

Abstract
Integrin αIIbβ3 is a member of the integrin family of transmembrane proteins present on the plasma membrane of platelets. Integrin αIIbβ3 is widely known to regulate the process of thrombosis via activation at its cytoplasmic side by talin and interaction with the soluble fibrinogen. It is also reported that three groups of interactions restrain integrin family members in the inactive state, including a set of salt bridges on the cytoplasmic side of the transmembrane domain of the integrin α- and β-subunits known as the inner membrane clasp, hydrophobic packing of a few transmembrane residues on the extracellular side between the α- and β-subunits that is known as the outer membrane clasp, and the key interaction group of the βA domain (located on the β-subunit head domain) with the βTD (proximal to the plasma membrane on the β-subunit). However, molecular details of this key interaction group as well as events that lead to detachment of the βTD and βA domains have remained ambiguous. In this study, we use molecular dynamics models to take a comprehensive outside-in and inside-out approach at exploring how integrin αIIbβ3 is activated. First, we show that talin’s interaction with the membrane-proximal and membrane-distal regions of integrin cytoplasmic-transmembrane domains significantly loosens the inner membrane clasp. Talin also interacts with an additional salt bridge (R734-E1006), which facilitates integrin activation through the separation of the integrin’s α- and β-subunits. The second part of our study classifies three types of interactions between RGD peptides and the extracellular domains of integrin αIIbβ3. Finally, we show that the interaction of the Arg of the RGD sequence may activate integrin via disrupting the key interaction group between K350 on the βA domain and S673/S674 on the βTD.
Introduction
Integrins are a family of extracellular matrix (ECM) binding proteins that consist of 25 heterodimeric members. Each integrin molecule is composed of a α and a β noncovalently associated subunits. Each integrin subunit consists of a large extracellular domain (ectodomain), a single-pass transmembrane helix, and a short cytoplasmic tail (1,2). Integrins play a host of crucial roles in a variety of cell functions, including linking focal adhesion assemblies to the ECM, facilitating cell migration, transmitting signals bidirectionally, and controlling cell growth, differentiation, and apoptosis (2–6). Additionally, clustering of integrins is known to be a critical regulator of the focal adhesion size (7–9). Integrin αIIbβ3, which is the major integrin type expressed on the surface of platelets, plays a critical role in platelet aggregation and blood clotting (10,11). Therefore, integrin αIIbβ3 malfunction is marked by several diseases such as thrombosis in myocardial infarction, ischemic stroke, peripheral arterial disease, and Glanzmann thrombasthenia bleeding disorder (11,12). Integrin αIIbβ3 is activated via inside-out and/or outside-in signaling mechanisms, and these two pathways are reported to be discrete while having mutual effects on each other (1,2,13).
The inside-out activation of integrins is defined as the process of activation that is triggered by a cytoplasmic signal and leads to increased affinity of the integrin head domain for the ligand. Inside-out activation processes modulate integrin αIIbβ3 affinity for extracellular ligands (i.e., fibrinogen) through binding of key cytoplasmic proteins (see Fig. 1). Among the various proteins that bind integrins and link them to the actin cytoskeleton, talin and kindlin are reported to activate integrin via associating with the cytoplasmic tail of the integrin β-subunit (4,14). The most well-known inside-out activation cascade for integrin αIIbβ3 is triggered by binding of cleaved thrombin to proteinase-activated receptor 1 (PAR1) located on the plasma membrane of human platelets. This binding leads to phospholipid hydrolysis, which results in the generation of inositol trisphosphate (IP3) and diacylglycerol (DAG), and an increase in cytosolic free Ca2+. The elevated concentration of Ca2+ and DAG, which in turn activates CALDAG-GEFl and protein kinase C (PKC), converts RAP1 from a GDP-bound to a membrane-attached, GTP-bound form. Activation of RAP1 leads to recruitment of its effectors, RAP1-GTP-interacting adaptor molecule (RIAM), and its binding partner, talin 1, to the plasma membrane. This facilitates access of talin to the integrin β3 tail and talin-induced activation of integrin αIIbβ3 (4). Two major interactions attach the transmembrane domains of the α- and β-subunits: the outer membrane clasp (OMC) embedded within the lipid bilayer and the inner membrane clasp (IMC) located near the membrane-proximal (MP), cytoplasmic tail between the integrin subunits (2). Upon release of these clasps, the transmembrane-cytoplasmic domains of integrin are separated and activated, causing the extracellular domains to become more receptive to ECM ligand binding. Binding of the integrin β3 subunit to talin-1 is one of the major factors that disrupts the interactions between the two integrin subunits, separating the transmembrane-cytoplasmic domains (14). The current study provides molecular-resolution simulations that show how talin head domain binding to the transmembrane-cytoplasmic domain of the integrin β3 subunit mechanically loosens the IMC.
Figure 1.
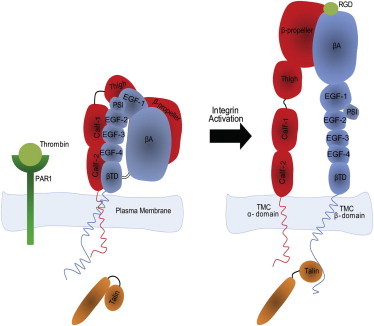
A schematic of one of the main pathways of integrin αIIbβ3 inside-out activation. Binding of thrombin to PAR1 causes separation of the talin head and tail domains. Consequently, the talin head domain associates with the β3 subunit cytoplasmic domain, activating integrin αIIbβ3.
On the other hand, the outside-in activation is identified by interactions of the integrin extracellular head domain with the ECM ligand or divalent cations that lead to partial unfolding of the extracellular domain and separation of integrin subunits. The integrin β3 subunit is known to be the major regulator of activation for integrin αllbβ3 and αvβ3 (15,16). Binding of divalent cations with the integrin head domain (i.e., βA and β-propeller domains on the extracellular side) is reported to determine the integrin activation state. There exist three divalent metal ion binding sites on the integrin βA domain. The first one, termed as the metal ion-dependent adhesion site (MIDAS) is coordinated with Mg2+, although the two other cation binding sites, called the adjacent to MIDAS (ADMIDAS) and ligand-induced metal binding site (LIMBS) located on the opposite sides of MIDAS, are coordinated with Ca2+ (15,17,18). High concentrations of Ca2+ reinforce the bent, inactive state of integrin αvβ3, whereas in the presence of Mn2+, a cation that activates integrins, most integrin αIIbβ3 molecules observed by electron microscopy were in an extended conformation (19). It is proposed by some researchers that the RGD ligand and divalent cation share a common binding pocket on the integrin β3 head domain on the interface of the βA and β-propeller domains (20,21). Importantly, external force has also been shown to directly activate certain types of integrin and prolong their ligand-bound lifetime (20,21). Last but not least, the integrin head domain interaction with the ligand at the metal ion-dependent binding sites is shown to activate integrins though its details are not yet fully agreed upon. Furthermore, whether an interaction between the CD loop of the βTD and the β6-α7 loop of the βA domains regulates integrin activation remains highly controversial (22,23). Hence, we explored potential effects of RGD-binding to integrin αllbβ3 through positioning two soluble RGD peptides proximal to the metal ion-dependent binding sites and one near the βA-βTD domains interface.
Two mechanistic models are widely invoked to describe the behavior of integrins: the switchblade and the dead-bolt model (24). According to the switchblade model, integrins take on three distinct conformations, corresponding to their inactive, active, and ligand-bound states. The inactive state of integrin is featured by a bent-over configuration wherein the molecule shows a low affinity for ECM ligands (see Fig. 1) (24). Furthermore, in the inactive state, IMCs and OMCs hold the transmembrane domains of integrin together (1). Integrin activation results in a stretched conformation and breakage of the IMCs and OMCs, most likely via presence of a competing molecule (e.g., talin and kindlin) that occupies these binding sites on the transmembrane domain of the integrin β-subunit (2,4). Activated integrin is known to have a significantly higher affinity for ECM ligands (19). Finally, the ligand-bound state takes place when the integrin head domain engages in an interaction with an ECM molecule (e.g., fibronectin and fibrinogen). The ligand-bound conformation differs from the nonbonded active state in that the integrin head domain introduces an opening between the α- and β-subunits when in the ligand-bound state (23,25). Alternatively, the deadbolt model suggests a significantly smaller conformational change, wherein a hairpin loop between β-strands C and D of the β3 tail domain (i.e., the CD loop), located on the β-subunit proximal to the plasma membrane, functions as a dead bolt. Upon release of the CD loop, the integrin molecule is activated, as the bonding between the βTD (proximal to the plasma membrane) and the βA domains on the integrin head domain is lost (23,26,27).
Despite numerous experimental studies conducted to shed light on the elaborate mechanism of integrin αllbβ3 activation from inside-out as well as outside-in perspectives, the atomistic details of either of these mechanisms have remained ambiguous (4,13). In this study, we developed molecular dynamics (MD) models to probe details of inside-out and outside-in activation of integrin αIIbβ3 via integrin-talin interactions. It has been shown previously that outside-in signaling pathways of integrin αIIbβ3 are spatially distinct from those of inside-out activation in platelets (13). Hence, this study chose to dissect two of the most established pathways for integrin αIIbβ3 activation in molecular details: Talin binding to the cytoplasmic domain of the β3 subunit to represent the inside-out activation, and RGD binding to the ectodomain to simulate the outside-in activation. First, we estimated the strength of the integrin transmembrane bonds (i.e., IMCs and OMCs) and showed that the presence of the talin head domain bound to the cytoplasmic domain of the integrin β-subunit markedly loosens the IMC such that a lower external force is required to separate the transmembrane domains of the α- and β-subunits. We focused our attention on the talin-integrin β3 interaction as the last step of a major cascade of events that leads to integrin activation to show, in molecular details, the contribution of this critical event in dissociating the integrin transmembrane domains. Furthermore, using the full-length crystal structure of integrin including the ectodomain and transmembrane-cytoplasmic domains for the first time to the best of our knowledge, we identified a number of potential binding sites of the soluble RGD ligand to the integrin head domain. Based on these results, three likely mechanisms are proposed for integrin-ligand binding. Finally, we explored the dead-bolt model in atomic details, mapping out three key residues that act as the dead bolt.
Methods
Interactions of RGD peptides with full-length integrin
All MD simulations were carried out with the program NAMD (28), using CHARMM27 force field. The integrin αllbβ3 ectodomain (PDB ID: 3FCS chains A and B (29)) and the transmembrane-cytoplasmic domains (PDB ID: 2KNC (46)) were obtained from the Protein Data Bank (PDB) and combined to build the full-length integrin αIIbβ3 structure (except residues 954–959 of the α-subunit and 684–690 of the β-subunit), assuming covalent bonds between residues C959 and G955 on the α-subunit and G684 and G690 on the β-subunit. The integrin structure was embedded in a patch of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayer with dimensions of 152 × 113 Å, using the software VMD (30). In all simulations, lipid chains overlapping with the transmembrane domain were removed afterward. PDB files of three RGD peptides were manually produced and positioned in proximity of the integrin head domain as shown in Fig. 2. The system was solvated afterward in a water box with an ionic KCl concentration of 150 mM (31).
Figure 2.
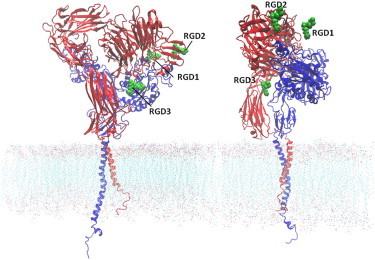
Crystal structure of integrin αIIbβ3 embedded in the plasma membrane. Three RGD peptides (green) are positioned in potential binding regions of integrin. The only RGD sequence that interacted with the integrin molecule was the one located near the βTD-βA pocket (RGD3). The αIIb and β3 subunits are depicted in red and blue, respectively.
The pressure and temperature of the system were held constant at 1 atm and 310 K, respectively, using Langevin’s piston and Hoover’s method (28). The time step was taken as 2 fs. The cutoff distance for nonbonded interactions was 1.2 nm. For all simulations, the particle mesh Ewald method was used for electrostatic force calculations (28). The cutoff distance for nonbonded interactions was 1.2 nm. The hydrogen atom bond length was constrained using the SHAKE method. The SHAKE method fixes bond lengths between large atoms and hydrogen atoms, preventing unnecessary calculation of irrelevant interactions (32). The system was minimized, at 2000 steps, and equilibrated six times with RGD peptides present and once with no RGD peptide in the system as control, each time for ∼13 ns.
Integrin-talin binding
The crystal structure of the transmembrane and cytoplasmic domains of integrin αIIbβ3 was embedded in a patch of POPC. The first step in our simulations was to induce the talin-1 FERM head domain to bind to integrin αIIbβ3, which are known to bind to each other (33,34). A model was created, using the molecular graphics software VMD, with the transmembrane-cytoplasmic regions of integrin αIIbβ3 (PDB ID: 2KNC (35)) embedded in a POPC lipid bilayer (30). The Talin-1 FERM head domain (PDB ID: 3IVF (36)) was placed with the F3 (PTB-like) domain, facing the integrin β3 subunit with a 27Å distance between the center of mass of the F3 domain (L320) to β3 residue W739. The system was solvated, then neutralized and ionized to a concentration of 150 mM NaCl. The system underwent minimization (2000 steps) and equilibration for 7 ns, before each center of mass in the F0–F3 talin domains was given a nudge for 1 ns (k = 0.07 kcal/mol/Å2; velocity = 17.5 Å/ns) in the direction that minimizes the mentioned 27 Å distance. Following that, the system was allowed 28 ns to equilibrate. At the end of the total 36 ns, we nudged the F0 domain of talin for 1 ns (k = 0.06 kcal/mol/Å2, velocity = 12.5 Å/ns) to tilt the F3 domain of talin toward the integrin β3 subunit. This was performed to promote the MP interactions between F727 and F730 on the β3 subunit to the S1-S2 loop of talin F3 (33). Following that, the system was equilibrated for 9.5 ns.
Simulation and analysis of integrin-talin interactions using full-length integrin structure
Having obtained a full-length integrin αIIbβ3 structure, we placed it inside a membrane, solvated and ionized it with 150 mM NaCl, and ran a minimization and equilibration simulation for 6 ns. Next, with our equilibrated full-length integrin structure, a similar model consisting of integrin and talin-1 FERM head domain was built, with a similar orientation and initial distance maintained between the integrin β3 subunit and F3 domain of talin-1. This model was minimized and equilibrated for 4 ns, 10 times to get a random sample of velocities and trajectories. From the simulations, we measured the final distance (at ∼4 ns) between R734-E1006, between the αIIb and β3 extracellular domains, between the βTD and βA domains, and between the center of masses of selected regions of the β3 subunit of integrin and talin-1 F3 domain. Finally, the Spearman’s tests of dependence were performed to further pinpoint which region of the β3 subunit tail is correlated to the weakening of this additional interaction and whether the degree of talin binding to the integrin β3 subunit affected integrin’s extracellular conformation. (Details for the comparison of the IMC interactions and other results in integrin versus integrin-talin systems are included in the Supporting Material.)
Results
Interactions between the soluble RGD and integrin potentially activates integrin αIIbβ3
To probe likely binding sites of the soluble ligand, RGD sequences were placed at three locations proximal to the integrin head domain previously reported as potential binding spots of integrin for fibrinogen (15,18,23). Depicted in Fig. 2 are locations of the three soluble RGD ligands relative to the integrin molecule. Two RGD peptides were positioned in the vicinity of the interface between the β-propeller domain of the α-subunit and the βA domain of the β-subunit, whereas another RGD peptide was located in proximity of the pocket formed by the βA and βTD domains of the β-subunit. Fibrinogen is a large macromolecule with a low transformational and rotational diffusion coefficient (37). Therefore, identifying the potential binding sites of the fibrinogen on the integrin head domain requires an excessive amount of computational time, i.e., several seconds or even longer, which is beyond the timescale of current MD models. Hence, merely a functional part of the fibrinogen molecule, i.e., the RGD sequence, was located close to the integrin head domain and allowed to diffuse freely.
The integrin molecule embedded in the lipid bilayer was solvated in a water box, minimized in 2000 steps of 2 fs, and equilibrated for no less than 13 ns. The three RGD peptides diffused throughout the water box until they formed interactions at various regions on the integrin head domain. The simulation was repeated seven times, corresponding to a total of ∼104 ns. RGD peptide 3 (RGD3) interacted with various binding sites located on the integrin head domain and/or within the inner pocket formed by the bent molecule (see Fig. S1). However, RGD peptide 1 and 2 (RGD1 and RGD2) combined interacted with a more limited number of binding sites on the interface of the βA and β-propeller domains. Intriguingly, RGD3 was confined to the βA-βTD pocket region in all simulations, whereas RGD1 and RGD2 remained associated with the head domain only in half of the simulations. Interactive residues on each subunit are shown in Table S1.
Interactions that lasted for a minimum of ∼3 ns after their initial formation were called permanent bonds as opposed to those formed only intermittently, termed temporary bonds hereinafter. Depicted in Fig. S1 is the distance between interacting residues of integrin within the βA-βTD pocket region and RGD3. It is noteworthy that interactions between the RGD peptide and extracellular domain of integrin are mainly salt bridges, as the RGD peptide is equipped with a strong basic residue and a highly acidic one (i.e., Asp and Arg) with long side chains at its two ends that are separated by a small hydrophobic spacer (i.e., Gly). For this reason, all permanent bonds shown in Fig. S1 are of ∼4.5 Å bond length, which is the characteristic length of a salt bridge (38,39).
According to the nature of bonds in the simulated trajectories, integrin-RGD bindings were classified into three major groups; the first group (i.e., type1 bond) comprised interactions with only a single, permanent interaction between the RGD peptide and integrin as shown in Fig. S1 a and b. Permanent bonds were formed as a result of type1 bonds with minimal fluctuations in all cases. The bond distance between the Arg of RGD3 and D817 on the Calf2 domain of the α-subunit is presented in Fig. S1, whereas Fig. S1 b shows the bond distance between the Arg of RGD3 and D127 on the βA domain of the β-subunit.
The second type of binding that occurred in two of our simulations included bindings in which one side of the soluble RGD peptide interacted permanently with the βA domain on the integrin head domain, whereas the other end only showed temporary engagement with membrane proximal domains of integrin. In one simulation, the Arg side of the soluble RGD formed a permanent bond with E312 on the βA domain (see Fig. S1 c). This interaction served as an anchorage for the ligand while the other side interacted temporarily with K650 on the βTD. Another permanent interaction formed between the Arg of RGD3 and D232 of the β-propeller domain of the αIIb subunit, whereas the Asp side of RGD3 formed a temporary interaction with N313 on the βA domain (see Fig. S1 d).
The other, and the most complex, type of interactions involved one permanent binding site and two temporary interactions on the integrin molecule that competed with each other. This group of interactions featured one end of the free RGD peptide tethering to the β-subunit permanently with other short-term bindings also taking place at the other end of the RGD peptide. For instance, Fig. S1 e shows a permanent bond between the Arg of RGD3 and D628 on the Calf1 domain of the α-subunit (blue) as well as the temporary bonds between the Asp of RGD3 with R897 on the Calf1 domain of the α-subunit (green), and K600 on the linker region between the EGF-1 and βTD (red). Although partial detachments were seen in a few of the simulations, a full separation of the βTD and βA domains was only observed in one out of six RGD-included simulations, when RGD3 interacted with three binding sites as depicted in Fig. S1 f. A permanent, polar interaction (as shown in blue in Fig. S1 f) was formed between the Arg of RGD3 and E671 on the βTD. Importantly, it was observed that the electrostatic interactions of S673 and S674 on the βTD with K350 serve as a key for activation of integrin αIIbβ3, as depicted by Fig. 3. This simulation was run for ∼21 ns. The key residues on the βTD (i.e., S673 and S674) continued to interact with K350 until t = 7.5 ns (see Fig. 3 c). However, as the Arg of the RGD peptide reached the vicinity of S674 at t = 7.5 ns, the Arg of the RGD peptide competed with K350 for S674 and the key interaction was highly disturbed (Fig. 3 d). Finally, the key interaction was fully removed as the Asp of the RGD peptide engaged in a competitive interaction with K600 and K611, pulling the βTD away from the βA domain (Fig. 3 e). As a result of the RGD peptide replacing K350 on the βA domain, the βTD swung open, separating from the βA domain (see Fig. 3 a and Fig. 4 b). The open conformation of the βTD appeared to be stable insofar as the βTD remained open for another 4.5 ns until the end of the simulation, as shown in Fig. S2. Finally, we repeated the simulation one more time without any RGD peptides included. As expected, the βTD and βA domains remained attached in the absence of an RGD peptide.
Figure 3.
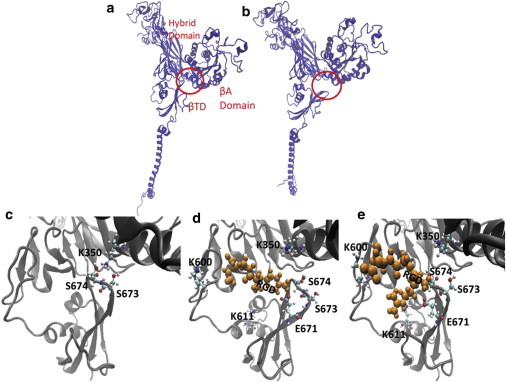
The Arg of the RGD peptide disrupts the βA-βTD interactions via competing with K350 on the βA domain for S673. (a) The βA and βTD are attached in the inactive, bent state of integrin. (b) The βTD swings open and separates apart from the βA domain, which leads to integrin activation. (c) Focusing further into the βTD-βA pocket shows the key interaction between K350 and S673/S674 that keeps integrin inactive. (d) The RGD peptide approaches S674 and interacts with it, disrupting the key interaction between the βTD and βA domains. (e) Although the Arg of the RGD peptide is bound to E671 and S674, the Asp of the RGD peptide switches back and forth between K600 and K611. This competitive interaction on the Asp side of the RGD peptide shifts the βTD further toward the EGF3/4 domains and apart from the βA domain.
Figure 4.
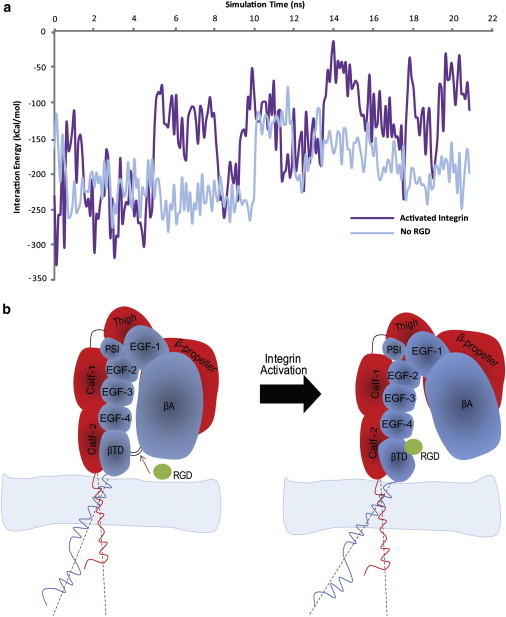
(a) The interaction energy between the integrin transmembrane-cytoplasmic domains of the α- and β-subunits as the simulation progresses for the integrin activation case as well as for the simulation with no RGD peptide. The initial 5 ns of the simulation shows the preequilibration phase, where both systems show similar interaction energies of ∼250 kCal/mol. This energy finally reaches a stable state at ∼200 kCal/mol in the absence of an RGD peptide. However, for the case where separation between the integrin βTD and βA domains occurs, the energy of interaction between the transmembrane-cytoplasmic domains of integrin significantly drops to ∼100 kCal/mol. This difference highlights a potential underlying mechanism that could loosen and eventually break apart the electrostatic interaction that holds the transmembrane-cytoplasmic domains of integrin together, as a result of an RGD-induced outside-in activation. (b) A schematic of integrin αIIbβ3 immediately after activation via RGD binding to the βTD. The integrin βTD and βA domains are bound by the key interaction group. Insertion of an RGD peptide into the βTD-βA pocket leads to a separation between the βTD and βA domains that somehow unbend integrin and loosens the electrostatic interaction between the integrin transmembrane domains via rotating the transmembrane domain of the β-subunit.
We observed that the dissociation of the βTD and βA domains caused the βTD to snap back, which induced a slight rotation in the β-subunit transmembrane domain (∼3° over the course of our simulations) in a lever-like motion. This rotation increased the angle between the α- and β-subunit transmembrane domains. We conducted energy analyses to measure the electrostatic energy between the α- and β-subunit transmembrane domains for the case where there was no RGD peptide as well as for the case where the integrin activation was observed. Interestingly, the electrostatic energy of interaction appeared to decrease dramatically after the βTD and βA domains separated (see Fig. 4 a). A tiny rotation of the long, β-subunit transmembrane domain increased the distance between acidic and basic residues on the α- and β-subunit transmembrane domains that considerably weakened the IMC.
Integrin-talin binding
To study the effect of talin on integrin activation, we first needed to create two separate conditions: the experimental condition, integrin with talin bound, and the control, integrin alone. To create our experimental condition, we promoted the binding of talin to integrin through a series of equilibration and steering simulations. After the 48 ns of equilibration and nudging, we observed binding between integrin and talin. Our results match that of the suggested mode of binding in the literature (2).
First, hydrophobic packing is evident between conserved residues and motifs, W739 and NPLY (744 to 747), on the integrin β3, and residues on the F3 domain of talin. The identified residues on the F3 domain of talin that bind to W739 on the integrin β3 subunit are the residues R358 and A360 on the S5 β-sheet on talin, which form a pocket for W739 (40). Additionally, the NPLY motif interacts with these S5 residues and additional residues, A389, Q390, A393, and I398, on the final H1 helix of the F3 domain (Fig. 5). Finally, after the nudges to tilt talin, as expected, F727 on the β3 subunit embedded itself into the talin S1-S2 loop region strand, consisting partly of residues K318, M319, and K320 (33).
Figure 5.
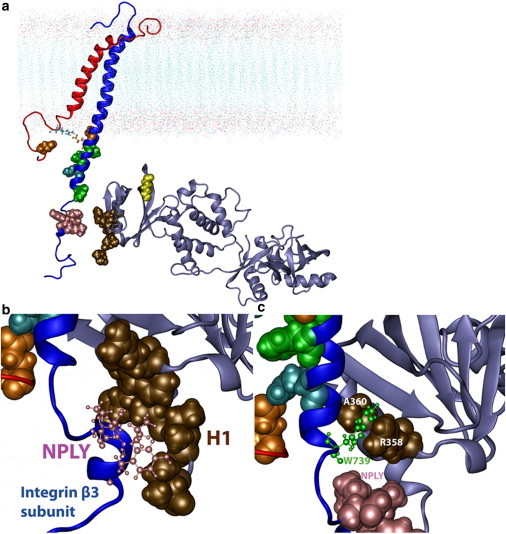
(a) Initial simulation position of the integrin αIIbβ3 transmembrane-cytoplasmic domains (PDB ID: 2K9J) and talin-1 FERM domain (PDB ID: 3IVF). For the new-cartoon representations, in red is the αIIb subunit of integrin, in blue is the β3 subunit of integrin, and in silver is the talin-1 FERM domain. For van der Waals sphere representations, in orange are the acidic residues E1006 (on αIIb) and D723 (on β3) of integrin, in cyan are the basic residues and R734 (on β3), in green are the residues F727, F730, and W739 on β3, in pink is the NPLY motif, in brown are residues R358, A360, A389, Q390, A393, and I398 on the talin F3 domain, and in yellow is K320 of the S1-S2 loop on the talin-1 F3 domain. Note that the atoms shown with the CPK representation in cyan (R995, αIIb) and orange (E726, β3) are tightly bound to hold the IMC together. These highlighted residues play an important role as shown by our simulations. (b and c) Talin-1 F3 domain (F0–F2 not shown) binding with the NPLY motif and W739 on the integrin β3 subunit. The left rendering (b) highlights NPLY motif binding to the H1 helix; whereas the right rendering (c) highlights W739 binding in the pocket formed by R358 and A360 in talin. The picture was taken 24 ns after the start of the simulation.
Effect of talin binding on integrin
Using the integrin-talin and integrin-alone models, we randomized the initial velocities in 12 repetitions, equilibrated for 5 ns, and pulled apart the IMC for 5 ns. The IMC of integrin changed its conformation upon binding talin. Three parameters were used to quantify the state of the IMC. The average distance between the carbon-α atoms of D723–R995 and E726–R995 shows that talin induces a bulge in the IMC even before the steered separation. The mean distances, as indicated by distance d in Fig. S3, for both the integrin-talin and integrin alone conditions were 9.8 Å and 13.5 Å, respectively, and this difference was significant using a permutated two-sample, one-tailed, t-test (p < 0.0003) (see the Supporting Material). As a result of this increase in distance, the interaction energy between the acidic β3 and basic αIIb IMC residues was also diminished (corresponding to less negative energy, see Fig. 6, a and b). The mean nonbonding energy for the integrin-talin systems was −93.4 kcal/mol and for the integrin-only system was −133.4 kcal/mol (Fig. 6), and again this difference was significant (p < 0.002). More interestingly, in all 12 simulations, the interaction energy between the IMC residues was never stronger than −129.7 kcal/mol in integrin-talin simulations, whereas the strongest observed energy was −163.6 kcal/mol in the integrin-only simulations.
Figure 6.
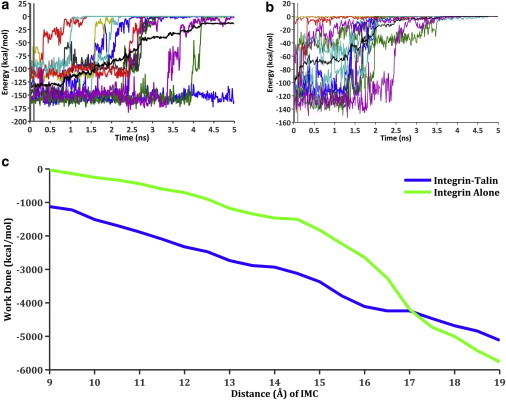
(a and b) Total nonbonding (van der Waals and electrostatic) energy between the IMC residues R995 on the αIIb, and D723 and E726 on the β3 subunits for the steered molecular dynamics (SMD) simulations, where the αIIb R995 carbon-α atom was pulled (k = 0.05 kcal/mol/Å2, velocity = 10 Å/ns) and the β3 D723 and E726 carbon-α atoms were fixed. The colored lines represent the raw data from each of the 12 trials per condition, whereas the dark black line indicates the average of these trials. The two vertical dark gray bars within the first 100 ps indicate the averaged region to obtain the initial nonbonding energy before performing SMD. The graph on the left (a) is the integrin-alone condition, whereas the right (b) is with talin bound to integrin. (c) Work done by the SMD simulations to separate the IMC, as a function of the amount of distance between the carbon-α atoms.
The last parameter investigated was the force required to break apart the IMC. As expected, with talin bound to integrin, the force required to break the IMC was lower than without talin. The mean force required, given the spring constant of k = 0.05 kcal/mol/Å2 and the constant-pulling velocity 10 Å/ns of the carbon-α atom of R995 while fixing the carbon-α atoms of D723/E726, was 224.6 ± 9.75 pN with talin bound and 249.8 ± 12.2 pN with integrin only (mean ± standard error). This difference was slightly significant (p < 0.04). These differences in initial energies, distances, and forces between the integrin-talin and integrin-alone conditions were confirmed with the difference in work of ∼1000 kcal/mol required to separate the IMC, up to 16 Å of separation (Fig. 6 c). In addition, with the integrin-talin simulations, a basic residue, K320, on talin’s S1-S2 loop interacted with the integrin β3 IMC residue D723 before and after the breakage of the IMC, with interaction energies of −75.2 kcal/mol and −84.5 kcal/mol, respectively.
One interesting difference between the integrin-talin versus the integrin-only system is that there is an additional salt bridge interaction between R734 on the β3 and E1006 on the αIIb subunit with the integrin-talin system (Fig. 6). Quantitatively, we observed that the distance between the most terminal carbons on R734 and E1006 was significantly different between the integrin-talin (5.25 Å) and the integrin only (16.1 Å) conditions, with p < 0.0003 (see the Supporting Material).
Effect of talin binding on full integrin structure
Using the full-length integrin structure, we assessed the effects of the F3 domain of talin binding to specific local regions on the integrin β3 cytoplasmic tail. Focusing on this additional interaction, R734-E1006, we found an overall negative correlation between the center of mass distances of the integrin β3 cytoplasmic tail to the talin F3 domain and distance between R734 and E1006. In other words, as the talin F3 domain approaches the integrin tail, the distance between R734 and E1006 enlarges, which also results in lower interaction energy. Upon closer analysis (see Table S2), a stronger correlation is observed when selecting the dependent variable as the center of mass of a subdomain of the cytoplasmic β3 subunit, specifically the region from the NPLY motif onward (termed NPLY-T762), to measure the distance to the center of mass of the F3 domain of talin (rho = −0.82, p = 0.0038). This suggests a greater significance in the membrane-distal binding of the β3 integrin to the F3 domain of talin-1. Similarly, we also examined the potential conformational changes in the integrin extracellular domain followed by the F3 domain of talin binding to the integrin β3 cytoplasmic tail. The degree of talin binding did not show any significant effects to alter integrin extracellular conformation (Table S3).
Discussion
Although integrin has been a subject of intensive study over the past two decades, molecular details of its function have remained ambiguous (3,23,24). As shown in Fig. 1, interaction of PAR1 with thrombin triggers the integrin αIIbβ3 activation pathway, which finally leads to thrombosis (1,4,41). Therefore, understanding how integrin αIIbβ3 functions paves the road for deciphering pathways that regulate thrombosis (41,42). We have successfully used MD techniques to model mechanosensitive focal adhesion proteins (7,43–49). Here, we performed a series of MD simulations to observe how exposing integrin αIIbβ3 to soluble RGD peptides as well as the talin head domain may affect the integrin structure. Integrin αIIbβ3 is a highly studied member of the integrin family, which is present in platelets, enabling them to interact with soluble fibrinogen (a specialized ligand of integrin αIIbβ3 that exists abundantly in the blood stream). It is widely known that soluble fibrinogen binds the head domain of integrin αIIbβ3, initiating the blood clotting process by forming a network of fibrinogen ligands with platelets trapped inside it (42,50,51).
Approaching integrin αIIbβ3 from the intracellular perspective, to our knowledge, we believe that we have uncovered new biomolecular, mechanistic details on the inside-out activation process by talin. Although it is known that talin is required for inside-out activation of integrin, the specific mechanism of how integrin is activated by talin is uncertain (52). The current working model suggests that talin binding to the cytoplasmic tail of the β3 subunit would occur first at the membrane-distal region, which then promotes a MP interaction to take place between talin and integrin β3 subunit. This MP interaction is where the mechanism of activation arises (53). Our data suggest a similar mechanism, in that β3 subunit F727 embedding in the talin S1-S2 loop may help the IMC salt bridge to loosen its grip because this allows the basic K320 residue to energetically compensate for the IMC salt bridge loss. Furthermore, in the timescale of our simulations, we did not observe significant conformational changes in the integrin extracellular domains though previous work suggested that breakage of the IMC salt bridge by talin is necessary but not sufficient to fully activate integrin (53).
Additionally, we observed a salt bridge interaction between the most membrane distal regions of the αIIb and β3 integrin subunits with residues E1006 and R734, respectively. Interestingly, this interaction appeared in two of the four separate simulation conditions, forming in the integrin-talin bound simulations using only the cytoplasmic-transmembrane domains of integrin, and in the integrin-alone simulations using the full-length integrin αIIbβ3. We make two propositions, the first of which states that this interaction may be an artifact of using only the cytoplasmic-transmembrane domains of integrin. Second, the default presence of this interaction using full-length integrin suggests that talin plays an even more important role in integrin activation. This artifact in the cytoplasmic-transmembrane simulations may cause the force and work required to separate the IMC under the integrin-talin conditions to be overestimated. Without this interaction, the integrin-talin conditions may be even more favorable toward IMC separation and integrin activation. More interestingly, it seems that the membrane-distal interactions of talin and integrin are important for more than solely providing an anchor for the MP interactions to weaken the IMC (53). Instead, we propose that the binding of talin to the membrane-distal region of the β3 subunit cytoplasmic tail weakens this salt bridge E1006-R734 interaction, which is the first step in integrin inside-out activation.
Integrin is also activated via association with an extracellular ligand, a process called outside-in activation. It is reported that integrin αIIbβ3 has a high affinity for the RGD sequence of fibrinogen (3,11,54). Interactions between integrin αIIbβ3 and fibrinogen are reported to occur on the interface of the β-propeller domain of the αIIb and the βA domain of the β3 subunit. In particular, RGD peptides are shown to associate with a region of the β3 defined by D109 and E171 as well as another region limited to S211 and G222 (20,21). Nonetheless, a number of studies showed that the integrin αIIbβ3 head domain interacts solely with a C-terminal region of fibrinogen (i.e., γC) rather than the RGD sequences on the fibrinogen α-subunit (15,18). To address this controversy making best use of the computational time, instead of modeling the entire fibrinogen molecule, we placed two RGD sequences at regions of the integrin αIIbβ3 head domain that had been reported to have high affinity for fibrinogen (3,15). An RGD peptide was positioned in the vicinity of the βTD-βA domain interface, another sensitive region that is suggested to contribute to integrin activation, to explore its potential interactions especially ones that could regulate integrin function. The small size of RGD peptides conferred upon them high diffusivity and capability to sweep the space in the vicinity of the sensitive areas of integrin αIIbβ3. We initiated our simulations with inactive integrin αIIbβ3, wherein the βA domain of the integrin head domain is attached to the MP βTD. This forms a pocket-like configuration between the integrin head domain and MP region, which is geometrically appropriate for the long, coiled-coil molecule of fibrinogen to insert into. Because the RGD motif of fibrinogen is located on the surface of the γ-subunit of fibrinogen, it has high potency for detecting RGD-sensitive regions of integrin αIIbβ3.
The association constant for the integrin-peptide interaction is
| (1) |
where [I], [P], and [IP] are concentrations of integrin, free RGD peptide, and integrin-RGD complex, respectively. The association constant is related to the free energy of association as follows:
| (2) |
where ΔG, kB, and T are the free energy of association per mol, Boltzmann constant, and temperature, respectively. Our simulations indicated that for the highly dynamic RGD binding sites in the βTD-βA pocket, 10 ns is a reasonable timescale for bonds to form and break down at least once (see Fig. S1). As the free energy per mol of a significant electrostatic bond in amino acids is typically taken as 5 kCal/mol (55,56), substituting that value for ΔG in Eq. 2 and solving at T = 310 K yields ka∼0.06 mol−1. According to the ergodic hypothesis in statistical thermodynamics, for a stochastic system, any average of thermodynamic parameters over the entire volume of the system at any instant in time equals the average of the same parameter taken over a long time span at a single point in the system (57). Hence, as a first approximation, we can assume the free concentrations of integrin and peptide (denoted as [I] and [P] in Eq. 1) are proportional to the sum of time spans that the system experiences the unbound state. On the other hand, the concentration of the complex (denoted by [IP] in Eq. 1) is proportional to the accumulative time period that the system spends in the bound state. Therefore, the minimum association constant for significant bond formations (ka = 0.06 mol−1) is corresponding to a binding time of at least 3 ns within a simulation scale of 10 ns. We defined bindings that remained associated for > ∼3 ns as permanent bonds and bindings with life spans any shorter than 3 ns as temporary bonds.
Our simulations highlighted several binding sites for the RGD peptide on integrin αIIbβ3, most of which were either inside or proximal to the βA-βTD pocket. Binding sites of integrin αIIbβ3 for RGD peptides in this region fell into three distinct categories, wherein permanent interactions always form at the Arg end of the RGD peptide. This could be attributed to the highly basic side chain of the Arg that facilitates formation of permanent salt bridges. The first category featured bonds formed between the Arg of the RGD peptide and integrin. Although this group included permanent bindings, these bindings did not contribute to detachment of the βA and βTD within our simulation time because they were not able to disturb the key interaction (i.e., K350 with S673 and S674). In the second type of interactions, the Arg of the RGD peptide formed a permanent bond with integrin αIIbβ3, whereas the Asp of the RGD peptide formed a temporary bond, which was highly fluctuating and unstable. Finally, the third and most complex type of RGD peptide-integrin interactions were composed of a permanent interaction of the Arg of the RGD peptide with integrin αIIbβ3 at one end and a competitive, temporary interaction of two other residues with the Asp of the RGD peptide at the other end. Within the timescale of our simulations (∼13 ns), one out of two observed, type3 interactions fully dethatched the βA domain from the βTD, which according to both switchblade and dead- bolt models is equivalent to integrin activation.
After ∼7.5 ns of equilibration and free diffusion of the RGD peptide in the vicinity of the βA-βTD pocket, as depicted in Fig. 3, the key βA-βTD domain interactions (i.e., K350 with S673 and S674) were disrupted as a result of the Arg of the RGD side chain taking over the OH-group on the side chain of S673. About 1 ns after the key interaction was disrupted, the βTD swung open and the conformational change continued as the Asp of the RGD peptide was engaged in two competing interactions with K600 on the EGF3-EGF4 domain linker region and K611 on the EGF4 domain of the β3 subunit (see Fig. 3 e). These competing interactions shifted the RGD peptide more toward the EGF3 and EGF4 domains, which furthered the distance between the βA and βTD domains. It is worthwhile mentioning that because Ser residues do not have significant avidity for Lys, the two Ser residues together still do not maintain K346 on the βA domain in a permanent salt bridge (i.e., a bond distance of ∼4 Å). Hence, this rather weak interaction combined with the strong basic identity of Arg side chains may explain how integrin αIIbβ3 is activated by a soluble, RGD-containing ligand (e.g., fibrinogen).
It has been shown that fibrinogen binds an extensive interface between the βA and β-propeller domains of the integrin αIIbβ3 head domain proximal to the metal ion-dependent sites (i.e., MIDAS and ADMIDAS), which leads to integrin activation (15,18,58). Aligned with previous studies, RGD1 and RGD2 interacted with the crevice region between the βA and β-propeller on the head domain (20,21). However, we did not observe any significant conformational changes induced in integrin as a result of RGD binding to the head domain. This is aligned with a more recent study that showed functional associations of fibrinogen with integrin occur at the RGD-lacking γC peptide of fibrinogen (18). Our study shed light on another important binding site for the RGD motif inside the βA-βTD pocket that activates integrin. Our simulations indicated that the pocket formed by the βTD and βA domains of integrin is home to several binding spots for the RGD peptide. Short-term interactions between these binding spots and the RGD peptide increase the residence time of the soluble RGD peptide near the pocket, which escalates odds of an interaction occurring between the RGD peptide and the key bonds between the βTD and βA domains (i.e., K350 with S673 and S674), which would lead to integrin activation via disrupting the key interactions.
Two major mechanisms are reported to unlock the integrin inhibitory interactions. Although the switchblade model proposes a drastic change, from bent to fully stretched conformation upon activation, the dead-bolt model suggests more local conformational changes in the molecule due to dissociation of the βTD and βA domains (13,23,34,42). The switchblade model suggests that the interaction of the integrin cytoplasmic domain with particular focal adhesion proteins leads to a 135° rotation of the integrin head piece relative to the tail piece. This rotation causes the βA-β-propeller interface to face away from the plasma membrane, thereby becoming available to interact with the ligand (13,19). The switchblade model is corroborated for different members of the integrin family by a number of previous studies. First, when recombinant integrin α5β1 was artificially constrained with disulfide bonds in a bent conformation it did not associate with immobilized ligands (59). For integrin αvβ3, a double mutation that held the head domain against the leg domains via a Cys bridge hindered activation (19). In addition, a significant increase in the protein Stokes radius has been recorded for cases where integrins were exposed to RGDFV peptides or Mn2+ (i.e., integrin activators) versus the case where they are in a Ca2+ buffer, which is an activation inhibitor (19). However, integrin αIIbβ3 activation paradigm appears to remain controversial, as detailed monoclonal antibody epitope maps for inactive and active integrin αIIbβ3 in vivo demonstrated similar compact forms consistent with the bent conformation (23,60). Furthermore, binding of different ligand mimics to integrin αIIbβ3 in intact platelets has shown distinct conformational changes in the receptor (61).
Another mechanistic explanation for integrin activation was given by the dead-bolt model. The dead-bolt model assumes that the elongated CD loop of the βTD locks the βA domain via forming bonds with it in the inactive state of the native structure, although the atomistic details of these interactions are not currently clear (23). It is important to note that the dead-bolt model does not necessarily exclude a subsequent ligand-induced switchblade-like swing-out. Interestingly enough, our simulations demonstrated that the disruption in the interactions between the βTD and βA domains could be a trigger for integrin activation, recognizing electrostatic interactions of K350 on the βA with S673 and S674 on the βTD as a potential regulatory lock.
We observed that the dissociation of the βTD and βA domains was succeeded by the βTD snap-back, which caused the β-subunit transmembrane domain to slightly rotate (∼3°) in a lever-like motion. This increase in the angle between the α- and β-subunit transmembrane domains is also reported by previous electron microscopy and biochemical data as a potential activating mechanism for integrin αIIbβ3 (21,52,54,62). However, another model is proposed for a number of other integrin family members (e.g., α5β1) that suggests the hinge region for the scissors-like motion of integrin, upon activation, is located on the interface of the α- and β-subunit head domains (59). We measured the electrostatic energy between the α- and β-subunit transmembrane domains for the simulation with no RGD peptides and compared it to the one in which the integrin activation had been observed. Intriguingly, the electrostatic energy of interaction appeared to decrease dramatically after the βTD and βA domains separated. A slight rotation of the long, transmembrane domain of the β-subunit could considerably increase the distance between charged residues on the α- and β-subunit transmembrane domains, which would weaken the IMC. Therefore, we believe RGD interaction with the βTD could be a potential trigger for the complete separation of the transmembrane domains. Although this set of data proposes a key interaction that might function as a regulating dead-bolt, the loosening of the transmembrane domain clasps as a result of the βTD-βA domain separation suggests subsequent, more drastic conformational changes, and explains how ligand-binding could lead to the transmembrane domain separation as proposed by the switchblade model. On the other hand, within the timescale of our simulations, we did not observe any significant conformational changes in the ectodomain nor did we see any decreases in the βA-βTD interaction energy upon talin binding with the β3 cytoplasmic tail. This suggests that outside-in activation via RGD association with the key residues embedded in the βA-βTD pocket occurs more rapidly than inside-out activation followed by talin binding. It is also noteworthy that distinct activation mechanisms and conformational changes might be at work for different combinations of integrin-ligand interactions.
Conclusion
The activated state of integrin is commonly characterized by three major dissociations: breaking of the IMC and OMC, and unbinding of the βA and βTD (1,4,23,63). This study attempted to explain the integrin αIIbβ3 function, via exploring how these key interaction groups are disrupted in specific pathways. Our observations of the transmembrane-cytoplasmic domains of integrin αIIbβ3 showed that the IMC is loosened with the presence of talin, most likely through MP interactions that result from the energetic compensation of the weakened IMC by K320 on the S1-S2 loop of talin. We also found, using the full-length crystal structure we built, that talin has a larger role in integrin activation by disrupting an additional stable clasp (R734-E1006) through the initial membrane-distal interactions that anchors talin to integrin (2). Furthermore, we used the full-length crystal structure of integrin αIIbβ3 to explore binding sites of soluble RGD ligands and their ability to activate integrin αIIbβ3 or maintain it in the activated state. We found three major groups of bindings with two specific binding sites corresponding to each group. According to our simulations, the major group of interactions that maintains integrin αIIbβ3 in the inactive, bent state occurs between K350 on the βA domain, and S673 and S674 on the βTD. Interestingly, we observed the full dissociation of the βTD and βA domains when this interaction group was disrupted and eventually removed as a result of a competition between the Arg of the RGD peptide with S674. Therefore, we proposed this molecular scenario as a potential trigger mechanism for outside-in activation of integrin αIIbβ3 by soluble RGD ligand.
Acknowledgments
Financial support by National Science Foundation through a CAREER award to M.R.K.M. (CBET 0955291) is gratefully acknowledged.
Fruitful discussions with members of the Molecular Cell Biomechanics Lab are highly appreciated.
Supporting Material
References
- 1.Lau T.L., Kim C., Ulmer T.S. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anthis N.J., Campbell I.D. The tail of integrin activation. Trends Biochem. Sci. 2011;36:191–198. doi: 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barczyk M., Carracedo S., Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shattil S.J., Kim C., Ginsberg M.H. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mofrad M.R.K., Kamm R.D. Cambridge University Press; Cambridge, New York: 2011. Cytoskeletal Mechanics: Models and Measurements in Cell Mechanics. [Google Scholar]
- 6.Mofrad M.R.K., Kamm R.D. Cambridge University Press; Cambridge, New York: 2010. Cellular Mechanotransduction: Diverse Perspectives from Molecules to Tissues. [Google Scholar]
- 7.Mehrbod M., Mofrad M.R. Localized lipid packing of transmembrane domains impedes integrin clustering. PLOS Comput. Biol. 2013;9:e1002948. doi: 10.1371/journal.pcbi.1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan J.L., Tien J., Chen C.S. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jamali Y., Jamali T., Mofrad M.R.K. An agent based model of integrin clustering: exploring the role of ligand clustering, integrin homo-oligomerization, integrin–ligand affinity, membrane crowdedness and ligand mobility. J. Comp. Phys. 2012;244:264–278. [Google Scholar]
- 10.Jones M.L., Harper M.T., Poole A.W. RGD-ligand mimetic antagonists of integrin alphaIIbbeta3 paradoxically enhance GPVI-induced human platelet activation. J. Thromb. Haemost. 2010;8:567–576. doi: 10.1111/j.1538-7836.2009.03719.x. [DOI] [PubMed] [Google Scholar]
- 11.Ma Y.Q., Qin J., Plow E.F. Platelet integrin alpha(IIb)beta(3): activation mechanisms. J. Thromb. Haemost. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- 12.Shattil S.J., Newman P.J. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 13.Zou Z., Chen H., Kahn M.L. Structure-function analysis reveals discrete beta3 integrin inside-out and outside-in signaling pathways in platelets. Blood. 2007;109:3284–3290. doi: 10.1182/blood-2006-10-051664. [DOI] [PubMed] [Google Scholar]
- 14.Vinogradova O., Velyvis A., Qin J. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 15.Xiao T., Takagi J., Springer T.A. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong J.P., Stehle T., Arnaout M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong J.P., Stehle T., Arnaout M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 18.Springer T.A., Zhu J., Xiao T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J. Cell Biol. 2008;182:791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takagi J., Petre B.M., Springer T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 20.D’Souza S.E., Haas T.A., Smith J.W. Ligand and cation binding are dual functions of a discrete segment of the integrin beta 3 subunit: cation displacement is involved in ligand binding. Cell. 1994;79:659–667. doi: 10.1016/0092-8674(94)90551-7. [DOI] [PubMed] [Google Scholar]
- 21.Plow E.F., Haas T.A., Smith J.W. Ligand binding to integrins. J. Biol. Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J., Boylan B., Springer T.A. Tests of the extension and deadbolt models of integrin activation. J. Biol. Chem. 2007;282:11914–11920. doi: 10.1074/jbc.M700249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong J.P., Stehle T., Arnaout M.A. New insights into the structural basis of integrin activation. Blood. 2003;102:1155–1159. doi: 10.1182/blood-2003-01-0334. [DOI] [PubMed] [Google Scholar]
- 24.Beglova N., Blacklow S.C., Springer T.A. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 2002;9:282–287. doi: 10.1038/nsb779. [DOI] [PubMed] [Google Scholar]
- 25.Jin M., Andricioaei I., Springer T.A. Conversion between three conformational states of integrin I domains with a C-terminal pull spring studied with molecular dynamics. Structure. 2004;12:2137–2147. doi: 10.1016/j.str.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Chen W., Lou J., Zhu C. Molecular dynamics simulations of forced unbending of integrin α(v)β3. PLOS Comput. Biol. 2011;7:e1001086. doi: 10.1371/journal.pcbi.1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnaout M.A., Mahalingam B., Xiong J.P. Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 28.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J., Luo B.H., Springer T.A. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 31.Lodish H.F., Berk A., Matsudaira P. W. H. Freeman; New York: 2008. Molecular Cell Biology. [Google Scholar]
- 32.Krautler V., Van Gunsteren W.F., Hunenberger P.H. A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- 33.Wegener K.L., Partridge A.W., Campbell I.D. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 34.Tadokoro S., Shattil S.J., Calderwood D.A. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 35.Yang J., Ma Y.Q., Qin J. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc. Natl. Acad. Sci. USA. 2009;106:17729–17734. doi: 10.1073/pnas.0909589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elliott P.R., Goult B.T., Barsukov I.L. The Structure of the talin head reveals a novel extended conformation of the FERM domain. Structure. 2010;18:1289–1299. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kollman J.M., Pandi L., Doolittle R.F. Crystal structure of human fibrinogen. Biochemistry. 2009;48:3877–3886. doi: 10.1021/bi802205g. [DOI] [PubMed] [Google Scholar]
- 38.Anslyn E.V., Dougherty D.A. University Science; Sausalito, CA: 2006. Modern Physical Organic Chemistry. [Google Scholar]
- 39.Kumar S., Nussinov R. Close-range electrostatic interactions in proteins. ChemBioChem. 2002;3:604–617. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 40.García-Alvarez B., de Pereda J.M., Liddington R.C. Structural determinants of integrin recognition by talin. Mol. Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 41.Furie B., Furie B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008;359:938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 42.Leng L.J., Kashiwagi H., Shattil S.J. RhoA and the function of platelet integrin alphaIIbbeta3. Blood. 1998;91:4206–4215. [PubMed] [Google Scholar]
- 43.Golji J., Collins R., Mofrad M.R. Molecular mechanics of the alpha-actinin rod domain: bending, torsional, and extensional behavior. PLOS Comput. Biol. 2009;5:e1000389. doi: 10.1371/journal.pcbi.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golji J., Lam J., Mofrad M.R. Vinculin activation is necessary for complete talin binding. Biophys. J. 2011;100:332–340. doi: 10.1016/j.bpj.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golji J., Mofrad M.R. A molecular dynamics investigation of vinculin activation. Biophys. J. 2010;99:1073–1081. doi: 10.1016/j.bpj.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Golji J., Wendorff T., Mofrad M.R. Phosphorylation primes vinculin for activation. Biophys. J. 2012;102:2022–2030. doi: 10.1016/j.bpj.2012.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shams H., Golji J., Mofrad M.R. A molecular trajectory of α-actinin activation. Biophys. J. 2012;103:2050–2059. doi: 10.1016/j.bpj.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mofrad M.R., Golji J., Kamm R.D. Force-induced unfolding of the focal adhesion targeting domain and the influence of paxillin binding. Mech. Chem. Biosyst. 2004;1:253–265. [PubMed] [Google Scholar]
- 49.Golji J., Mofrad M.R. The interaction of vinculin with actin. PLOS Comput. Biol. 2013;9:e1002995. doi: 10.1371/journal.pcbi.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parise L.V. Integrin alpha(IIb)beta(3) signaling in platelet adhesion and aggregation. Curr. Opin. Cell Biol. 1999;11:597–601. doi: 10.1016/s0955-0674(99)00018-6. [DOI] [PubMed] [Google Scholar]
- 51.Shattil S.J., Kashiwagi H., Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91:2645–2657. [PubMed] [Google Scholar]
- 52.Adair B.D., Yeager M. Three-dimensional model of the human platelet integrin alpha IIbbeta 3 based on electron cryomicroscopy and x-ray crystallography. Proc. Natl. Acad. Sci. USA. 2002;99:14059–14064. doi: 10.1073/pnas.212498199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Askari J.A., Buckley P.A., Humphries M.J. Linking integrin conformation to function. J. Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hantgan R.R., Paumi C., Weisel J.W. Effects of ligand-mimetic peptides Arg-Gly-Asp-X (X = Phe, Trp, Ser) on alphaIIbbeta3 integrin conformation and oligomerization. Biochemistry. 1999;38:14461–14474. doi: 10.1021/bi9907680. [DOI] [PubMed] [Google Scholar]
- 55.Loftus J.C., Liddington R.C. Cell adhesion in vascular biology. New insights into integrin-ligand interaction. J. Clin. Invest. 1997;99:2302–2306. doi: 10.1172/JCI119408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyce S.E., Mobley D.L., Shoichet B.K. Predicting ligand binding affinity with alchemical free energy methods in a polar model binding site. J. Mol. Biol. 2009;394:747–763. doi: 10.1016/j.jmb.2009.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carey V.P. Cambridge University Press; Cambridge, UK; New York: 1999. Statistical Thermodynamics and Microscale Thermophysics. [Google Scholar]
- 58.Kamata T., Tieu K.K., Takada Y. Amino acid residues in the alpha IIb subunit that are critical for ligand binding to integrin alpha IIbbeta 3 are clustered in the beta-propeller model. J. Biol. Chem. 2001;276:44275–44283. doi: 10.1074/jbc.M107021200. [DOI] [PubMed] [Google Scholar]
- 59.Takagi J., Erickson H.P., Springer T.A. C-terminal opening mimics ‘inside-out’ activation of integrin alpha5beta1. Nat. Struct. Biol. 2001;8:412–416. doi: 10.1038/87569. [DOI] [PubMed] [Google Scholar]
- 60.Calzada M.J., Alvarez M.V., Gonzalez-Rodriguez J. Agonist-specific structural rearrangements of integrin alpha IIbbeta 3. Confirmation of the bent conformation in platelets at rest and after activation. J. Biol. Chem. 2002;277:39899–39908. doi: 10.1074/jbc.M205886200. [DOI] [PubMed] [Google Scholar]
- 61.Cierniewski C.S., Byzova T., Plow E.F. Peptide ligands can bind to distinct sites in integrin alphaIIbbeta3 and elicit different functional responses. J. Biol. Chem. 1999;274:16923–16932. doi: 10.1074/jbc.274.24.16923. [DOI] [PubMed] [Google Scholar]
- 62.Gottschalk K.E., Adams P.D., Kessler H. Transmembrane signal transduction of the alpha(IIb)beta(3) integrin. Protein Sci. 2002;11:1800–1812. doi: 10.1110/ps.4120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim M., Carman C.V., Springer T.A. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.