

Abstract
Most human cancers arise either from epithelial cells or their progenitors. Epithelial cells possess a distinctive apical–basal polarity and loss of polarity is frequently assumed to be a common feature of cancer progression. In particular, cancer cell dissemination to ectopic sites, and metastatic growth at those sites, is often considered to require a mesenchymal transition in which the transformed epithelial cells lose their apical–basal polarity. However, many cancers retain epithelial characteristics, and until recently there has been little conclusive evidence for an involvement of the cell polarity machinery in tumour growth and metastasis. In this article, we discuss evidence that polarity proteins can be potent invasion suppressors but that loss of epithelial character is not essential either for tumour growth and invasion, or metastatic colonization.
Keywords: oncogene, tumour suppressor, epithelial–mesenchymal transition
1. Introduction
Epithelial tissues consist of sheets of cells that adhere to one another and possess plasma membranes with distinct apical and basolateral domains. Stratified epithelia consist of multiple layers, whereas in simple, single-layered epithelia, the apical membranes are unattached to other cells and face lumens that connect directly or indirectly to the external environment. The sheets can invaginate or fold into tubes to create diverse structures that grow in extent through cell divisions in the plane of the epithelial sheet. The majority of our organs are built from epithelial cells and most human cancers arise from these cells or their precursors. Acquisition of oncogenic mutations, and/or loss of tumour suppressor genes disrupts not only growth control but also the ability of cells to communicate appropriately with their neighbours. For this reason, the transformed cells cannot organize correctly into tissue structures, even though each individual cell might retain many normal epithelial characteristics. Epithelial cells can also lose cell–cell communication and apical/basal polarity through a process called the epithelial–mesenchymal transition (EMT), which occurs during both normal development and in tumorigenesis [1]. Thus, cancer can be considered a disease of intercell behaviour.
Many epithelial tissues arise from, and are maintained by, stem cells that self-renew while generating daughter cells that can also differentiate into a variety of fates. Daughter cells are allocated to specific lineages, often through progenitor stages that have more limited plasticity and controlled by transcriptional programmes that are not yet fully understood. Epithelial cancer cells are also believed to arise from progenitors or stem cells [2], and the intratumoural heterogeneity that is found in many epithelial cancers likely occurs in part from defects in lineage allocation. Furthermore, tumour initiation in different populations along the differentiation pathway may give rise to distinct cancer subtypes. This is best understood in the breast, where different breast cancer molecular subtypes have gene expression profiles that resemble different populations of cells along the stem, progenitor and differentiated cell spectrum [3]. This is directly supported by the finding that basal-like breast cancers of women with BRCA1 mutations have an enriched luminal progenitor population [4] and that targeted deletion of Brca1 in luminal progenitors in mice generated tumours with a histology that resembles human BRCA1 breast cancers, whereas targeting other lineages did not [5].
Some stem cells give rise to differentiated daughters through asymmetric cell divisions, but it remains unclear whether all epithelial stem cells use this mechanism or not [6]. For example, while epidermal stem cells can divide asymmetrically, Lgr5+ intestinal epithelial stem cells use a population asymmetry rather than a cell-autonomous asymmetry during mitosis [6]. Defects in asymmetric cell divisions might also be important for the progression of some cancers. In Drosophila, failure of embryonic neuroblasts to divide asymmetrically results in hyperplasia [7]. Murine oligodendrocyte progenitor cells also normally undergo asymmetric cell divisions, a property that is lost in gliomas [8]. Moreover, isolated mammary stem cells have been reported to divide asymmetrically in spheroid suspension cultures, but similar cells divide symmetrically when isolated from p53-null or ErbB2 breast cancer mouse models [9]. Therefore, disruption of asymmetric cell divisions might contribute to enhanced tumorigenesis by increasing the number of highly plastic stem cells.
2. Par polarity proteins in cancer progression
Neuronal stem cells in Drosophila embryos use a conserved set of polarity genes (par genes) to drive asymmetric mitoses, and the same set of genes is also required for the apical/basal polarity of epithelial cells throughout the animal kingdom [7,10]. This group of proteins includes Par1, 3, 4, 5 and 6, plus atypical protein kinase C (aPKC), and the Cdc42 GTPase. However, it remains ambiguous as to whether the par genes are necessary for asymmetric stem cell divisions in mammals, in the few cases where such divisions have been documented. For example, Par3 plays a role in radial glial progenitors [11] but is not required for stem cell maintenance in mammary glands [12], and aPKC is entirely dispensable for haematopoietic stem cell function [13]. It has also been unclear whether the Par genes play any role in cancer initiation or progression, despite the general assumption that defects in cell polarity occur during epithelial tumorigenesis.
Until recently, only Par4, a protein kinase also known as LKB1, had been identified as a bona fide tumour suppressor in mammals [14] and it remains uncertain whether carcinogenesis in patients with mutant LKB1 is caused by loss of its polarity function or is instead a result of perturbations in other downstream signalling processes, including metabolism. Par4/LKB1 is a master kinase that can phosphorylate and activate a group of 13 distinct but related downstream protein kinases that includes AMP-activated protein kinase (AMPK), which controls energy homoeostasis, and Par1, another polarity protein, which functions in microtubule stability and cell fate specification [15]. Which of these multiple signalling pathways—known and unknown—contribute to tumour suppression remains to be understood and is likely to be context-specific. In the pancreas, loss of LKB1 initiates precancerous lesions independently of AMPK, whereas in the intestine AMPK does appear to be involved [16]. Moreover, the LKB1–AMPK axis can act in tumour promotion rather than tumour suppression, by protecting cancer cells from oxidative stress [17]. In a c-Myc breast cancer model, loss of lkb1 promotes tumorigenesis and disrupts epithelial organization and polarity and basement membrane integrity, suggesting that LKB1 polarity functions are important [18]. Loss of lkb1 was also found to promote tumorigenesis in an ErbB2/neu breast cancer model with changes in metabolic signalling, implying that altered metabolism is important [19]. A closer examination of polarity and metabolism in both models would help to resolve whether the effects of loss of lkb1 are tumour-type dependent or, more likely, that multiple effectors of LKB1 contribute to tumorigenesis.
This context-dependent duality of effects is not unique to Par4/LKB1. Two forms of aPKC occur in vertebrates, aPKCζ and aPKC λ/ι, which appear to have opposing effects in cancer. The aPKC λ/ι isoform has been proposed to function as a tumour promoter in non-small cell lung cancer [20] and pancreatic cancer, and also stimulates epithelial–mesenchymal transitions. By contrast, the closely related isoform aPKCζ behaves as a tumour suppressor, through effects on glutamine metabolism [21]. As described below, the Par3 polarity protein also behaves as either an oncogene or tumour suppressor, depending on the tumour type.
Recently, three groups addressed the question of whether another Par polarity protein, Par3, functions in tumorigenesis (figure 1). The Collard group used a conditional knock-out mouse and a classical two-stage skin cancer model in which K-Ras mutations are induced by application of a carcinogen, DMBA, and tumour outgrowth is promoted by addition of phorbol esters [22]. Surprisingly—as the Par3 protein is likely necessary for the oriented cell divisions that occur during epidermal differentiation—deletion of the pard3 gene in the epidermis had no obvious phenotype. Loss of Par3 did, however, cause a substantial reduction in the number and size of papillomas, mediated by mislocalization of aPKC away from cell–cell junctions, which resulted in increased apoptosis and reduced cell proliferation (figure 1a) [22]. Increased apoptosis in response to loss of Par3 has also been reported previously for mammary glands [12].
Figure 1.

Tumorigenic functions of Par3 in skin and breast cancer. (a) Par3 has both oncogenic and tumour-suppressive functions in the skin. Loss of Par3 reduces papilloma induced by H-Ras and TPA (12-O-tetradecanoylphorbol-13-acetate) in mouse skin. Alternatively, loss of Par3 promotes formation of the highly aggressive keratoacanthomas. (b,c) In breast, loss of Par3 promotes invasion and metastasis. Loss of Par3 mislocalizes aPKC and cooperates with oncogenic Notch intracellular domain (NICD) to activate Jak/Stat signalling and induction of matrix metalloproteinases to degrade the extracellular matrix, and enable invasion and metastasis (b). Loss of Par3 can also delocalize the Rac-GEF Tiam to hyperactivate Rac1, which can cooperate with ErbB2 to promote invasion by altering cell–cell adhesions (c). (Online version in colour.)
These data would support a role for Par3 as a tumour promoter. However, the few papillomas that formed in the skin of mice deleted for Par3 were highly invasive and rapidly formed keratoacanthomas, an aggressive cancer that only rarely occurs in wild-type mice [22]. Paradoxically, therefore, Par3 functions as a tumour suppressor for keratoacanthomas. The cause for these opposing functions of Par3 is not known but could be owing to differences in stem or progenitor cells of origin for different forms of skin cancer.
The downstream signalling events involved in the promotion of keratoacanthomas remain unknown. However, in a mammary tumour model, loss of Par3 strongly promotes invasion and metastasis through an aPKC-dependent activation of Jak/Stat3 signalling, which induces expression of a metalloproteinase, MMP9 (figure 1b), with subsequent destruction of the extracellular matrix and invasion [23]. Silencing of Stat3 blocked lung colonization and expression of a constitutively active Stat3 mutant stimulated invasive behaviour even in the presence of Par3. Strikingly, human breast cancers frequently exhibit loss of Par3 expression, which correlates closely with increased aPKC and Stat3 phosphorylation [23]. This pathway seems to depend on oncogenic activation, because although loss of Par3 is in itself sufficient to cause mislocalization of aPKC, the kinase is not activated except in transformed cells. The molecular link between aPKC activation and Jak/Stat3 activation remains to be discovered.
A third study, by Muthuswamy and co-workers [24], investigated Par3 function in an ErbB2 model of breast cancer and also found that loss of Par3 potentiated metastasis, but in this case in the absence of increased primary tumour growth. They ascribed the increased invasiveness of the tumours to reduced adhesive activity by E-cadherin, mediated through constitutive activation of the Rac GTPase (figure 1c). Par3 is known to regulate an exchange factor for Rac, called Tiam1, and in most situations—both in mammals and Drosophila—restricts its activity [25,26]. Loss of Par3 leads to Rac hyperactivation through the inappropriate activation/mislocalization of Tiam1. This effect is apparently context dependent because in MDCK kidney epithelial cells, loss of Par3 disturbs tight junction formation but has little effect on adherens junctions [27]. Nonetheless, there is evidence from genomic sequencing of various human cancers that Rac activation might be an important driver of transformation [28], and early studies had shown that active Rac mutants could transform NIH 3T3 fibroblasts in vitro [29].
It remains to be established whether in any of these models, the loss of Par3 alters the ability of skin or mammary stem cells to undergo asymmetric cell divisions or alters self-renewal capacity. However, we know that Par3, in conjunction with leucine-guanine-asparagine (LGN), is required for spindle orientation during division of polarized epithelial cells [30]. Moreover, Par3 has also been reported to control the direction of basal cell mitoses [31,32]. Basal cells are progenitors for keratinocytes, generated by vertical divisions, perpendicular to the basal layer, whereas horizontal divisions provide self-renewal of the basal population. Nonetheless, deletion of Par3 from keratin-14 positive cells in the skin does not cause any phenotype [22]. Presumably, therefore, this polarity protein is not essential for basal cell function—or a compensatory mechanism rescues normal development in its absence.
3. Other polarity proteins in cancer
Certain groups of polarity proteins, such as Scribble and Crumbs, are less widely expressed than the Par proteins and are predominantly associated with epithelia [10,33]. Interestingly, although the Drosophila Scribble is essential for apical/basal polarity, in mammals there is little evidence of any such linkage [34], and instead the protein seems to be important in planar cell polarity and perhaps in Hippo signalling [35]. Loss of Scribble can promote primary tumour growth in a c-myc mouse model [36]. Additionally, mislocalization of Scribble has been linked to prostate cancer, and in a mouse model deletion of Scribble can predispose the animals to intra-epithelial neoplasia and cooperates with oncogenic K-ras to drive tumorigenesis, but the absence of Scribble from human prostate cancers does not correlate with poor prognosis [37].
Loss of Scribble also cooperates with Ras in fly models of cancer. Flies possess a quality control system called cell competition, through which defective cells are killed by their neighbours and replaced through compensatory proliferation. Mutation of polarity proteins, for instance in the eye epithelium, can trigger cell competition, resulting in JNK-dependent apoptosis of the mutant cells, which is activated through a TNF-α mediated inflammatory mechanism [38,39]. Strikingly, however, the coexpression of an oncogene, for instance Ras, within the mutant cells switches the response from apoptosis to hyperproliferation, resulting in the formation of metastatic tumours [39,40]. Even more remarkably, the same effect can occur even if Ras is expressed not in the mutant cell but in a neighbouring cell [41]. There are fascinating parallels to the effects of Par3 depletion in breast cancer, because overgrowth of the Scrb-mutant eye cells in the fly is triggered through a Jak/Stat pathway just as occurs in NICD-transformed mammary cells. However, while there is some evidence that cell competition can occur in mammalian cells [42], the underlying mechanisms are not fully understood and it remains to be determined whether this process plays any role in human cancers.
4. Epithelial–mesenchymal transition and metastasis
A widely accepted paradigm for cancer progression is that epithelial cells undergo a mesenchymal transition, during which they lose apical/basal polarity and intercellular adhesions, become highly migratory, and express a characteristic set of mesenchymal genes, such as N-cadherin and vimentin [43,44]. Single mesenchymal cells—which in some ways resemble stem cells—penetrate the basement membrane, cross the endothelium and enter the lymphatic system or bloodstream through which they are rapidly disseminated. At ectopic sites in the body, the cells extravasate and colonize surrounding tissue to form metastases. Control of the EMT involves multiple transcription factors and micro-RNAs. Snail, Slug, Twist, Prrx1 and Zeb1 are potent inducers of EMT in some epithelial cell types and are induced by signalling pathways that can activate EMT. For example, TGF-β induces Zeb1 expression [45]. In turn, Zeb1 suppresses a pro-epithelial miRNA, miR-200, which through a negative feedback look suppresses Zeb1 expression [46,47]. An array of epithelial polarity genes, including E-cadherin, Crumbs, Patj and Lgl are shut down by Zeb1. Most intriguingly, several recent studies have implicated EMT in the generation of cancer stem cells or tumour-initiating cells (TICs) [48]. Such cells are important for the outgrowth of metastases and are often considered to be resistant to chemotherapy. Breast cancer TICs express low levels of miR-200, for instance, which promotes expression of the stem-cell marker Bmi-1 [49], and Slug has been shown to induce a TIC phenotype [50]. Moreover, TICs isolated from mammary carcinomas express EMT markers.
There are, however, some concerns about this view of cancer progression through EMT, particularly in connection with breast cancer. First, and most importantly, normal mammary basal/myoepithelial cells—which are believed to arise with luminal epithelial cells from a common progenitor—lack E-cadherin and express mesenchymal markers such as vimentin and Slug [51]. In addition, mammary progenitor cells reside within the myoepithelial population. Therefore, it seems quite likely that the so-called mesenchymal cancer stem cells are in fact derived from or have reverted to basal cells. Another critical issue is that stem cell differentiation is plastic and is perturbed by experimental manipulations. In adult mice, lineage tracing shows that keratin-14 positive (i.e. myoepithelial) mammary cells are not multi-potent and give rise only to myoepithelial cells, whereas keratin-8 positive luminal progenitor cells only give rise to luminal cells [52]. However, the keratin-14 positive cells, when isolated from adult mammary glands and transplanted into recipient mice, are multi-potent and give rise to both luminal and myoepithelial cell types. This difference could arise if a different, dormant population of stem cells is activated by transplantation, or if the keratin-14 positive cells dedifferentiate into an embryonic multi-potent state during transplantation. This plasticity might be triggered by the inflammatory response caused by transplantation surgery or may reflect the presence of active and reserve stem cell pools within the mammary gland. Stem cell plasticity might extend to cancer cells, highlighted by recent findings that cells with EMT properties also have stem cell properties and that cancer cells can interconvert between more and less differentiated phenotypes [53]. Therefore, careful interpretation of in vivo assays used for quantifying stem cells from isolated, dissociated tumour tissues is necessary.
An additional complication arises from observations that, in spite of the belief that EMT is necessary for metastasis, mesenchymal cells are in fact very inefficient at colonizing ectopic sites to form metastases, and often are impoverished in TICs [54]. In a human cancer cell line model, epithelial tumour subpopulations were highly metastatic while expression of Snail induced EMT and suppressed metastasis [54]. Consistent with these observations, the overexpression of miR-200, which promotes epithelialization, increases rather than decreases metastasis. Moreover, the expression of Prrx1, an EMT-inducing transcription factor, inhibits proliferation, strongly reduces the efficiency of lung colonization in a mouse cancer model and reduces TIC frequency [55]. The loss of Prrx1—which promotes epithelialization—is, additionally, coupled to the acquisition of cancer stem cell properties and correlates with poor prognosis in breast and lung cancer patients.
The studies on Prrx1, and an independent study using an inducible Twist expression model of skin cancer [56], suggest, however, that EMT can potentiate the initial dissemination of cancer cells, even though such cells do not efficiently colonize ectopic sites. Yet, EMT is not essential for dissemination. Tumour cells can migrate through three-dimensional matrices by collective migration while retaining epithelial markers. Indeed, epithelial cells are well known to migrate into wounded areas as sheets rather than as individual mesenchymal-like cells. We found that loss of the Par3 polarity protein strongly promotes invasion of transformed mammary epithelial cells through collagen or Matrigel but does not induce EMT [23], and the cells migrate as clusters that retain E-cadherin-based adhesions, rather than as individual cells (figure 2).
Figure 2.
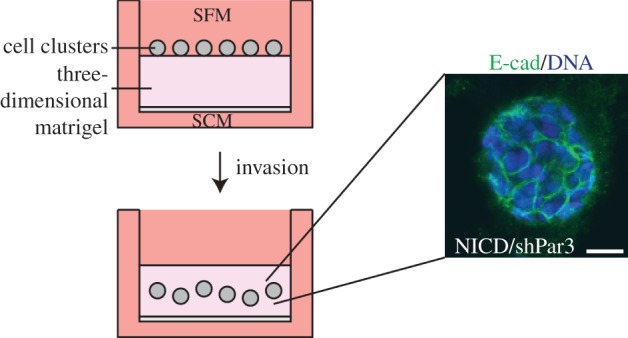
Collective invasion of Par3-depleted mammary cancer cells. Invasion assay chambers were established with clusters of tumour cells on top of a three-dimensional extracellular matrix pad in serum-free medium (SFM). Cells invaded towards the lower chamber with serum containing medium (SCM). Invading tumour cell clusters expressing NICD and either a control shRNA or Par3 shRNA were fixed and stained for E-cadherin and DNA. A representative cluster is shown. Scale bar, 10 µm. (Online version in colour.)
Collective migration, however, is not unique to epithelia: mesenchymal cells express cell adhesion molecules, including cadherins, and can migrate collectively. Neural crest cells, for example, undergo EMT, but then collectively migrate as chains of cells in vivo [57,58]. In an important recent study, human-circulating tumour cells were captured from breast cancer patients and analysed for several epithelial and mesenchymal markers [59]. Although one conclusion was that such circulating cells are highly enriched for the mesenchymal phenotype, in fact those from lobular type cancers were predominantly epithelial, whereas those from triple-negative cancers (which possess a basal/myoepithelial cell phenotype) were—unsurprisingly—mostly mesenchymal. Strikingly, disease progression was highly correlated with mesenchymal cell clusters, not single cells. However, HER2+ tumours also generated mesenchymal cells in the circulation, suggesting that EMT might be important for dissemination of this class of cancers. Taken together, these data suggest that tumours can access multiple mechanisms for dissemination, including but not limited to EMT, and that loss of epithelial character is not a necessary event during cancer progression and metastasis.
5. Questions for the future
Several questions still remain regarding the relationship between cell polarity, stem cells and cancer progression. A major unknown is how the microenvironment, or niche, regulates stem cell behaviour in mammalian epithelia. The niche of epithelial stem cells often is comprised of polarized epithelial cells. In cases, such as the intestine, where stem cells divide symmetrically then stochastically migrate away from the niche, do the niche cells control this? Also, what effect does loss of cell polarity in niche cells have on stem-cell regulation? Is loss of asymmetry in stem cell division an important component of human tumorigenesis? Although EMT-inducing programmes indeed disrupt apical–basal cell polarity, disrupting cell polarity does not induce EMT, in either normal or transformed cells. If EMT occurs during tumour cell dissemination, how and why does the reverse process, MET, occur at metastatic sites? A better understanding of cell autonomous and non-cell autonomous mechanisms that regulate stem cell renewal, differentiation and plasticity, and EMT will not only provide a deeper understanding of different stem and progenitor cell populations within a tissue, but also their deregulation and transformation in cancer progression, which may lead to enhance therapeutic opportunities.
References
- 1.Thiery JP. 2003. Epithelial–mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 15, 740–746 (doi:10.1016/j.ceb.2003.10.006) [DOI] [PubMed] [Google Scholar]
- 2.Liu C, et al. 2011. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (doi:10.1016/j.cell.2011.06.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visvader JE. 2009. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 23, 2563–2577 (doi:10.1101/gad.1849509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim E, et al. 2009. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 15, 907–913 (doi:10.1038/nm.2000) [DOI] [PubMed] [Google Scholar]
- 5.Molyneux G, et al. 2010. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7, 403–417 (doi:10.1016/j.stem.2010.07.010) [DOI] [PubMed] [Google Scholar]
- 6.Simons BD, Clevers H. 2011. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862 (doi:10.1016/j.cell.2011.05.033) [DOI] [PubMed] [Google Scholar]
- 7.Knoblich JA. 2008. Mechanisms of asymmetric stem cell division. Cell 132, 583–597 (doi:10.1016/j.cell.2008.02.007) [DOI] [PubMed] [Google Scholar]
- 8.Sugiarto S, et al. 2011. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell 20, 328–340 (doi:10.1016/j.ccr.2011.08.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cicalese A, et al. 2009. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138, 1083–1095 (doi:10.1016/j.cell.2009.06.048) [DOI] [PubMed] [Google Scholar]
- 10.Goldstein B, Macara IG. 2007. The PAR proteins: fundamental players in animal cell polarization. Dev. Cell 13, 609–622 (doi:10.1016/j.devcel.2007.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bultje RS, Castaneda-Castellanos DR, Jan LY, Jan YN, Kriegstein AR, Shi S-H. 2009. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 63, 189–202 (doi:10.1016/j.neuron.2009.07.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCaffrey LM, Macara IG. 2009. The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 23, 1450–1460 (doi:10.1101/gad.1795909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengupta A, et al. 2011. Atypical protein kinase C (aPKCζ and aPKCλ) is dispensable for mammalian hematopoietic stem cell activity and blood formation. Proc. Natl Acad. Sci. USA 108, 9957–9962 (doi:10.1073/pnas.1103132108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. 2009. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol. Rev. 89, 777–798 (doi:10.1152/physrev.00026.2008) [DOI] [PubMed] [Google Scholar]
- 15.Alessi DR, Sakamoto K, Bayascas JR. 2006. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 75, 137–163 (doi:10.1146/annurev.biochem.75.103004.142702) [DOI] [PubMed] [Google Scholar]
- 16.Lo B, Strasser G, Sagolla M, Austin CD, Junttila M, Mellman I. 2012. Lkb1 regulates organogenesis and early oncogenesis along AMPK-dependent and -independent pathways. J. Cell Biol. 199, 1117–1130 (doi:10.1083/jcb.201208080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon SM, Chandel NS, Hay N. 2012. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (doi:10.1038/nature11066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Partanen JI, et al. 2012. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc. Natl Acad. Sci. USA 109, E388–E397 (doi:10.1073/pnas.1120421109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrade-Vieira R, Xu Z, Colp P, Marignani PA. 2013. Loss of LKB1 expression reduces the latency of ErbB2-mediated mammary gland tumorigenesis, promoting changes in metabolic pathways. PLoS ONE 8, e56567 (doi:10.1371/journal.pone.0056567) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. 2005. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 65, 8905–8911 (doi:10.1158/0008-5472.CAN-05-2372) [DOI] [PubMed] [Google Scholar]
- 21.Ma L, et al. 2013. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell 152, 599–611 (doi:10.1016/j.cell.2012.12.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iden S, van Riel WE, Schafer R, Song JY, Hirose T, Ohno S, Collard JG. 2012. Tumor type-dependent function of the Par3 polarity protein in skin tumorigenesis. Cancer Cell 22, 389–403 (doi:10.1016/j.ccr.2012.08.004) [DOI] [PubMed] [Google Scholar]
- 23.McCaffrey LM, Montalbano J, Mihai C, Macara IG. 2012. Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 22, 601–614 (doi:10.1016/j.ccr.2012.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK. 2012. Loss of Par3 promotes breast cancer metastasis by compromising cell–cell cohesion. Nat. Cell Biol. 15, 189–200 (doi:10.1038/ncb2663) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Macara IG. 2006. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat. Cell Biol. 8, 227–237 (doi:10.1038/ncb1368) [DOI] [PubMed] [Google Scholar]
- 26.Georgiou M, Baum B. 2010. Polarity proteins and Rho GTPases cooperate to spatially organise epithelial actin-based protrusions. J. Cell Sci. 123, 1089–1098 (doi:10.1242/jcs.060772) [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Macara IG. 2005. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat. Cell Biol. 7, 262–269 (doi:10.1038/ncb1226) [DOI] [PubMed] [Google Scholar]
- 28.Kawazu M, et al. 2013. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc. Natl Acad. Sci. USA 110, 3029–3034 (doi:10.1073/pnas.1216141110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. 1995. An essential role for Rac in Ras transformation. Nature 374, 457–459 (doi:10.1038/374457a0) [DOI] [PubMed] [Google Scholar]
- 30.Hao Y, Du Q, Chen X, Zheng Z, Balsbaugh JL, Maitra S, Shabanowitz J, Hunt DF, Macara IG. 2010. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr. Biol. 20, 1809–1818 (doi:10.1016/j.cub.2010.09.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lechler T, Fuchs E. 2005. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 437, 275–280 (doi:10.1038/nature03922) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams SE, Beronja S, Pasolli HA, Fuchs E. 2011. Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature 470, 353–358 (doi:10.1038/nature09793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tepass U. 2012. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu. Rev. Cell Dev. Biol. 28, 655–685 (doi:10.1146/annurev-cellbio-092910-154033) [DOI] [PubMed] [Google Scholar]
- 34.Qin Y, Capaldo C, Gumbiner BM, Macara IG. 2005. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J. Cell Biol. 171, 1061–1071 (doi:10.1083/jcb.200506094) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skouloudaki K, et al. 2009. Scribble participates in Hippo signaling and is required for normal zebrafish pronephros development. Proc. Natl Acad. Sci. USA 106, 8579–8584 (doi:10.1073/pnas.0811691106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK. 2008. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878 (doi:10.1016/j.cell.2008.09.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearson HB, et al. 2011. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J. Clin. Invest. 121, 4257–4267 (doi:10.1172/JCI58509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. 2009. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell 16, 458–465 (doi:10.1016/j.devcel.2009.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M. 2010. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell 18, 999–1011 (doi:10.1016/j.devcel.2010.05.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brumby AM, Richardson HE. 2003. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779 (doi:10.1093/emboj/cdg548) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu M, Pastor-Pareja JC, Xu T. 2010. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 463, 545–548 (doi:10.1038/nature08702) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogan C, Kajita M, Lawrenson K, Fujita Y. 2011. Interactions between normal and transformed epithelial cells: their contributions to tumourigenesis. Int. J. Biochem. Cell Biol. 43, 496–503 (doi:10.1016/j.biocel.2010.12.019) [DOI] [PubMed] [Google Scholar]
- 43.Scheel C, Weinberg RA. 2012. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol. 22, 396–403 (doi:10.1016/j.semcancer.2012.04.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Weinberg RA. 2008. Epithelial–mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829 (doi:10.1016/j.devcel.2008.05.009) [DOI] [PubMed] [Google Scholar]
- 45.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. 2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 (doi:10.1038/ncb1722) [DOI] [PubMed] [Google Scholar]
- 46.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. 2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589 (doi:10.1038/embor.2008.74) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. 2008. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial–mesenchymal transition. Cancer Res. 68, 7846–7854 (doi:10.1158/0008-5472.CAN-08-1942) [DOI] [PubMed] [Google Scholar]
- 48.Mani SA, et al. 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (doi:10.1016/j.cell.2008.03.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimono Y, et al. 2009. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138, 592–603 (doi:10.1016/j.cell.2009.07.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo W, et al. 2012. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028 (doi:10.1016/j.cell.2012.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kendrick H, Regan JL, Magnay FA, Grigoriadis A, Mitsopoulos C, Zvelebil M, Smalley MJ. 2008. Transcriptome analysis of mammary epithelial subpopulations identifies novel determinants of lineage commitment and cell fate. BMC Genomics 9, 591 (doi:10.1186/1471-2164-9-591) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, Sharma N, Dekoninck S, Blanpain C. 2011. Distinct stem cells contribute to mammary gland development and maintenance. Nature 479, 189–193 (doi:10.1038/nature10573) [DOI] [PubMed] [Google Scholar]
- 53.Chaffer CL, et al. 2011. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl Acad. Sci. USA 108, 7950–7955 (doi:10.1073/pnas.1102454108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Celia-Terrassa T, et al. 2012. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Invest. 122, 1849–1868 (doi:10.1172/JCI59218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. 2012. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (doi:10.1016/j.ccr.2012.10.012) [DOI] [PubMed] [Google Scholar]
- 56.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. 2012. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (doi:10.1016/j.ccr.2012.09.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKeown SJ, Wallace AS, Anderson RB. 2013. Expression and function of cell adhesion molecules during neural crest migration. Dev. Biol. 373, 244–257 (doi:10.1016/j.ydbio.2012.10.028) [DOI] [PubMed] [Google Scholar]
- 58.Kasemeier-Kulesa JC, Kulesa PM, Lefcort F. 2005. Imaging neural crest cell dynamics during formation of dorsal root ganglia and sympathetic ganglia. Development 132, 235–245 (doi:10.1242/dev.01553) [DOI] [PubMed] [Google Scholar]
- 59.Yu M, et al. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (doi:10.1126/science.1228522) [DOI] [PMC free article] [PubMed] [Google Scholar]