptors CXCR4 (infecting mostly T-cells) or CCR5 (infecting mostly macrophages) (6–11). The molecular recognition of chemokine receptors by the V3 loop results in a series of rearrangements in the envelope glycoprotein, leading to the fusion of the virus and the cell membranes (12).
At the beginning of the 1990s, the V3 loop was identified as the primary determinant of cell tropism in HIV-1 (13). Since the discovery of the key role of V3 loop in HIV-1 infection, with regard to the binding to chemokine receptors CXCR4 and CCR5 (6,14,15) and the determination of cell-tropism (13), recognizing CXCR4 or CCR5 or both (referred to as “dual tropic”), several experimental studies aimed at elucidating the key interacting residues of chemokine receptors involved in the V3 loop binding through the mapping of the chemokine receptors’ binding sites (16–26). These studies employed site-directed mutagenesis or chimeric substitutions, and identified specific residues or residue moieties of the chemokine receptors that are critical to, or correlate with, viral infection.
The HIV-1 gp120 V3 loop is sustained in a loop conformation through a disulfide bridge between its N- and C-terminal ends, is encountered in a large sequence variability, is positively charged, and is predominantly composed of 35 residues (27–29). Owing to its highly dynamic character (27,29,30), the V3 loop is absent in the majority of gp120 crystallographic structures; nevertheless, it was resolved in two crystallographic Protein Data Bank (PDB) entries (4,5). Numerous studies aimed at understanding the physicochemical properties of the V3 loop and elucidating its viral tropism (5,11,19,26,31–34). It has been suggested that charge complementarity and electrostatic interactions among the N-terminal, extracellular loop 2 (ECL2) coreceptor domains, and the V3 loop (5,11,19,26,31–33), are associated with the viral tropism. Furthermore, it has been proposed that the interchange from coreceptor CCR5 to CXCR4, as the disease progresses, is linked to 1), The increase of the net charge of the V3 loop (10,31); 2), The presence of positively charged residues at one or more of positions 11, 24, and 25, known as the 11/24/25 rule (9); and 3), The absence of the glycosylation motif N6X7T8|S8X9 (where X = Pro) (8).
Recently, molecular dynamics (MD) simulations showed that V3 loops undergo common correlated motions, in association with specific charged interactions between residues on opposite stems (27). Understanding the unbound properties of gp120 domains is important for delineating the mechanism of conformational changes from unbound to bound structures, related to gp120:CD4 binding (35,36). Similarly, the identification of unbound V3 loop conformations associated with electrostatic-driven correlated motions (27) could prove significant for the elucidation of the gp120 (V3 loop):CXCR4 binding.
Despite the numerous studies related to the V3 loop and the chemokine receptors, the basic biological knowledge on the specific interactions between the V3 loop and the chemokine receptors is limited due to the absence of a complete V3 loop:coreceptor complex structure (34). This could be associated with the high flexibility of the V3 loop leading to absence of electron density in the gp120 crystal structures, as in Liao et al. (37).
A comprehensive attempt to computationally derive a V3 loop:CXCR4 complex structure to enlighten the role of the key interacting V3 loop and CXCR4 residues has never before been reported, according to our knowledge. In this study, we exploit both the CXCR4 crystallographic structure (11) and one of the V3 loop crystallographic structures (5) to theoretically derive what is, to our knowledge, the first V3 loop:CXCR4 complex structure using a combination of primarily binding/interaction free-energy calculations and MD simulations. The computational protocol applied was not biased by any experimental evidence regarding the key interacting residues, and interestingly, our results are in remarkable agreement with previous experimental findings (see Table 1; marked in boldface are CXCR4 residues reported in experimental findings) (16–21,23–25). Thus, the reported V3 loop:CXCR4 complex structure sheds light on the functional role of V3 loop and CXCR4 residues, which are experimentally determined as critical for the HIV-1 coreceptor activity.
Table 1.
Important intermolecular polar and nonpolar interaction free energies, hydrogen bonds, and salt bridges, between V3 loop and CXCR4 residue pairs within the MD simulation of the complex with the lowest average binding free energy
| V3 loop residueb | CXCR4 residues (polar, nonpolar interaction free energies)c | Salt bridgesd | Hydrogen bondse |
|---|---|---|---|
| Arg3 |
Asp20 (−0.0, −1.6)e Tys21 (1.1, −2,4) Asp22 (−9.3, −2.5) Glu268 (−29.3, 2.6) |
Arg3:Asp22 Arg3:Glu268 |
|
| Asn5 |
Asp20 (−2.9, −1.9) Tys21 (−1.1, −1.5) Ser23 (−2.2, −1.3) |
Asn5 ND2:Asp20 OD∗ Asn5 ND2:Tys21 O |
|
| Asn6 | Ser23 (−3.3, −2.3) | Asn6 N:Ser23 OG Asn6 ND2:Ser23 OG Asn6 OD1:Ser23 OG |
|
| Asn7 | Asn11 (−0.1, −0.8) Glu14 (−1.9, −0.9) |
Asn7 OD1:Asn11 ND2 Asn7 ND2:Glu14 OE∗ |
|
| Thr8 |
Tys21 (0.3, −2.9) Asp22 (−3.1, −0.7) Lys25 (0.5, −2.1) |
Thr8 OG1:Asp22 O | |
| Arg9 | Met1 (0.0, −1.9) Tys7 (−7.3, −1.2) Ser9 (0.2, −1.6) Asp10 (−2.8, −1.0) Tys12 (−4.4, −5.0) |
Arg9 NH∗:Tys7 O Arg9 NH∗:Asp10 O |
|
| Lys10 |
Tys12 (−9.5, −3.0) Asp20 (−8.5, −1.0) Tys21 (−1.0, −3.4) Asp193 (−17.8, −0.9) Leu266 (−6.7, −2.3) |
Lys10:Asp193 | Lys10 N:Tys12 SO∗ Lys10 NZ:Asp20 O Lys10 NZ:Leu266 O |
| Arg11 | Met1 (−7.9, −2.1) Glu2 (−0.9, −1.9) Gly3 (−1.2, −1.7) Tys7 (−1.0, −4.5) Tys12 (−2.3, −6.3) Phe189 (0.1, −1.7) |
Arg11 NH∗/NE:Met1 O Arg11 NH∗:Glu2O Arg11 N:Tys12 SO∗ |
|
| Val12 |
Phe189 (0.1, −2.3) Tyr190 (−0.0, −3.2) |
||
| Ser13 | Met1 (−5.8, −1.9) | Ser13 OG:Met1 N | |
| Leu14 |
Glu2 (−1.2, −1.4) Gly3 (−0.2, −1.7) Ser178 (−0.1, −1.7) Ala180 (−0.0, −2.4) Arg183 (−1.6, −1.0) Ile185 (0.1, −2.5) Asp187 (−3.8, −3.4) Phe189 (0.1, −1.6) |
Leu14 N:Glu2O Leu14 O:Arg183 NH∗ |
|
| Gly15 | Ile185 (−0.1, −1.7) Asp187 (−6.3, 0.1) |
Gly15 N:Asp187 OD∗ | |
| Pro16 | Arg30 (−0.3, −2.0) Asn37 (−0.1, −2.2) |
Pro16 N:Asn37 ND2 | |
| Gly17 |
Tyr45 (−0.8, −0.6) Trp94 (−1.2, −1.7) Asp97 (−2.2, −0.1) Glu288 (−2.8, −0.1) |
Gly17 N:Tyr45 OH Gly17 N:Asp97 OD∗ |
|
| Arg18 | Trp94 (−0.1, −2.0) His113 (−3.2, −1.3) Tyr116 (5.1, −2.5) Thr117 (−2.1, −1.0) Asp171 (−37.2, −0.3) Asp187 (−1.1, −1.4) Arg188 (8.27, −3.1) His203 (−18.1, 0.4) Tyr255 (2.8, −0.5) Glu288 (−65.6, 2.6) |
Arg18:Asp171 Arg18:Glu288 |
Arg18 NH∗:Thr117 OG1 Arg18 O:Arg188 NH∗/NE Arg18 NH∗:His203 NE2 Arg18 NH∗:Tyr255 OH |
| Val19 | Glu288 (−2.7,−1.0) | ||
| Trp20 |
Arg188 (−4.8, −3.2) Tyr190 (−1.5, −1.7) Val196 (0.1, −2.2) Phe199 (−0.0, −1.6) Gln200 (−1.5, −3.4) His281 (−0.9, −0.9) |
Trp20 NE1:Arg188 NH∗ Trp20 NE1:Tyr190 OH Trp20 O:His281 ND1 |
|
| Tyr21 | Arg30 (0.2, −3.3) Glu277 (−0.2, −2.0) Asn278 (−1.2, −2.8) His281 (0.2, −4.3) |
Tyr21 OH:Asn278 ND2 | |
| Thr22 | Leu266 (−0.0, 1.8) Glu277 (−2.8, −0.8) |
Thr22 OG1:Glu277 OE∗ | |
| Thr23 | Met1 (−0.0, −2.1) Cys28 (−0.6, −1.8) |
Thr23 OG1:Cys28 O | |
| Gly24 |
Lys25 (−0.4, −1.8) Cys28 (−3.2, −2.1) |
Gly24 N:Cys28 O Gly24 O:Cys28 N |
|
| Gln25 | Met1 (0.1, −1.6) Pro27 (0.0, −2.7) Cys28 (−0.4, −1.5) |
Gln25 NE2:Cys28 O | |
| Ile26 | Lys25 (−0.0, −1.7) | ||
| Val27 | Ser9 (0.1, −1.7) Asp10 (−0.1, −1.7) Asn11 (−0.0, −1.9) |
||
| Arg31 |
Tys12 (−1.8, −1.2) Glu14 (−16.8, −0.5) Ser18 (0.2, −3.0) Asp20 (−8.4, −1.4) |
Arg31:Glu14 Arg31:Asp20 |
Arg31 NH∗:Ser18 OG |
| Lys32 | Gly17 (−3.4, −0.8) Ser18 (−3.1, −0.6) Asp20 (16.4, 0.4) |
Lys32:Asp20 | Lys32 NZ:Gly17 O Lys32 NZ:Ser18 O |
| His34 | Asp20 (−1.0, −1.5) | His34 ND1:Asp20 OD2 |
CXCR4 residues marked in boldface are experimentally associated with HIV-1 coreceptor activity (see Discussion). The results presented correspond to analysis of 1000 snapshots. The asterisk (∗) symbol used after any V3 loop/CXCR4 atom in the hydrogen bonding pair denotes that any of the atoms in the charged, carboxyl or amide, side-chain group can participate in the hydrogen-bond formation.
aPrincipal interacting V3 loop.
Principal interacting CXCR4 residue pairs.
For each pair, the average polar and nonpolar average interaction free energies (polar, nonpolar), are provided in parentheses next to each CXCR4 residue; all energies are in kcal/mol.
Salt bridges between V3 loop and CXCR4 residue pairs.
Hydrogen bonds between V3 loop and CXCR4 atom pairs.
Methods
Modeling, free energy calculations, and molecular dynamics simulations
The methodology used in this study to derive the V3 loop:CXCR4 complex structure consists of the seven following principal steps.
Step 1: Modeling and selection of the initial V3 loop and CXCR4 structural conformations
V3 loop. The initial backbone structural conformation of the V3 loop corresponds to PDB:2QAD, one of the two intact crystal structures of gp120 in the Protein Data Bank (PDB) (5). V3 loop residues 296–331 of PDB:2QAD were renumbered, starting from 1 and ending at 35. The gp120 residues outside the V3 loop were not considered for investigation in this study because the V3 loop region is the principal determinant of chemokine receptor specificity (2), and the presence of a disulfide bridge between the N- and C-terminal residues of V3 loop base constitutes a physical constraint for the preservation of the structure in the base region. In addition, according to a recent MD study, the V3 loop does not have any strong concerted motion with other parts of the gp120 protein but only within residues of the V3 loop (29). To identify a representative dual-tropic CXCR4/CCR5 V3 loop to examine in this study, we utilized the CD-HIT (38) to perform a sequence-based clustering on the dual tropic V3 loops, which was deposited in the Los Alamos National Laboratory (Los Alamos, NM) database (http://www.hiv.lanl.gov). According to the results, we identified the following sequence of subtype B as the centroid of the most populated dual tropic cluster (extracted from a Chinese patient),
which obeys the 11/24/25 rule (8). In what follows, we use this particular sequence to derive the V3 loop:CXCR4 structure.
CXCR4. The initial structural templates used to construct the CXCR4 conformation correspond to PDB:3OE0 (11) and PDB:2K05 (39) (human fragment containing the missing N-terminal residues 1–24 of PDB:3OE0 (11)); the two fragments were combined using pairwise alignment on their overlapping regions. The FREAD loop modeling algorithm, was applied to model the missing loops (40), and the I-TASSER server was used to model the missing C-terminal residues (41).
Step 2: Production of flexible templates for V3 loop and CXCR4 using MD simulations
V3 loop. We performed replica exchange MD simulations of the V3 loop so as to enhance its conformational sampling (42–45). Solvent effects were taken into account by the FACTS19 implicit solvent model (46). The implementation of implicit solvent models (47), including the particular one used in this study, FACTS (46), in conjunction with the replica exchange MD scheme, successfully balances fast exploration of the conformational space with accuracy, to reproduce results from explicit solvent MD simulations (48,49). The simulations were conducted with the molecular mechanics program CHARMM, Ver. c35b6 (50). The average root mean-square deviation (RMSD) of the conformations produced at 300 K with regard to the starting conformation upon minimization is 5.1 ± 1.3 Å. The result demonstrates that this dual tropic V3 loop encounters a high flexibility in the unbound simulations, and the computational protocol used has successfully sampled the conformational space in order to produce representative flexible templates for docking. Additional information regarding the simulation protocol and parameterization is provided in the Supporting Material.
CXCR4. We employed MD simulations to produce multiple flexible templates for the human CXCR4 protein receptors and to refine the modeled regions of CXCR4. Because the goal was not only to refine the structure but also to produce flexible templates that could constitute docking receptors, we considered that a preliminary docking of V3 loop on CXCR4 would be beneficial for the subsequent docking procedure. Thus, we used the software CLUSPRO 2.0 (51) to primarily dock a V3 loop, corresponding to the PDB:2QAD conformation (5), on the modeled CXCR4, and we produced three conformations with different poses of the V3 loop proximal to the experimentally defined binding site. For each of the three complex structures yielded, we performed two independent MD simulations to produce flexible template structures for CXCR4. Within the MD simulations, the system was immersed in an implicit membrane represented by the switching-function generalized Born (GBSW) module (52,53). The membrane thickness Tmemb was set to 36 Å with one-half of the membrane switching length equal to 2.5 Å; the choice of these values complies with the approximate intramembrane helical length of CXCR4 (11), and was shown to provide optimal protein stability compared to test simulations with larger or smaller. The surface tension coefficient was set to 0.03 kcal/(mol∗Å2); all other parameters were set to default values (54). The simulations were conducted with the molecular mechanics program CHARMM, Ver. c35b6 (50). Additional information regarding the simulation protocol and parameterization is provided in the Supporting Material.
Step 3: Docking of selected V3 loop structures on selected CXCR4 structures
We clustered the structures produced in the V3 loop replica exchange MD simulations as well as the CXCR4 structures produced from the six independent aforesaid MD simulations using the quality clustering method of the command line utility WORDOM (55). We extracted the 20 most populated clusters for the V3 loop, and 17 clusters for CXCR4, including the initially modeled CXCR4 structure. Additional information is provided in the Supporting Material.
Subsequently, we used the parallel LINUX version of the ZDOCK Ver. 3.0.2 (56) to dock the 20 V3 loop clustered structures on the 17 CXCR4 clustered structures. For each run of ZDOCK, 2000 docked structures were produced with a dense rotational sampling and a masking applied on the region with protein coordinates z < 0 Å, so as to exclude the nonpotential binding region from the docking calculations. As a result, 680,000 complex structures were produced from docking.
Step 4: First round of energy minimization and binding free energy calculation of the docked complexes using the membrane-GBSA approximation
All 680,000 complexes were subjected to 100 steps of steepest-descent minimization to alleviate bad contacts and, the binding free energy was evaluated subsequently for all complexes using the generalized Born (GB) solvent-accessible (SA) approximation in a heterogeneous water-membrane-water environment, modeled by GBSW (53). The binding free energy is evaluated via the expression
where EX is the total (free) energy of molecule X (complex PL:CXCR4:V3 loop, free protein P:CXCR4, or free ligand L:V3 loop) as in the literature (57–59). The protein and ligand conformations were assumed identical in the complex and in their free (unbound) states as in the literature (60–64). With this assumption, any bonded-energy contributions to ΔG are canceled in the equation. The solvation free energy components of the complex and unbound protein were computed in the heterogeneous membrane/water environment, while the solvation free energy component of the unbound ligand was computed in a homogeneous aqueous environment, also modeled by the implicit GBSW model (52,53).
Step 5: Second round of energy minimization and binding free energy calculations of the docked complexes using the membrane-PBSA approximation
Out of the 680,000 complexes, we selected the 9000 V3 loop:CXCR4 complexes with the lowest GBSA binding free energy, and subsequently, we performed an additional round of 100 steps steepest-descent minimization and calculated the binding free energy using the Poisson-Boltzmann (PB) SA approximation (57). At the end of this procedure, we identified the complex structure with the lowest binding free energy −144.2 kcal/mol, and additionally, we selected all the complex structures within a 10-kcal/mol range of the lowest binding free energy (−144.2:−134.1 kcal/mol) for subsequent investigation. As a result, the total number of complex structures selected for subsequent investigation was 17. Table S1 in the Supporting Material presents the binding free energies of the 17 different complex structures produced in Step 5. Additional information regarding the PB calculations and the results is provided in the Supporting Material.
Step 6: MD simulations of the docked complexes acquiring the lowest binding free energy
We performed 17 independent MD simulations of the complexes with the lowest PBSA binding free energies, as identified in the previous step. The MD simulations comprised a 400-ps heating procedure and an additional 700-ps equilibration procedure at which the harmonic restraints were gradually removed from the protein and the peptide. No restraints were imposed during the production run at 300 K, the duration of which was equal to 20 ns for every individual complex. The simulation methodology and force-field parameterization used was identical to Step 2, at which point we also performed MD simulations in implicit membrane to produce flexible templates for CXCR4.
Step 7: Binding free energy calculations of the complex structures produced in the MD simulations to identify the complex structure with the lowest average binding free energy
We extracted 1000 snapshots, corresponding to 20-ps intervals, from the 17 MD 20-ns simulations and reevaluated the binding free energy by employing the MM PBSA approximation (57) (using the parameters presented in Step 5). Interestingly, the MM PBSA calculation of the average binding free energies for each complex expanded the free energy range from [−144.2:−134.1 kcal/mol] in step 5 to [−367.2:−275.2 kcal/mol] in Step 7 (see Table S1). The complex with the lowest average binding free energy in this step, according to both MM PBSA and supplementary MM GBSA calculations (58,59,60–64), corresponds to the complex structure of Step 5, which also possesses the lowest binding free energy (−144.2 kcal/mol; see Table S1). The change in the binding free energy ranking among Steps 5 and 7, as well as the improvement of interactions leading to lower binding free energies in Step 7 compared to Step 5, emphasize the key role of conducting MD simulations a posteriori to docking.
In what follows, we present the methodology used to investigate pairwise residue interaction free energies of the complex structure possessing the lowest average binding free energy.
Analysis of interaction free energies of V3 loop:V3 loop and V3 loop:CXCR4 residue pairs
The interaction free energies between two residues (R and R′) were computed by the relation
| (1) |
The first and second group of terms on the right-hand side of Eq. 1 describe, respectively, polar and nonpolar interactions between R and R′. For the investigation of V3 loop:CXCR4 intermolecular interactions, R corresponds to a V3 loop residue and R′ to a CXCR4 residue. For the investigation of V3 loop intramolecular interactions, both R and R′ correspond to V3 loop residues. To compute the EijGB term in Eq. 1, we included all protein (CXCR4) and ligand (V3 loop) atoms and set the charges of atoms outside the two residues R and R′ under investigation to zero. The generalized-Born energies and the atomic accessible-surface areas (ΔSi) entering in Eq. 1 depend on the location of R and R′ in the complex. The polar component contains a Coulombic term and a GB contribution, modeling the interaction between group R and the solvent polarization potential induced by R′. Similarly, the nonpolar component contains a van der Waals interaction between R, R′ and a surface term, expressing cavity contributions and nonpolar interactions with the surrounding solvent. The last term contains the difference in solvent-accessible surface areas of residues R and R′ in the complex and unbound states (62–64). The analysis was performed using all 1000 snapshots from Complex 1. The nonpolar and polar solvation terms were calculated using the heterogeneous membrane-water GBSW using the same parameters of step 4. The sum of the two components reflects the total direct interaction between R and R′ in the solvated complex (62,63,64).
We decomposed the polar and nonpolar interaction free energy contributions and present the results of the average interaction free energies of the lowest binding free energy complex in two-dimensional density maps in Fig. S1 and Fig. S2 in the Supporting Material. In addition, we calculated the average interaction energy per V3 loop residue, by summing up the interaction energies of the specific residue with all of its possible CXCR4 interacting residues, so as to investigate the critical gp120-V3 loop residues with regard to coreceptor binding. The results are presented in Table S4.
Results
We employed a comprehensive set of computational tools, primarily comprising binding free energy calculations and MD simulations, and identified Complex 1 as the MD simulation with the lowest average binding free energy (Fig. 1, and see Table S1). In all 17 complexes, the V3 loop residues lying outside the chemokine receptor experience higher flexibility; the average backbone RMSD without alignment with respect to the starting simulation conformation is 3.9 ± 0.3 and 2.9 ± 0.1 Å for the entire V3 loop and the embedded region (8:26), respectively, in Complex 1 (see Table S2). In general, in the lowest binding free energy complexes (1, 4, and 12) the V3 loop possesses a relatively low RMSD average and standard deviation values, specifically within the residue moiety 8–26 (see Table S2). On the contrary, the unbound V3 loop is considered to be highly flexible as shown in Methods, in line with (27,29,30). Therefore, the results of this study indicate that the coreceptor binding stabilizes the conformation of the V3 loop, especially for the V3 loop residue moiety 8:26. In addition, at least for the lowest binding free energy complexes (1, 4, and 12), the V3 loop binding also provides stabilization for the N-terminal end of CXCR4 (see Table S2).
Figure 1.
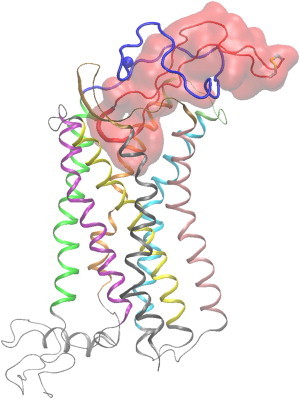
Molecular graphics image of the entire simulation system corresponding to the complex with the lowest average binding free energy. The V3 loop is shown in tube and transparent surface representation in red color, and its 16–20 residue moiety is shown in thick tube representation. The CXCR4 is shown in cartoon representation, and the coloring used for different protein domains is as follows: (blue) N-terminal domain; (green) intramembrane helix 1 (IH1); (light gray) intracellular loop 1 (ICL1); (purple) IH2L; (light gray) ECL1; (yellow) IH3; (light gray) ICL 2; (medium gray) IH4; (ochre) ECL2; (pink) IH5; (light gray) ICL3; (cyan) IH6; (lime) ECL3; (orange) IH7; and (light gray) C-terminal domain. (van der Waals sphere) N-terminal Cα atom of CXCR4. (Thick transparent licorice representation) V3 loop disulfide bridge. The definition of CXCR4 and V3 loop domains is presented in the Supporting Material (colors appear in the online version only).
The nonpolar binding free energy of Complex 1 is comparable to Complex 4. However, according to both GB and PB approximations, the polar component of Complex 1 is more favored than Complex 4. According to the analysis of hydrogen-bond interactions of all complexes (see Table S3), this is mainly due to the absence of salt bridges between V3 loop:CXCR4 residues Arg3:Glu268, Arg18:Asp171, and Arg31:Glu14 in Complex 4 (see Table S3). Also, the lower polar binding free energy of Complex 12, compared to Complex 1, according to both GB and PB calculations, could mainly be attributed to the presence of additional salt bridges between V3 loop:CXCR4 residues Arg11:Asp10 and to a lesser extent Arg9:Asp10, Lys10:Tys12 (see Table S3); in addition, the salt bridge of Arg31 with residue Glu14 in Complex 1 is interchanged with a lower occupancy salt bridge with residue Tys12 in Complex 12. On the contrary, Arg3:Asp22 and Arg3:Glu268 have lower occupancy in Complex 12 compared to 1. Nevertheless, despite the lower polar binding free energy of Complex 12 compared to Complex 1, the nonpolar component of Complex 1 is more favored compared to Complex 12 by ∼24 kcal/mol, and overall Complex 1 is the most favored among all complexes. In what follows, we oriented our analysis to Complex 1, as its binding free energy is optimum compared to other complexes with regard to the sum of polar and nonpolar binding components, and provide a detailed investigation of the structural and physicochemical properties encountered within the 1000 frames extracted from the simulation.
Structural properties of the bound V3 loop
Residues 8–26 of the V3 loop are buried within CXCR4, whereas residue moieties 1–7 and 27–35 mainly lie upon the N-terminal end of CXCR4 (all V3 loop residues are renumbered, starting from 1 and ending at 35). The V3 loop conformation is twisted, as shown in Fig. 1 and, two consecutive antiparallel β-sheets among the following residue moieties, 12–13:22–21 and 3–9:32–26, are observed in the trajectory. A β-turn or bend is observed within the core of the tip comprising residues 16:20, which is the mostly buried region of the V3 loop within the binding pocket, as shown in Fig. 1. The β-sheets provide a compact-thin shape and a relatively stable conformation of the V3 loop within the simulation. The two primary hydrogen bonds involving side chains of opposite V3 loop stems are Arg9 NH1/2:Gln25 OE1 and Thr8 O:Arg31 NH1/2. The relatively tight binding of a highly flexible loop can be attributed to both the cooperativity of intramolecular interactions in the bound structure, shown in Fig. S1, and the intermolecular interactions, analyzed below.
Investigation of the V3 loop: CXCR4 complex structure
We present a detailed overview of the structural and physicochemical properties of the complex structure, which is based on the assessment of the intermolecular pairwise residue interaction free energies, shown in Fig. S2. Fig. 2 presents the most important intermolecular polar interactions in the trajectory, using the software VMD (65). Table 1 extracts information from Fig. S2 as well as Table S3 and summarizes the key interactions between V3 loop:CXCR4 residues, over all 1000 snapshots, and features in boldface the agreement with experimental findings.
Figure 2.
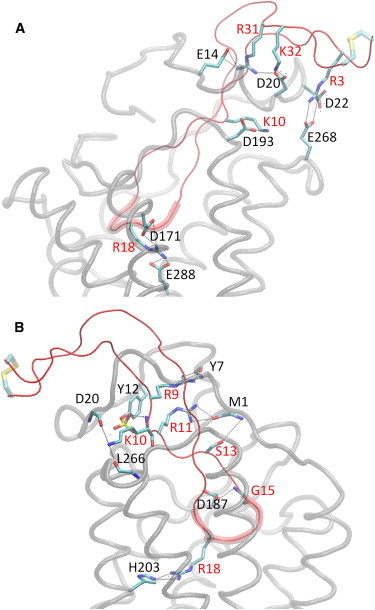
Molecular graphics images of important polar interactions corresponding to the complex with the lowest average binding free energy. Panels (A) and (B) depict the salt bridges and the most important hydrogen bonds, respectively, according to residue pair-wise interaction free energies. Panel (B) is rotated by approximately 180° with respect to (A) around the z (membrane) axis. The V3 loop is shown in tube and in red color, and its 16–20 residue moiety is shown in fat tube representation. The CXCR4 is shown in light gray transparent tube representation. The salt bridge and hydrogen bonds in panels (A) and (B) are denoted in dashed lines and the participating V3 loop and CXCR4 residue moieties are shown in licorice; V3 loop and CXCR4 residues are annotated in red and black, color respectively. Hydrogen atoms are omitted for clarity and the V3 loop disulfide bridge is shown in fat transparent licorice representation. (colors appear in the online version only).
The coordinates of structures in Complex 1, extracted every 2 ns, are provided (see Supporting Material). Within the simulation, the conformation of the coreceptor is very well retained with regard to the starting simulation structure. The average backbone RMSD of the intramembrane helical residues is equal to 1.3 ± 0.0 Å, and the average RMSD of the entire backbone is 2.2 ± 0.2 Å. The larger value of the latter is attributed to the higher flexibility of the non-intramembrane domains. It is worth noting that in all simulations, the average backbone RMSD of the intramembrane helical residues is low (see Table S2), showing the ability of the implicit solvent model used to preserve the integrity of the x-ray structure (11).
Interactions of V3 loop residues 1:15 with CXCR4
V3 loop residues 3–15 are mainly engaged to intermolecular interactions with the N-terminal end of CXCR4 and to a lesser extent with ECL2 and ECL3 residues of CXCR4. Cys1, Thr2, and Pro4 of the V3 loop are predominantly solvent-exposed. Arg3 of the V3 loop forms a highly interacting salt bridge with Glu268, a salt bridge with CXCR4 residue Asp22 (Fig. 2 A), and is also proximal to the negatively charged Asp20 and Tys21. Asn5 of the V3 loop intercalates between CXCR4 residues Asp20, Tys21, Asp22, and Ser23; two simultaneous hydrogen bonds are observed: Asn5 ND2:Tys21 O and Asn5 ND2:Asp20 OD1/2. Asn6 of the V3 loop mainly interacts with CXCR4 residue Ser23; as a result, two simultaneous hydrogen bonds are frequently formed among atom pairs Asn6 N:Ser23 OG and Asn6 ND2/OD1:Ser23 OG. Because position 6 is an N-linked glycosylation site, we assume that apart from the loss of the aforementioned hydrogen-bond interactions, the N-linked glycosylation will not interfere with the binding as, according to the simulation, the side chain of Asn6 points toward the aqueous environment. Asn7 of the V3 loop is mainly interacting with CXCR4 residues Glu14 and Asn11 through weak Asn7 OD1:Asn11 ND2, Asn7 ND2:Glu14 OE1/2 hydrogen bonds. Thr8 of the V3 loop intercalates between the backbone moieties of CXCR4 residues Tyr21, Asp22, Ser23, and Lys25; Thr8 OG1 is hydrogen-bonded to Asp22 O consistently during approximately the last half of the simulation. Arg9 of the V3 loop interacts with the backbone moieties of residues Met1, Tys7, Thr8, Ser9, Asp10, and Asn11, as well as the side chain of CXCR4 residue Tys12. Two hydrogen bonds are formed between the charged amide group of Arg9 and the backbone carbonyl groups of Tys7 (Fig. 2 B) and Asp10; also, the proximity of the positively charged amide group of Arg9 to the negatively charged Tys12 results in a polar interaction between the two.
Lys10 of the V3 loop participates in a highly interacting salt bridge with CXCR4 residue Asp193 (Fig. 2 A) and in the following hydrogen bonds: Lys10 N:Tys12 SO(2/3), Lys10 NZ:Asp20 O, and Lys10 NZ:Leu266 O (Fig. 2 B). In addition, the nonpolar part of the Lys10 side chain is proximal to the backbone of Ser18, Gly19, and the aromatic ring of Tys21. Arg11 of the V3 loop is buried in a diverse polar/nonpolar binding pocket comprising CXCR4 residues Met1 (Fig. 2 B), Glu2, Gly3, Tys7, Tys12, Phe189, and Pro191; its nonpolar side-chain part is proximal to the aromatic rings of Tys7, Tys12, and Phe189, and its backbone amide is hydrogen-bonded to Tys12 SO3; moreover, its charged amide is close to the backbone of the first three CXCR4 N-terminal residues and is hydrogen-bonded to Met1 O and Glu2 O. Val12 of the V3 loop forms nonpolar contacts with nearby CXCR4 residues Phe189, Tyr190, and Val196. Ser13 of the V3 loop is interacting mainly with Met1 through the Ser13 OG:Met1 N hydrogen bond (Fig. 2 B). Leu14 of the V3 loop is positioned between N-terminal and ECL2 of CXCR4 residues. Its backbone is mainly interacting with CXCR4 residues Glu2, Gly3 and, as a result, a low frequency hydrogen bond is observed between Leu14 N and Glu2 O; in addition the backbone carbonyl group of Leu14 is hydrogen-bonded to the charged side-chain NH1 or NH2 Arg183 in more than half of the simulation snapshots. The Leu14 side chain intercalates around residues Ser178, Ala180, Ile185, Asp187, and Phe189. The placement of the charged Asp187 in the vicinity of the hydrophobic Leu14 is attributed to the hydrogen bond among Asp187 OD1 or OD2 with the V3 loop atom Gly15 N (Fig. 2 B).
Interactions of V3 loop residues 16:22 with CXCR4
Pro16 of the V3 loop is buried within a binding pocket composed of CXCR4 residues Arg30, Phe36, Asn37, and Leu41, and its amide group forms a weak hydrogen bond with atom Asn37 ND2. Gly17 of the V3 loop is mainly involved in a hydrogen bond with Asp97 OD1/2 in approximately the last-quarter of the simulation; in addition it is involved in a nonpolar interaction with residue Trp94 and in a weak hydrogen bond with Tyr45 OH. Arg18 of the V3 loop forms the most highly interacting polar interactions with a group of CXCR4 residues because it is simultaneously engaged in two high occupancy salt bridges with residues Asp171 and Glu288 (Fig. 2 A). In addition, Arg18 NH1/2 is hydrogen-bonded to His203 NE2 (Fig. 2 B), Thr117 OG1, Tyr255 OH, and the Arg18 backbone carbonyl group is hydrogen-bonded to the charged amide of Arg188. The nonpolar part of the Arg18 side chain is involved in nonpolar contacts with residues Trp94 and Tyr116, and the nonpolar moieties of Asp187 and Arg188. Val19 is involved in mainly nonpolar interactions with its neighboring CXCR4 residues Ile284 and Ser285. Trp20 of the V3 loop is embedded in a binding pocket that mainly consists of residues Arg188, Tyr190, Val196, Phe199, Gln200, and Ile284; the interactions with Arg188 and Tyr190 are polarly driven due to the Trp20 NE1:Arg188 NH1/2 and Trp20 NE1:Tyr190 OH hydrogen bonds, whereas the latter interactions mainly involve nonpolar contacts. The backbone carbonyl group of Trp20 is weakly hydrogen-bonded to CXCR4 atom His281 ND1. Tyr21 of the V3 loop is surrounded by CXCR4 residues Phe29, Arg30, Asn278, Glu277, and His281; its side-chain hydroxyl group is hydrogen-bonded to CXCR4 atom Asn278 ND2/OD1 and the aromatic ring predominantly participates in nonpolar interactions with the nonpolar moieties of these residues. Thr22 of the V3 loop interacts with CXCR4 residues Leu266 and Glu277. The former residue interacts with the nonpolar moiety of Thr22, whereas the latter residue is involved in a Thr22 OG1:Glu277 OE1 hydrogen bond during approximately the last-third of the simulation.
Interactions of V3 loop residues 23:35 with CXCR4
V3 loop residues 23–35 interact solely with the CXCR4 N-terminal region. Thr23 of the V3 loop forms nonpolar contacts with CXCR4 residues Met1, the disulfide bridge residue Cys28, and the backbone of Phe29, and in addition, its side-chain hydroxyl group is weakly hydrogen-bonded to the CXCR4 atom Cys28 O. Gly24 of the V3 loop is in proximity with the nonpolar moiety of CXCR4 residue Lys25 and interacts with the disulfide bridge CXCR4 residue Cys28 via the Gly24 O:Cys28 N or Gly24 N:Cys28 O hydrogen bonds. Gln25 of the V3 loop is in the vicinity of CXCR4 residues Met1, Pro27, Cys28, and Phe29; its interaction with residue Cys28 is facilitated through a low-occupancy hydrogen bond between Gln25 NE2 and Cys28 O. Ile26 of the V3 loop is primarily interacting with the nonpolar moieties of CXCR4 residues Ser23 and Lys25, and in a similar fashion, Val27 of the V3 loop is buried within the nonpolar moieties of CXCR4 residues Ser9, Asp10, and Asn11. Residues Gly28, Asp29, and Ile30 of the V3 loop are solvent-exposed and are not engaged in intermolecular interactions. Arg31 of the V3 loop forms two simultaneous salt bridges with CXCR4 residues Glu14 and Asp20, with the former being stronger (Fig. 2 A), and is proximal to oppositely charged residue Tys12 and Ser18 of CXCR4; as a result its charged amide is weakly hydrogen-bonded to Ser18 OG. Lys32 of the V3 loop forms a highly interacting salt bridge with CXCR4 residue Asp20 (Fig. 2 A), and its charged amide group is hydrogen-bonded to the backbone carbonyl groups of CXCR4 residues Glu17 and Ser18. Residue Arg31 of the V3 loop is proximal to the oppositely charged CXCR4 residue Tys12, and residue Lys32 of the V3 loop participates in a hydrogen bond through its charged amide group with the backbone carbonyl group of CXCR4 Ser18. Residues Ala33 and Cys35 of the V3 loop are solvent-exposed and are not engaged in any interactions with CXCR4 residues, whereas the side-chain amide group of V3 loop His34 is weakly hydrogen-bonded to the charged carboxyl group of CXCR4 Asp20. The disulfide bridge points toward the aqueous environment throughout the simulation, as would be expected if it was covalently bonded to the entire gp120 protein.
Discussion
In the MD simulation, the bound V3 loop is twisted, and its compactness is associated with the formation of β-sheets and the Arg9 NH1/2:Gln25 OE1 hydrogen bond. These data are in agreement with recently published results from MD simulations of unbound V3 loops, according to which the compactness was linked to an electrostatic interaction between the side chains of residues Arg9 and Glu25 in one of the V3 loops (27). The compact-thin shape of the V3 loop bound structure suggests it is necessary for it to adopt a compact-thin shape before coreceptor binding. The compactness is related the maximization of the tip-base distance, as suggested in López de Victoria et al. (27), and provides the ability to residue Arg18 for the V3 loop to be buried deep inside the membrane and form highly interacting polar interactions with CXCR4 residues Asp171 and Glu288.
The average backbone RMSD of the unbound dual tropic V3 loop conformations of this study compared to the most representative, CCR5 tropic PDB:2QAD structures in solution (Fig. 7B, basins 1–3 of López de Victoria et al. (27)) is 6.2 ± 0.7, 5.4 ± 1.4, and 7.7 ± 0.8 Å, respectively. The corresponding values calculated for the residue moiety 8–26 are 5.9 ± 0.7, 4.6 ± 1.6, and 7.7 ± 1.0 Å. The relatively large values are in agreement with very recent results of Chandramouli et al. (30) showing, respectively overall structural dissimilarity between different tropic unbound V3 loops, at the level of crown region, defined as residues 11–25. Nevertheless, upon alignment, the average backbone RMSD between the bound dual tropic V3 loop conformations of Complex 1 for the last 5 ns, and the most representative, CCR5 tropic PDB:2QAD structure in solution with maximized tip-base distance (Fig. 7B, basin 2 of López de Victoria et al. (27)) is 3.8 ± 0.1 and 2.6 ± 0.1 Å, respectively, for the entire V3 loop and the 8–26 residues moiety. Thus, despite 1), the relatively large sequence dissimilarity between the two V3 loops in 9 out of 35 positions, and 2), the different simulation properties with regard to the V3 loop, bound versus unbound, the specific maximized tip-base conformation (which can both adopt and is required at least for CXCR4 binding) is interestingly comparable, specifically for the 8–26 residue moiety.
Key interacting V3 loop residues for CXCR4 recognition
The sum of the interaction energies per V3 loop residue, presented in Table S4, presents the V3 loop residues associated with CXCR4 intermolecular interactions, in descending order. The 10 most highly interacting V3 loop residues, with average total interacting free energies lower than −20 kcal/mol are: Arg18, Lys10, Arg3, Arg31, Arg11, Arg9, Trp20, Leu14, Lys32, and Tyr21. Seven out of ten are positively charged and are highly interacting, predominantly owing to strong polar interactions that involve salt bridges and/or hydrogen bonds with CXCR4 residues. The specific dual tropic V3 loop possesses a +6 net charge, and within the simulation of this study we observe an abundance of highly interacting specific salt bridges and hydrogen bonds between positively charged V3 loop residues and negatively charged CXCR4 residues. This also supports: 1), The rule implying that the increase of the net charge of the V3 loop is associated with CXCR4 tropism (10,31); and 2), The suggestion that electrostatics likely dictate coreceptor choice (30).
Furthermore, it is worth noting that residues Arg18, Lys10, Arg3, Arg31, and Arg9 possess the highest residue propensity for their corresponding positions regarding both CXCR4 and dual tropic 35-residue-V3 loops, while residues Arg11 and Lys32 are the second most probable residues for positions 11 and 32 (see Table S4). Residues Trp20, Leu14, and Tyr21 are highly interacting, predominantly owing to nonpolar interactions with CXCR4 residues. Residue Tyr21 possesses the highest residue propensity for position 21 for both CXCR4 and dual tropic 35-residue-V3 loops, while residues Trp20 and Leu14 are the second most probable residue for positions 20 and 14, respectively, after residues Phe20 and Ile14 which are first, and possess similar physicochemical properties (see Table S4). In addition, the residues Pro16, Gly15, Ser13, Asn5, Gly17, Gln25, Gly24, Val12, Thr8, Val19, Thr22, Thr23, Asn6, Val27, Ile26, and Asn7 possess interaction free energies, in descending order of strength, within the range of −14 to −5 kcal/mol (see Table S4). Moreover, this study provides what is, to our knowledge, the first reported molecular recognition-based piece of evidence on the 11/24/25 rule (8), as Arg11 NH1/2 of V3 loop is hydrogen-bonded to Met1 O of CXCR4. Taking into consideration that Met1 is the first N-terminal residue, and that interactions between the V3 loop and CXCR4 N-terminal are critical for the molecular recognition (19–21,24), this high-occupancy hydrogen bond could be invaluable for reducing the high flexibility of the N-terminal end (11) upon binding (see Table S2).
Since 1997, a series of experimental studies aimed at exploring the key CXCR4 residues related to the V3 loop interaction (16–22,24–26). A fraction of these studies also aimed at comparing the CXCR4 residues associated with V3 loop binding to the CXCR4 residues associated with the stromal cell-derived factor1 (SDF1α) binding, because SDF1α constitutes a natural ligand of CXCR4 with blocking capacity of HIV-1 (66,67). In addition, some studies have compared the CXCR4 residues associated with the V3 loop binding to the CXCR4 residues involved in the binding of potential therapeutic candidates that act as V3 loop antagonists (68). The primary limitation of all studies reported until now is the lack of information regarding the interacting V3 loop:CXCR4 key residue pairs, due to the absence of a published complex structure.
Role of the N-terminal of CXCR4
The deletion of CXCR4 residues 2–25 is associated with at least 60% reduction of HIV-1 coreceptor activity (17). The same residue moiety is also critical for SDF1α and DV1 dimer binding, where DV1 dimer is a CXCR4 HIV-1 inhibitor (68). Our results show that more than half of the V3 loop residues, mainly residues of the opposite stems, interact with the 2–25 CXCR4 residue sector. Alanine substitutions to sulfated tyrosines 7 and 12 correlate with or markedly impair the coreceptor activity for HIV-1 for different dual tropic V3 loops (19,24). According to our results, Tys7 is proximal to oppositely charged residues Arg9 and Arg11 of the V3 loop, and its backbone carbonyl is hydrogen-bonded to the charged side-chain group of Arg9. It is worth noting that the functional role of some CXCR4 residues could be associated with their intramolecular interactions because they can be fundamental in stabilizing the coreceptor structure. Within the simulation, we observe that Tys7 SO2−4 is mainly hydrogen-bonded with the CXCR4 backbone amide groups of Ile4 and Ser5, contributing to the stabilization of the 3–6 β-turn in CXCR4. Tys12 is attracted to oppositely charged residues Arg9, Lys10, and Arg11 of the V3 loop, and is stabilized in proximity to Lys10 and Arg11 via simultaneous hydrogen bonds among its charged SO4 side-chain group and the backbone amide groups of Lys10 and Arg11.
Additional alanine substitutions on CXCR4 N-terminal residues Glu2, Asp10, Glu14, Glu15, Asp20, Tys21, Asp22, Ser23, Lys25, Glu26, Cys28, and Glu32 lead to a decrease of HIV-1 coreceptor activity, showing that they are associated with V3 loop binding (19–21,24). In the MD simulation, Glu2 of CXCR4 is proximal to the oppositely charged Arg11 of the V3 loop, and owing to its position, it contributes to the stabilization of the polar interaction among the latter and CXCR4 residue Met1. Asp10 of CXCR4 is polarly attracted to oppositely charged Arg9 of the V3 loop and its position is stabilized by a hydrogen bond through its backbone carbonyl group with the side-chain amide of V3 loop residue Arg9. The charged side-chain carboxyl group of CXCR4 Glu14 forms a highly interacting salt bridge with the V3 loop residue Lys32 and a hydrogen bond with V3 loop atom Asn7 ND2. Glu15 of CXCR4 points toward the opposite side of the binding site and is not directly related to the binding with V3 loop; nevertheless, as both Glu14 and Glu15 correlate with HIV-1 coreceptor activity, the interactions formed by Glu14 can be interchanged by its neighboring Glu15. Asp20 of CXCR4 forms two highly interacting salt bridges with residues Arg31 and Lys32, as well as hydrogen bonds with residues Asn5, Lys10, and His34. Residue Tys21 of CXCR4 is proximal to V3 loop residues Thr8 and Lys10, forming noteworthy nonpolar interactions. The critical role of CXCR4 residue Asp22 could mainly be attributed to its high-occupancy salt bridge with V3 loop residue Arg3. Ser23 of CXCR4 participates in hydrogen bonds between its side-chain hydroxyl group with main- and side-chain atoms of V3 loop residue Asn6. Residue Asp25 of CXCR4 forms noteworthy nonpolar interactions with V3 loop residues Thr8, Gly24, and Ile26. Residue Glu26 of CXCR4 is weakly interacting with V3 loop residues Gly24, Gln25, and Ile26, while residue Glu32 of CXCR4 is not part of the binding pocket.
We observe the presence of intramolecular hydrogen bonds and salt bridges between the N-terminal side chains and ECL3 side/main chains of CXCR4 residue pairs Tys21:Gly272, Asp22:Lys271, Glu26:Gly273, and Glu32:Lys282. The aforementioned interactions involve the Tys21, Asp22, Glu26, and Glu32 N-terminal CXCR4 residues, which are associated with HIV-1 coreceptor activity. The interactions can be fundamental and constitute a sufficient condition for the proper connection of the N-terminal end and ECL3 of CXCR4, as the deletion of the Cys28-Cys274 disulfide bridge affects, but not to a large extent, the HIV-1 coreceptor activity (20). Therefore, the side chains of CXCR4 residues Tys21, Asp22, Glu26, and Glu32 can play a key role in the preservation of the appropriate CXCR4 structure required for the gp120 binding through its V3 loop.
The interactions reported in this study between the V3 loop and the CXCR4 N-terminal end are not similar to the predicted interactions between the V3 loop and the CCR5 N-terminal end by Huang et al. (5) using molecular docking. This variation can mainly be attributed to the facts that 1), the N-terminal conformations of CXCR4 (39) and CCR5 (5) are different, and 2) CXCR4 and CCR5 V3 loops mainly differ owing to their stem-region residues, which predominantly interact with the N-termini of the coreceptors. In line with this suggestion, a recent analysis indicated that the critical sites of features informative of viral tropism belong to stem regions (69).
Role of intramembrane helices 1 and 2 of CXCR4
Alanine substitutions showed a decrease in HIV-1 coreceptor activity for Tyr45 (17,68), Asp97 (17,18,20), to a lesser extent for His79 (17,18,68) and Phe87 (18), but not for Lys110 and Tyr121 (18). In line with this, Lys110 and Tyr121, are not involved in interactions with the V3 loop in the simulation. On the contrary, both Tyr45 and Asp97 of CXCR4 interact through their side chains with mainly the carbonyl backbone group of Gly17. According to our simulation, residue Phe87 of CXCR4 is indirectly involved in the V3 loop binding by forming π-π interactions with residue CXCR4 residue Tyr116, which directly interacts with Arg18; the Phe87-Tyr116 interaction is also present in the crystallographic structure (11). According to both the crystal structure (11) and our findings, His79 is far from the binding site and it is hydrogen-bonded through its side-chain amide group to Tyr157 OH of CXCR4.
Role of ECL2 of CXCR4
Studies have shown that alanine substitution on residues Trp161 and Pro163 correlate with the HIV-1 activity (17,18). In both the crystallographic structure (11) and our simulation, residues Trp161 and Pro163 are positioned in the center of the membrane, away from the binding site; we hypothesize that these residues play a role in the preservation of the helical shape of intramembrane helix 4. Asp171 of CXCR4 is experimentally considered one of the most important residues involved in the HIV-1 coreceptor activity (17,18) because an alanine substitution at this position reduces by >60% the activity of both HIV-1 (17) and of DV1 (68). Within our simulation, Asp171 of CXCR4 forms a highly interacting salt bridge with V3 loop Arg18. Despite the fact that Asp182 of CXCR4 is not part of the binding site, also according to the CXCR4 crystal structure (11), its alanine substitution results in a reduction of HIV-1 coreceptor activity (18). Asp182 can play a key role in attracting the positively charged V3 loop before its binding, because Asp182 possesses the peak position in ECL2 facing toward the aqueous extracellular region in our simulation and the crystallographic structure (11). Alanine substitutions revealed that Arg183 (21,23), and to a lesser extent Tyr184 (21,23) of CXCR4, are related to HIV-1 coreceptor activity. Within the simulation trajectory, the charged amide side chain of Arg183 is involved in a hydrogen bond with the backbone carbonyl group of V3 loop residue Leu14, while its nonpolar side-chain moiety is proximal to Leu14 and Gly15 of the V3 loop.
In line with the experimental structure (11), residue Tyr184 is not in the binding site and it faces toward the aqueous extracellular environment, in similar fashion to Asp182; consequently it can play a role in attracting the positively charged V3 loop before its binding through its side-chain OH group. Furthermore, alanine substitutions at CXCR4 residues Asp187 and Phe189 are experimentally associated with a >60% reduction for HIV-1 coreceptor activity, suggesting the critical role of both residues (17,23). In line with it, the charged carboxyl group of Asp187 is hydrogen-bonded to atom Gly15 N of the V3 loop throughout the entire simulation trajectory and its nonpolar moiety is interacting with the proximal V3 loop Leu14 side chain. In the simulation, Phe189 is buried in a nonpolar core composed by V3 loop side chains of residues Arg11 (nonpolar moiety), Val12 and Leu14. Moreover, site-directed mutagenesis studies have depicted an involvement of the CXCR4 residues Arg188, Tyr190, and Pro191 (17,18,20,21,23) in the HIV-1 binding. In our simulation, the Arg188 charged amide group is hydrogen-bonded atoms Arg18 O and Trp20 NE1 atom of the V3 loop, the Tyr190 side chain is also hydrogen-bonded to Trp20 NE1 and forms nonpolar interactions with Val12 of the V3 loop, and Pro191 is also proximal to Val12 of the V3 loop. The coreceptor activity is experimentally markedly impaired by an alanine substitution at 193 for a specific HIV-1 strain (19); in agreement with this, in our simulation Asp193 forms a highly interacting salt bridge with V3 loop residue Lys10.
Role of intramembrane helices 5, 6, 7 and ECL3 of CXCR4
An alanine substitution on CXCR4 residue Gln200 affects significantly the HIV-1 coreceptor binding (21), and our results show that Gln200 forms a strong nonpolar interaction with the aromatic ring of the V3 loop residue Trp20. This interaction is additionally stabilized by an intramolecular hydrogen bond between atoms Gln200 NE2 and Asp262 OD2, and an alanine substitution to Asp262 also reduces the HIV-1 coreceptor activity (17,18). Furthermore, an alanine substitution of CXCR4 residue Trp252 is related to a decrease of the binding affinity of the V3 loop, SDF1α, and the DV1 inhibitor (17,68). The results of our simulation and the crystallographic structure (11) show that Trp252 is positioned in the center of the membrane, being away from the binding site. Nevertheless, in both the experimental structure and the simulation, the aromatic group of Trp252 forms aromatic nonpolar interactions with CXCR4 residues Phe248, Tyr255, Tyr256, and Phe192, which can be most important for the stabilization of the nucleus of aromatic residues and consequently, the hydrogen bond formation between Tyr255 OH of CXCR4 and Arg18 NH1/2 of the V3 loop. The latter interaction should be significant, owing to the experimentally determined critical role of Tyr255 and Tyr256 (17,18,68).
Our results justify the decrease of activity upon Glu268 substitution to alanine, because Glu268 participates in a highly interacting salt bridge with V3 loop residue Arg3 (21). Glu288 of CXCR4 is determined to be one of the most critical residues for HIV-1 coreceptor activity, because even an aspartic acid substitution at this position, which preserves the physicochemical properties of the former, results in a considerable 30–60% loss of HIV-1 coreceptor activity (17,18); also, alanine substitutions on Glu288 resulted in a significant decrease of coreceptor activity related to SDF1α, DV1 and the V3 loop (18,68). Within our simulation, Glu288 forms the most highly interacting salt bridge with Arg18 of the V3 loop. Because Arg18 of the V3 loop is simultaneously forming two highly interacting salt bridges with CXCR4 residues Asp171 and Glu288 residues, our data suggest that a substitution to Asp288 could not permit Arg18 to form the two concurrent salt bridges due to the shorter size of aspartate compared to glutamate (Fig. 2 A). Alanine substitutions at CXCR4 residues His294 and Asn298 showed that these residues correlate with HIV-1 coreceptor activity (17,18). Their presence below the center of the membrane, far from the binding site, also in the crystallographic structure (11), suggests that their role should be attributed to their involvement in intramolecular interactions within CXCR4.
Conclusions
We consider that the success of having remarkable accordance with experimental results is due to the systematic methodology employed, which includes the following features:
-
1.
The modeling of the entire CXCR4 structure and the use of an extensive set of computational tools and methods to produce a variety of structural templates of V3 loop and CXCR4 for docking;
-
2.
The large number of docked complexes investigated;
-
3.
The heterogeneous dielectric solvation models used to rank the complex structures according to their binding free energy;
-
4.
The employment of MD simulations for the most promising complexes, with regard to their binding free energy, in order to improve the conformational sampling and interactions; and
-
5.
The selection of the final complex acquiring the lowest average binding free energy affinity throughout the MD simulations.
These steps constitute a systematic methodology, and our study suggests that a similar computational framework can be applied for the elucidation of subsequent V3 loop:CXCR4/CCR5 complexes, or more generally, a broader series of ligand-G protein coupled receptor complexes.
The identification of the most highly interacting V3 loop and CXCR4 residues, as well as the specificity of interacting residue pairs accomplished in this study, could provide gateways for the de novo design of novel inhibitors targeting key CXCR4 residues and serving as antagonists against HIV-1 (70). According to the results of recent experiments and computational modeling, the novel HIV-1 inhibitor, DV1, partially meets the above hypothesis as it participates in polar interactions with CXCR4 residues Asp171 and Glu288 (68), the two most highly interacting polar residues identified in this study. Guided by the findings of the present study, and within this direction, de novo design methods (62,71,72) in conjunction with binding free energy calculations and MD simulations, as in Tamamis et al. (62), can be implemented to design new peptides acquiring augmented interactions with the key CXCR4 residues for HIV-1 coreceptor activity.
We employed computational methods, predominantly comprising MD simulations, as well as binding and interaction free energy calculations, to investigate the molecular recognition of CXCR4 by a dual receptor HIV-1 gp120 V3 loop. The reported complex structure, despite acquiring absolutely no bias toward experimental results, is in a remarkable accordance with experiments. To our knowledge, it is the first reported V3 loop:CXCR4 complex structure to shed light on the functional role of the HIV-1 gp120 V3 loop and chemokine receptor residues, which are experimentally associated with HIV-1 coreceptor activity.
Acknowledgments
All MD simulations and free energy calculations were performed on SESAME and TIGER computer clusters at the TIGRESS high performance computer center at Princeton University.
We thank the anonymous referees for constructive comments and suggestions.
C.A.F. acknowledges funding from the National Institutes of Health (grant No. R01 GM052032).
Supporting Material
References
- 1.Chan D.C., Fass D., Kim P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 2.Kwong P.D., Wyatt R., Hendrickson W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwong P.D., Wyatt R., Hendrickson W.A. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8:1329–1339. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 4.Huang C.C., Tang M., Kwong P.D. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C.C., Lam S.N., Kwong P.D. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dittmar M.T., McKnight A., Simmonds P. HIV-1 tropism and co-receptor use. Nature. 1997;385:495–496. doi: 10.1038/385495a0. [DOI] [PubMed] [Google Scholar]
- 7.Xiao L.H., Owen S.M., Lal R.B. CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 is independent of phylogenetically distinct global HIV-1 isolates: delineation of consensus motif in the V3 domain that predicts CCR-5 usage. Virology. 1998;240:83–92. doi: 10.1006/viro.1997.8924. [DOI] [PubMed] [Google Scholar]
- 8.Pollakis G., Kang S., Paxton W.A. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J. Biol. Chem. 2001;276:13433–13441. doi: 10.1074/jbc.M009779200. [DOI] [PubMed] [Google Scholar]
- 9.Cardozo T., Kimura T., Zolla-Pazner S. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res. Hum. Retroviruses. 2007;23:415–426. doi: 10.1089/aid.2006.0130. [DOI] [PubMed] [Google Scholar]
- 10.Edo-Matas D., van Dort K.A., Kootstra N.A. Comparison of in vivo and in vitro evolution of CCR5 to CXCR4 coreceptor use of primary human immunodeficiency virus type 1 variants. Virology. 2011;412:269–277. doi: 10.1016/j.virol.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Wu B., Chien E.Y.T., Stevens R.C. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan D.C., Kim P.S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 13.Hwang S.S., Boyle T.J., Cullen B.R. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 14.Weiss R.A., Clapham P.R. Hot fusion of HIV. Nature. 1996;381:647–648. doi: 10.1038/381647a0. [DOI] [PubMed] [Google Scholar]
- 15.Doranz B.J., Orsini M.J., Doms R.W. Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J. Virol. 1999;73:2752–2761. doi: 10.1128/jvi.73.4.2752-2761.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dragic T. An overview of the determinants of CCR5 and CXCR4 co-receptor function. J. Gen. Virol. 2001;82:1807–1814. doi: 10.1099/0022-1317-82-8-1807. [DOI] [PubMed] [Google Scholar]
- 17.Choi W.T., Tian S., Huang Z. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J. Virol. 2005;79:15398–15404. doi: 10.1128/JVI.79.24.15398-15404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian S., Choi W.T., Huang Z. Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1alpha on CXCR4 transmembrane helical domains. J. Virol. 2005;79:12667–12673. doi: 10.1128/JVI.79.20.12667-12673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brelot A., Heveker N., Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 2000;275:23736–23744. doi: 10.1074/jbc.M000776200. [DOI] [PubMed] [Google Scholar]
- 20.Chabot D.J., Zhang P.F., Broder C.C. Mutagenesis of CXCR4 identifies important domains for human immunodeficiency virus type 1 X4 isolate envelope-mediated membrane fusion and virus entry and reveals cryptic coreceptor activity for R5 isolates. J. Virol. 1999;73:6598–6609. doi: 10.1128/jvi.73.8.6598-6609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou N., Luo Z., Huang Z. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 2001;276:42826–42833. doi: 10.1074/jbc.M106582200. [DOI] [PubMed] [Google Scholar]
- 22.Chabot D.J., Chen H.D., Broder C.C. N-linked glycosylation of CXCR4 masks coreceptor function for CCR5-dependent human immunodeficiency virus type 1 isolates. J. Virol. 2000;74:4404–4413. doi: 10.1128/jvi.74.9.4404-4413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brelot A., Heveker N., Alizon M. Effect of mutations in the second extracellular loop of CXCR4 on its utilization by human and feline immunodeficiency viruses. J. Virol. 1999;73:2576–2586. doi: 10.1128/jvi.73.4.2576-2586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kajumo F., Thompson D.A., Dragic T. Entry of R5X4 and X4 human immunodeficiency virus type 1 strains is mediated by negatively charged and tyrosine residues in the amino-terminal domain and the second extracellular loop of CXCR4. Virology. 2000;271:240–247. doi: 10.1006/viro.2000.0308. [DOI] [PubMed] [Google Scholar]
- 25.Lu Z., Berson J.F., Doms R.W. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc. Natl. Acad. Sci. USA. 1997;94:6426–6431. doi: 10.1073/pnas.94.12.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S., Fan S., Sun Z. Structural and functional characterization of the human CCR5 receptor in complex with HIV gp120 envelope glycoprotein and CD4 receptor by molecular modeling studies. J. Mol. Model. 2003;9:329–336. doi: 10.1007/s00894-003-0154-9. [DOI] [PubMed] [Google Scholar]
- 27.López de Victoria A., Tamamis P., Morikis D. Insights into the structure, correlated motions, and electrostatic properties of two HIV-1 gp120 V3 loops. PLoS ONE. 2012;7:e49925. doi: 10.1371/journal.pone.0049925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley O., Klasse P.J., Moore J.P. V3: HIV’s switch-hitter. AIDS Res. Hum. Retroviruses. 2005;21:171–189. doi: 10.1089/aid.2005.21.171. [DOI] [PubMed] [Google Scholar]
- 29.Chandramouli B., Chillemi G., Desideri A. Importance of V3 loop flexibility and net charge in the context of co-receptor recognition. A molecular dynamics study on HIV gp120. J. Biomol. Struct. Dyn. 2012;29:879–891. doi: 10.1080/07391102.2012.10507416. [DOI] [PubMed] [Google Scholar]
- 30.Chandramouli B., Chillemi G., Desideri A. Structural dynamics of V3 loop with different electrostatics: implications on co-receptor recognition: a molecular dynamics study of HIV gp120. J. Biomol. Struct. Dyn. 2013;31:403–413. doi: 10.1080/07391102.2012.703068. [DOI] [PubMed] [Google Scholar]
- 31.López de Victoria A., Kieslich C.A., Morikis D. Clustering of HIV-1 subtypes Based on gp120 V3 loop electrostatic properties. BMC Biophys. 2012;5:3. doi: 10.1186/2046-1682-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Napier K.B., Wang Z.X., Trent J.O. CCR5 interactions with the variable 3 loop of gp120. J. Mol. Model. 2007;13:29–41. doi: 10.1007/s00894-006-0117-z. [DOI] [PubMed] [Google Scholar]
- 33.Morikis D., Rizos A.K., Krambovitis E. Electrostatic modeling of peptides derived from the V3-loop of HIV-1 gp120: implications of the interaction with chemokine receptor CCR5. Int. J. Mol. Med. 2007;19:343–351. [PubMed] [Google Scholar]
- 34.Xiang S.H., Pacheco B., Sodroski J. Characterization of a dual-tropic human immunodeficiency virus (HIV-1) strain derived from the prototypical X4 isolate HXBc2. Virology. 2013;438:5–13. doi: 10.1016/j.virol.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan Y., Ma B., Nussinov R. Characterization of the conformational state and flexibility of HIV-1 glycoprotein gp120 core domain. J. Biol. Chem. 2004;279:30523–30530. doi: 10.1074/jbc.M404364200. [DOI] [PubMed] [Google Scholar]
- 36.Pan Y., Ma B., Nussinov R. CD4 binding partially locks the bridging sheet in gp120 but leaves the β2/3 strands flexible. J. Mol. Biol. 2005;350:514–527. doi: 10.1016/j.jmb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Liao H.X., Lynch R., Haynes B.F. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W., Godzik A. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 39.Veldkamp C.T., Seibert C., Volkman B.F. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi Y., Deane C.M. FREAD revisited: accurate loop structure prediction using a database search algorithm. Proteins. 2010;78:1431–1440. doi: 10.1002/prot.22658. [DOI] [PubMed] [Google Scholar]
- 41.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansmann U. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997;281:140–150. [Google Scholar]
- 43.Sugita Y., Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999;314:141–151. [Google Scholar]
- 44.Nymeyer H., Gnanakaran S., García A.E. Atomic simulations of protein folding, using the replica exchange algorithm. Methods Enzymol. 2004;383:119–149. doi: 10.1016/S0076-6879(04)83006-4. [DOI] [PubMed] [Google Scholar]
- 45.Pieridou G., Avgousti-Menelaou C., Hayes S.C. UV resonance Raman study of TTR(105–115) structural evolution as a function of temperature. J. Phys. Chem. B. 2011;115:4088–4098. doi: 10.1021/jp107519b. [DOI] [PubMed] [Google Scholar]
- 46.Haberthür U., Caflisch A.J. FACTS: fast analytical continuum treatment of solvation. J. Comput. Chem. 2008;29:701–715. doi: 10.1002/jcc.20832. [DOI] [PubMed] [Google Scholar]
- 47.Tamamis P., Adler-Abramovich L., Archontis G. Self-assembly of phenylalanine oligopeptides: insights from experiments and simulations. Biophys. J. 2009;96:5020–5029. doi: 10.1016/j.bpj.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamamis P., Kasotakis E., Archontis G. Amyloid-like self-assembly of peptide sequences from the adenovirus fiber shaft: insights from molecular dynamics simulations. J. Phys. Chem. B. 2009;113:15639–15647. doi: 10.1021/jp9066718. [DOI] [PubMed] [Google Scholar]
- 49.Tamamis P., Archontis G. Amyloid-like self-assembly of a dodecapeptide sequence from the adenovirus fiber shaft: perspectives from molecular dynamics simulations. J. Non-Cryst. Solids. 2011;357:717–722. [Google Scholar]
- 50.Brooks B.R., Brooks C.L., 3rd, Karplus M.J. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Comeau S.R., Gatchell D.W., Camacho C.J. CLUSPRO: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics. 2004;20:45–50. doi: 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- 52.Im W., Lee M.S., Brooks C.L., 3rd Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 53.Im W., Feig M., Brooks C.L., 3rd An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michino M., Chen J., Brooks C.L., 3rd FoldGPCR: structure prediction protocol for the transmembrane domain of G protein-coupled receptors from class A. Proteins. 2010;78:2189–2201. doi: 10.1002/prot.22731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seeber M., Cecchini M., Caflisch A. WORDOM: a program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- 56.Pierce B.G., Hourai Y., Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE. 2011;6:e24657. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000;18:113–135. [Google Scholar]
- 58.Gilson M.K., Zhou H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomed. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 59.Zoete V., Irving M.B., Michielin O. MM-GBSA binding free energy decomposition and T cell receptor engineering. J. Mol. Recognit. 2010;23:142–152. doi: 10.1002/jmr.1005. [DOI] [PubMed] [Google Scholar]
- 60.Gohlke H., Case D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 2004;25:238–250. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 61.Page C.S., Bates P.A. Can MM-PBSA calculations predict the specificities of protein kinase inhibitors? J. Comput. Chem. 2006;27:1990–2007. doi: 10.1002/jcc.20534. [DOI] [PubMed] [Google Scholar]
- 62.Tamamis P., López de Victoria A., Archontis G. Molecular dynamics in drug design: new generations of compstatin analogs. Chem. Biol. Drug Des. 2012;79:703–718. doi: 10.1111/j.1747-0285.2012.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamamis P., Morikis D., Archontis G. Species specificity of the complement inhibitor compstatin investigated by all-atom molecular dynamics simulations. Proteins. 2010;78:2655–2667. doi: 10.1002/prot.22780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamamis P., Pierou P., Archontis G. Design of a modified mouse protein with ligand binding properties of its human analog by molecular dynamics simulations: the case of C3 inhibition by compstatin. Proteins. 2011;79:3166–3179. doi: 10.1002/prot.23149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 66.Oberlin E., Amara A., Moser B. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 67.Bleul C.C., Farzan M., Springer T.A. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 68.Choi W.T., Kumar S., An J. A novel synthetic bivalent ligand to probe chemokine receptor CXCR4 dimerization and inhibit HIV-1 entry. Biochemistry. 2012;51:7078–7086. doi: 10.1021/bi2016712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bozek K., Lengauer T., Domingues F.S. Analysis of physicochemical and structural properties determining HIV-1 coreceptor usage. PLOS Comput. Biol. 2013;9:e1002977. doi: 10.1371/journal.pcbi.1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cocchi F., DeVico A.L., Lusso P. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 1996;2:1244–1247. doi: 10.1038/nm1196-1244. [DOI] [PubMed] [Google Scholar]
- 71.Bellows M.L., Taylor M.S., Floudas C.A. Discovery of entry inhibitors for HIV-1 via a new de novo protein design framework. Biophys. J. 2010;99:3445–3453. doi: 10.1016/j.bpj.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bellows-Peterson M.L., Fung H.K., Woodruff T.M. De novo peptide design with C3a receptor agonist and antagonist activities: theoretical predictions and experimental validation. J. Med. Chem. 2012;55:4159–4168. doi: 10.1021/jm201609k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.