

Abstract
The budding yeast Saccharomyces cerevisiae has been used extensively for the study of cell polarity, owing to both its experimental tractability and the high conservation of cell polarity and other basic biological processes among eukaryotes. The budding yeast has also served as a pioneer model organism for virtually all genome-scale approaches, including functional genomics, which aims to define gene function and biological pathways systematically through the analysis of high-throughput experimental data. Here, we outline the contributions of functional genomics and high-throughput methodologies to the study of cell polarity in the budding yeast. We integrate data from published genetic screens that use a variety of functional genomics approaches to query different aspects of polarity. Our integrated dataset is enriched for polarity processes, as well as some processes that are not intrinsically linked to cell polarity, and may provide new areas for future study.
Keywords: functional genomics, polarity, genetics, microscopy, proteomics, Saccharomyces cerevisiae
1. Introduction
Cell asymmetry, or polarity, refers to spatial differences in shape, structure or function of cellular components. Cell polarity is a defining feature of almost all cells, and is used differently based on cell type to modulate cell behaviour and define distinct cellular domains. The budding yeast Saccharomyces cerevisiae is an attractive model for studying the establishment of cell polarity for two main reasons: (i) core biological processes in S. cerevisiae are conserved in higher eukaryotic cells, allowing inference of function; and (ii) yeast is an experimentally tractable organism that is amenable to genetic manipulation [1].
The field of functional genomics aims to define gene (and protein) functions and interactions, using data derived from genome-scale experiments. As noted above, model organisms like yeast have been essential for annotating gene function and for developing tools and approaches that have driven major advances in functional genomics and genome biology. In this review, we highlight research that has made use of functional genomics approaches to study polarity in S. cerevisiae. We then describe general trends identified through the analysis of 35 genome-wide genetic screens from different groups, which suggest that there are several essential biological processes required for cell polarity in yeast. Finally, we discuss our view of future directions for cell polarity research using functional genomics approaches in yeast. It is beyond the scope of this review to give a detailed description of the biology of polarity in yeast, but we refer the reader to excellent reviews recently published by the YeastBook project [2,3].
Saccharomyces cerevisiae cells become polarized during three discrete phases: budding, mating (shmoo formation) and filamentous growth. Each of these modes of polarized cell growth is regulated by different spatio-temporal and biological cues, but all hinge on a common series of molecular polarity determinants beginning with the small guanosine triphosphatase (GTPase) Cdc42. Budding is internally induced at the time of cell cycle commitment in late G1 (figure 1a), which is regulated by the cyclin-dependent protein kinase Cdc28 [4–6]. Cortical spatial landmarks inherited from the previous round of cell division determine the bud site and lead to activation of small GTPase molecules and their regulators: Rsr1, Cdc24 and Cdc42 (figure 1b). Active cycling of Rsr1 GTPase between GDP- and GTP-bound forms is hypothesized to concentrate the activated form of Cdc42 and Cdc24, the guanine nucleotide-exchange factor (GEF) of Cdc42, at the selected bud site [2,7,8]. Polarization of Cdc42 triggers the switch from isotropic, symmetrical growth to non-symmetrical growth along an axis dictated by the septins and the actin cytoskeleton [5,9]. First, Cdc42 activates Bni1, a formin that together with Bnr1 assembles the filamentous (F) actin cables [10]. Polarized F-actin cables function as tracks along which the myosin motor, Myo2, moves exocytic vesicles from the Golgi [11,12], as well as numerous other organelles [13–16], to the incipient bud site, promoting growth of the bud. The polarisome complex (consisting of Spa2, Pea2, Bni1, Bud6, Msb3 and Msb4) promotes this actin-mediated exocytosis by coupling the extension of F-actin cables with vesicle fusion [17]. Cortical actin patches are also polarized in response to Cdc42 activation and are nucleated by the highly conserved Arp2/3 complex following its activation by the Cdc42-regulated kinases, Cla4 and Ste20 [18,19]. Actin patches play an integral role in endocytosis, allowing for regulation of lipid and protein levels at the membrane of the emerging bud [20]. Second, septins are recruited to the bud neck following activation of the Rho-like GTPases Gic1 and Gic2 by Cdc42, and form a ring-shaped cytoskeletal complex that associates with the plasma membrane, providing a diffusion barrier between the mother and the bud [21,22]. The septins also provide a scaffold upon which the contractile actomyosin ring is assembled. During telophase, triggered by the mitotic exit network, the septin hourglass shape splits into two rings, surrounding the cytokinetic machinery [23]. As the actomyosin ring contracts, the cytoskeletal and exocytic machinery, which earlier in the cell cycle promoted bud growth, is redirected from the bud cortex to the bud neck, delivering post-Golgi vesicles to the actomyosin ring [24].
Figure 1.
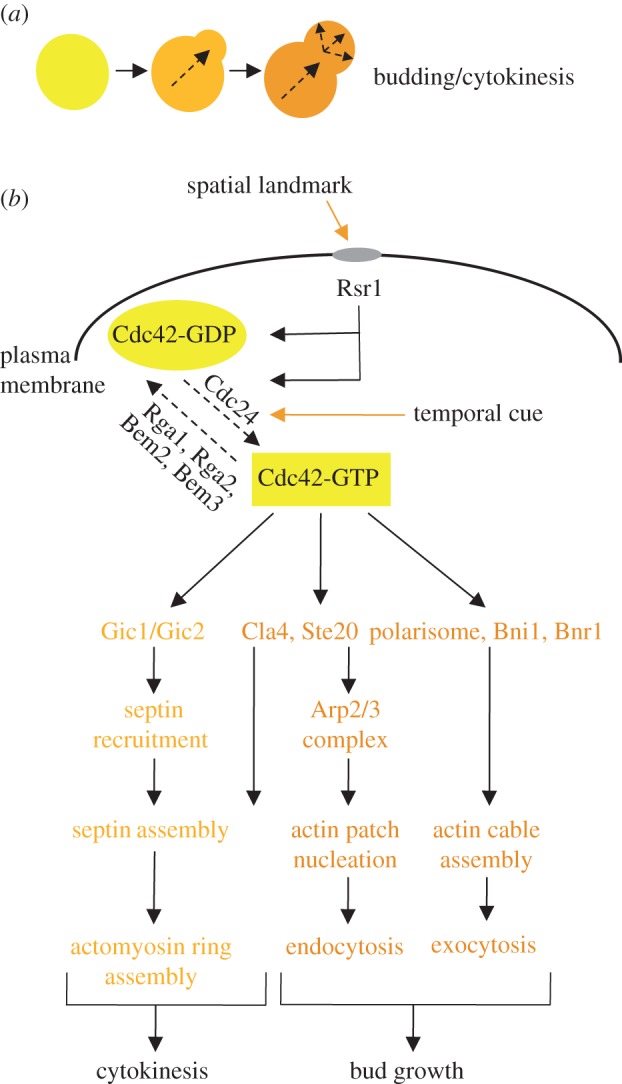
An overview of Cdc42 regulation during polarized bud growth. (a) Budding occurs when the cell switches from isotropic to apical cell growth, leading to the formation of a bud. (b) Bud-site selection protein Rsr1 activates the GEF Cdc24, which converts Cdc42 to its active GTP-bound state. Activated Cdc42 binds to numerous effectors, promoting actin patch nucleation, actin cable assembly and septin/actomyosin ring assembly.
Alternatively, polarity in both mating and filamentous growth is induced by external cues. Mating is initiated upon exposure to the mating pheromone from cells of the opposite mating type, which leads to the outgrowth of a mating projection (shmoo), allowing for growth toward the source of the pheromone [25,26] (figure 2a). As in the process of budding, Cdc42 also plays a central role in polarized cell growth and shmoo formation during mating. In addition to driving polarized secretion to the growing shmoo tip, Cdc42 also regulates the Ste20 kinase, which triggers activation of the mating mitogen-activated protein kinase (MAPK) signalling pathway to turn on mating-specific genes required for cell fusion and Far1, an inhibitor of Cdc28 (figure 2b). Filamentous growth is initiated in the absence of a nutrient-rich environment and involves cell elongation and enhanced cell adhesion, leading to a switch from spherical form to filamentous form and enabling expansion to new environments [27,28] (figure 3a). During filamentous growth, Cdc42 interacts with two key proteins to promote filamentation: (i) Cdc42 binds to the membrane protein Msb2, which, together with Sho1, senses nitrogen starvation; and (ii) Cdc42 interacts with the Ras2 GTPase, which also regulates the nitrogen starvation response. These interactions stimulate actin filamentation and secretion in the direction of growth, and also activate the MAPK pathway. The MAPK pathway consists of many of the same components that are involved in the mating pheromone response and leads to the activation of Flo11, a flocculin that allows for fibrous interconnections to form between cells (figure 3b). While the molecular basis of cellular polarity has been intensively studied for many years, the highly dynamic nature of the polarity process suggests that many players may remain to be discovered. Below, we summarize functional genomics efforts that aim to uncover new regulators of cell asymmetry and polarized growth in S. cerevisiae.
Figure 2.
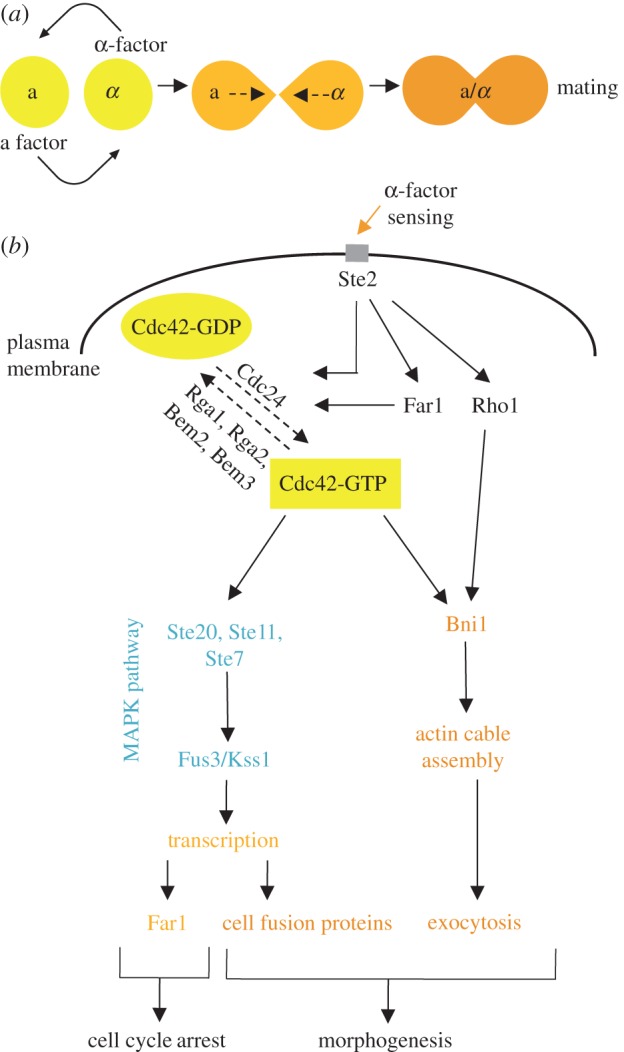
An overview of Cdc42 regulation during mating. (a) Mating begins after exposure to mating pheromone from cells of the opposite mating type, which leads to the outgrowth of a mating projection, or ‘shmoo’. (b) The same Cdc42-based machinery used in budding drives polarized cell growth and shmoo formation during mating. In addition to regulating polarized secretion, Cdc42 also regulates the mating MAPK signalling pathway to turn on mating-specific genes required for cell fusion and the cyclin-dependent kinase inhibitor Far1, leading to G1 arrest.
Figure 3.
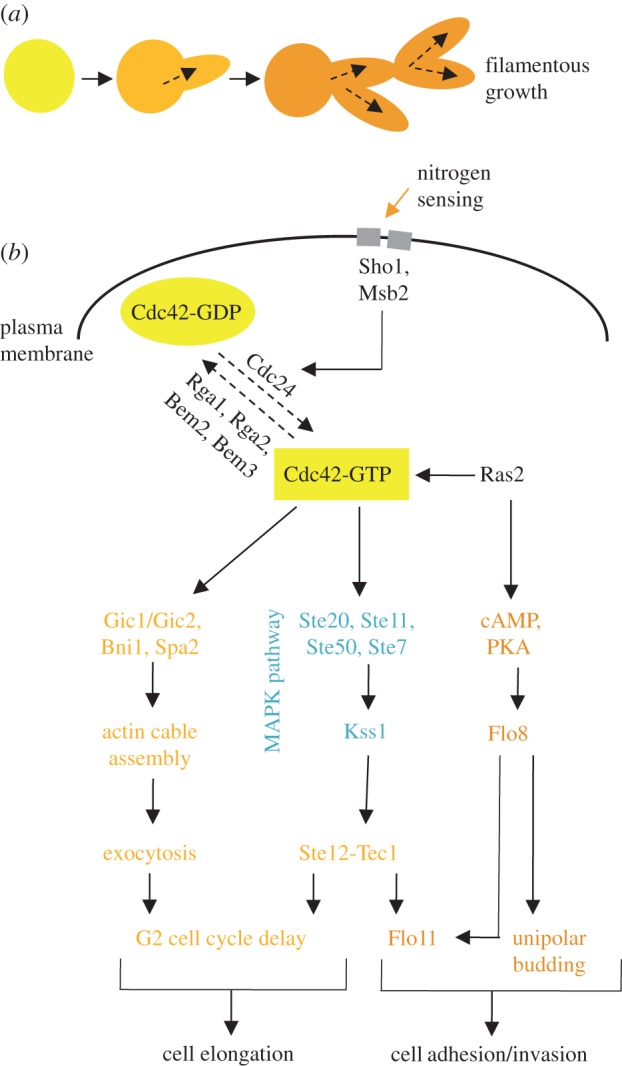
An overview of Cdc42 regulation during filamentous growth. (a) Filamentous growth occurs when cells elongate and exhibit enhanced cell adhesion, leading to a switch from yeast cell form to filamentous form. (b) Filamentation occurs when nitrogen starvation is sensed by Sho1 and Msb2, leading to the activation of Cdc42 and the downstream activation of the MAPK pathway. This leads to both cell elongation via a G2 delay as well as the expression of the flocculin Flo11 and other adhesion proteins.
2. High-throughput methods for polarity studies
The ease of genetic manipulation in S. cerevisiae has enabled the creation of a wealth of large-scale collections of strains with deleted [29,30], hypomorphic [31–34], tagged [35–37] or over-expressed genes [38–43], as well as the development of new methods for performing cost-effective and straightforward systematic analyses. Here, we give an overview of methodological advances in the fields of yeast genomics, microscopy and proteomics that have contributed to our understanding of cell polarity (figure 4).
Figure 4.

An overview of functional genomics approaches in the study of polarity. This review focuses on the use of genomic, cell biological and proteomic assays to study polarity in yeast.
(a). Genetic assays
Yeast researchers have used forward genetic screens productively for many years to discover regulators of cell polarity. For example, CDC42 was first identified in classical genetic screens for temperature-sensitive mutants that arrest their cell cycle with a uniform morphological phenotype [19,44]. More recently, so-called reverse genetic approaches, which involve assessment of the phenotypic consequences of a known genetic mutation, have provided a means to immediately link genotype to phenotype. The budding yeast heterozygous deletion collection is composed of a set of diploid yeast strains in which each of the approximately 6000 genes is individually deleted and replaced with a drug resistance cassette [29,30]. The deletion collection was the first genome-scale reagent produced for reverse genetics screens and was used to generate the haploid non-essential deletion collection (consisting of strains harbouring deletion mutations in 80% of yeast genes), inspiring the development of numerous methods for the manipulation of these collections. In particular, synthetic genetic array (SGA) analysis automates yeast genetics and has enabled high-throughput genetic studies in yeast. The SGA method involves a set of replica pinning and serial selection steps, allowing facile introduction of any marked allele into any set of arrayed strains in a high-throughput manner [45].
A major application of SGA analysis has involved systematic assessment of genetic interactions (GIs) between two partial or complete loss-of-function alleles [46–53]. A GI can be defined as an unexpected deviation in double mutant growth rate, using colony size as a proxy for cellular fitness [54]. A negative GI, in which the double mutant has a more severe fitness defect than would be predicted based on the fitness of the two single mutants, suggests that the two genes have a redundant role as components of parallel pathways. A positive GI, in which the double mutant is more fit than expected, suggests that the two gene products may function in the same pathway. A global survey of GIs between approximately 5.4 million gene pairs revealed an interesting relationship between GIs and the essentiality of protein complex members; genes encoding components of non-essential complexes show predominantly positive GIs, whereas negative GIs are more often found among genes encoding components of essential complexes [55]. This observation suggests that essential complexes contain internal redundancy, allowing retention of function after loss of a single complex member. Additionally, GI profiles (the set of GIs for a particular gene) can be used to infer gene function through a ‘guilt by association’ principle of analysis: genes that have similar GIs are likely to encode proteins that are part of the same pathway or complex. The first proof-of-principle work validating SGA analysis as a method for mapping synthetic lethal (negative) GIs included a focus on cell polarity genes and revealed new components of pathways known to regulate actin cable nucleation [45]. Alternative methods of assaying GIs involve competitive growth assays of pooled yeast strains (for example, deletion mutants) that each contain a unique ‘molecular barcode’. Flanked by common PCR priming sequences, all barcodes can be amplified simultaneously, allowing for analysis by either microarray hybridization or sequencing [56–61].
Other types of GIs have proved useful for exploring cell polarity pathways. In particular, complex haploinsufficiency (CHI) occurs when a heterozygous double mutant exhibits a more severe phenotype than the corresponding heterozygous single mutants. CHI is thought to identify genes that function within the same pathway or structure and provides useful insight in predicting multigenic influences in human disorders. CHI has been heavily used to explore GIs specific to strains heterozygous for different alleles of the actin gene [62,63]. For example, these screens revealed inappropriate and excessive actin assembly in strains deleted for genes that encode components of the endosomal sorting complex required for transport (ESCRT), leading to an unusual relocalization of F-actin to prevacuolar endocytic compartments called E-bodies [62]. Although actin's involvement with endocytosis is well established, this discovery marked the first instance of F-actin accumulation on E-bodies. Interestingly, CHI screens have also drawn a convincing link between the actin cytoskeleton and several proteasome components including numerous members of both the 19S regulatory particle and the 20S core particle. This observation suggests that loss of proteolytic activity may indirectly affect actin and cell polarity, and that there may in fact be a direct physical interaction between the proteasome and actin filaments [63].
Genetic interactions caused by changes in gene dosage have also been systematically explored using gene overexpression libraries. Synthetic dosage lethality (SDL), wherein overproduction of one gene product exacerbates a mutant phenotype, has been used to identify kinase targets whose regulated phosphorylation is important for cell polarity [52,64]. For example, follow-up of an SDL screen revealed that phosphorylation of Rga2, a GTPase-activating protein for Cdc42, by the cyclin-dependent protein kinase Pho85 inhibits Rga2 activity to ensure appropriate activation of Cdc42 during cell polarity establishment [64]. Dosage suppression, wherein overproduction of one gene rescues a mutant phenotype of another gene, often reveals downstream components of the pathway and has been used productively by many groups to study cell polarity pathways [65–68]. Finally, numerous polarity screens have assessed the effects of single gene perturbations, either mutation or overexpression, in specific conditions including chemical or environmental stress [69–75].
In addition to colony size, more specific colony-based assays have been used to discover genes involved in polarity and morphogenesis. For example, regulators of the mating pathway have been identified by screening yeast mutant strains for defects in activation of a relevant reporter gene [76–79]. Additionally, as filamentous growth is in essence a colony phenotype distinguished by filaments of interconnected cells, screens for colony morphology have been used with great success to implicate genes in filamentous differentiation [3,80–88]. Interestingly, the vast majority of mutants identified in colony morphology screens do not show compromised fitness [87], highlighting the importance of morphology-based assays as a complement to fitness-based assays to uncover cell polarity mechanisms.
(b). Microscopy-based assays
In order to fully dissect the spatio-temporal dynamics of the intricate biological systems controlling cell polarity, numerous qualitative and quantitative cellular imaging and analysis techniques have been developed, which can be used alongside the sophisticated genetics outlined in §2a. As polarized growth is characterized by distinct subcellular changes in morphology, systematic image-based approaches to directly observe cell morphology during budding, shmoo formation and hyphal outgrowth are very powerful. Global analysis of protein localization in S. cerevisiae is possible owing to the availability of an enormously useful collection of strains each expressing a unique endogenously tagged green fluorescent protein (GFP)-fusion protein [35,36]. The original analysis of this collection identified almost 250 proteins that localize to the primary compartments involved in polarization (bud, bud neck, actin cytoskeleton and cell periphery), comprising approximately 4% of the total number of tagged proteins [35]. So far, the GFP library has seen limited use for exploring cell polarity pathways—one screen identified proteins that localize to the mating projection in pheromone-treated cells [89]. A more common approach has been to study changes in the localization of cell polarity-specific proteins fused to a fluorescent moiety in the deletion collection or in the context of gene overexpression [53,90–98]. For instance, the deletion mutant array was screened for mislocalization of modified Snc1-GFP, yeast synaptobrevin, to identify novel regulators of its internalization [53].
While extremely useful for live-imaging, fusion to a fluorescent protein tag may lead to mislocalization of some proteins owing to steric hindrance or inhibition of critical protein–protein interactions, or even reorganization of subcellular complexes [35]. Also, multimerization of the fluorescent protein moiety can lead to artefactual formation of foci [99]. These effects can be minimized by carefully choosing the size and position of fluorescent labels, and selecting those that best complement corresponding loss-of-function alleles in phenotypic assays or in known GIs, for example synthetic lethality. One alternative to fluorescently tagging full-length proteins is to use fluorescent dyes that bind to specific proteins or subcellular compartments. For instance, rhodamine–phalloidin binds to F-actin and is commonly used to visualize the actin cytoskeleton. Calcofluor-white, which binds to chitin in the cell wall and highlights cell shape and bud scars, has been successfully used to identify genes involved in polarized budding [91]. Lastly, the lipophilic dye FM4-64 can be used to track endocytosis and endosomal trafficking in live cells [92]. Despite various technical challenges, microscopy-based assays remain the premier method for gaining insights into subcellular morphological changes.
(c). Proteomics assays
In parallel with systematic genetic approaches to identify the components of the polarity machinery, important advances have been made in the study of cell polarization using various proteomic methods. The yeast two-hybrid assay involves detection of a protein–protein interaction between two proteins fused to portions of a transcription factor [100], and has been used widely to explore protein complexes important for cell polarity in yeast [101–104]. For instance, using approximately 70 cell polarity components as bait, Drees et al. [103] identified approximately 130 novel physical interactions which provided an integrated network of signalling, cytoskeleton and organellar proteins directing cell polarity development. More recently, the integrated membrane yeast two-hybrid (iMYTH) system has enabled detection of protein–protein interactions that occur at the membrane by taking advantage of two split halves of a ubiquitin variant that can be reconstituted by association of two interacting proteins. The cytosolic deubiquitination enzymes recognize this reassociated ubiquitin form and cleave off a chimeric transcription factor linked to the ubiquitin, allowing for its translocation to the nucleus and subsequent activation of reporter gene transcription [105,106]. A conceptually similar method called the protein complementation assay (PCA) involves reconstitution of enzyme activity by interaction of a tagged protein of interest with an incomplete portion of a reporter enzyme [107]. One advantage to PCA and iMYTH is that genes are expressed from their endogenous promoters, and the interaction can occur in the endogenous cellular compartment, even if that compartment is outside of the nucleus. For example, in a systematic study by Tarassov et al. [108], interactions among the components of the Arp2/3 actin assembly complex, the actin patch and the exocyst were found, identifying a potential protein network involved in bud polarization, bud-neck organization and cytokinesis. This description of new connectivity between distinct complexes provides insight into the dynamic movements of polarity and exocyst components between the bud tip and the bud neck during cell division.
In a complementary approach, protein–protein interactions can be directly queried at the proteomic level using affinity-purification or fractionation coupled with mass spectrometry (AP–MS) [109–111]. This approach can identify all members of a protein complex, although it does not distinguish between direct and indirect physical interactions. Proteomic profiles of various subcellular compartments involved in cellular polarization have been analysed by mass spectrometry, including the vacuole [112], cell wall [113] and plasma membrane [114]. Additionally, post-translational modifications (specifically phosphorylation) have been surveyed in cells treated with mating pheromone to identify phosphoproteome changes during mating [115]. Datasets describing the results of several global proteomics assays have revealed integral members of the polarity machinery and provide a rich resource for further analysis of polarity pathways [108,116,117].
3. Analysis and synthesis of genetic screens
As summarized above, many functional genomics approaches have been used to study cell polarity in yeast. We wondered whether particular approaches were more suited for the analysis of different aspects of cell polarity—for example, are specific types of genes best identified with a particular method (cell biological as compared to fitness-based readout, for example), and are different polarity processes best queried with a certain approach (budding as compared to filamentation)? To begin to address these questions, we aimed to compile an inclusive, unbiased list of existing genome-wide datasets that either assessed the GIs between genes involved in different aspects of cell polarity or analysed chemo-GIs using compounds with known effects on the processes that establish cell polarity. A survey of the literature uncovered 35 published works from numerous groups (see electronic supplementary material, table S1), which included data on 3492 unique open reading frames (roughly 58% of the entire genome). As a filtering metric only those genes that were observed in four or more screens were integrated into our polarity network (n = 485; figure 5 and electronic supplementary material, table S2). We used GO Slim enrichment analysis [118] to identify biological processes that were over-represented in this dataset. As expected, the dataset was significantly enriched for genes with annotated roles in processes essential for cellular polarization, including cytoskeletal organization (p < 5.9 × 10−6), cell wall organization (p < 2.9 × 10−10), cellular membrane organization (p < 9.4 × 10−8) and transport (p < 1.5 × 10−8). The dataset was also enriched for genes with roles in a number of processes that require the establishment of cell polarity, such as pseudohyphal growth (p < 1.3 × 10−14), conjugation (p < 2.1 × 10−5), cell budding (p < 5.5 × 10−5) and cytokinesis (p < 2.4 × 10−8). Thus a significant fraction of the genes identified in systematic polarity screens (26%) have well-characterized roles in cellular polarization.
Figure 5.

Biological processes enriched in genomic polarity screens. A network diagram was created with Cytoscape and BiNGO to visualize biological GO Slim processes that are enriched in a set of 35 genome-wide genetic polarity screens (n = 485).
We anticipated that our analysis would identify a set of core polarity genes required in all polarity assays. Indeed, genes encoding polarisome components (Bni1, Spa2 and Pea2) and actin cytoskeleton components (Sla1, Tpm1, Sac6, Abp1 and Rvs161) were identified in seven or more screens. We had further predicted that specific biological processes would be enriched depending on the way cell polarity was queried. To test this idea, we defined three general categories in which to bin the published datasets: (i) type of polarized growth (analysis of budding/cytokinesis, filamentous growth or mating); (ii) type of mutation or sensitized background (deletion, overexpression or chemical treatment); and (iii) phenotypic readout (colony size, colony morphology, single-cell analysis or reporter detection) (see electronic supplementary material, table S2). Surprisingly, the same subset of GO Slim functional categories was enriched irrespective of categorization. For example, OST3 (which encodes a subunit of the oligosaccharyltransferase complex important for N-glycosylation) was identified in a variety of phenotypic assays in all three types of polarized growth assays. Ost3 has not been previously recognized as a ‘core’ regulator of polarity although it has numerous synthetic sick and lethal interactions with polarity genes [50,69,70] and is required for pseudohyphal growth [87], response to alpha factor [60] and proper sorting of a secreted protein [78,79]. OST3 is also implicated in resistance to aureobasidin A, a chemical inhibitor of complex sphingolipid synthesis which disrupts filamentous actin cables in growing yeast cells [56,119], suggesting a central role of OST3 in polarized growth. Interestingly, OST6, a homologue of OST3, is not present in our polarity network (observed in only two screens, as opposed to 10 in the case of OST3). This observation agrees with several pieces of evidence indicating that Ost3 and Ost6 are alternative members of the OST complex [120,121], suggesting that the Ost3 inclusive complex may have a more central role in cell polarity, although further studies are needed to elucidate this potential functional difference. We reason that our compiled dataset identifies a core set of processes essential for all aspects of polarity, in addition to the previously annotated polarity proteins, for example the polarisome components. These processes include the coordination of vesicle-mediated transport (p < 4.9 × 10−16), cell wall (p < 2.9 × 10−10) and cell membrane organization (p < 9.4 × 10−8), and activities of the cytoskeleton (p < 5.9 × 10−6), regulated by response to stress (p < 3.8 × 10−5) and chemicals (p < 6.8 × 10−6) through transcription (p < 9.5 × 10−9) and protein modification (p < 9.6 × 10−7). In support of this general idea, many of the gene products identified in multiple screens are annotated as phenotypic capacitors (127 of 485 polarity genes, from the set of 502 identified capacitors), gene products that contribute to phenotypic robustness, and which when deleted cause a high amount of variance in several polarity-based phenotypes [122].
The integrated polarity dataset was also enriched for processes not obviously related to polarity, which may provide some insight into the interconnected nature of cellular architecture. Some biological processes, for instance mitochondrion organization, are not closely connected to cell polarity based on their correlation in global GI profiles [47] but appear frequently in genome-wide cell polarity screens. For example, ILM1 has a proposed function in mitochondrial maintenance and was identified in 11 of 35 polarity datasets [81]. Interestingly, while the GI profiles of most mitochondrial genes are not highly correlated to those for the polarity machinery in the global GI network [47], the GI profile of ILM1 is similar to the profiles of genes involved in cell wall synthesis and integrity, including SMI1 and FKS1 [47], consistent with a role for ILM1 in cell polarization. Moreover, an ilm1Δ mutant is highly sensitive to several drugs that inhibit polarity-related processes, including latrunculin (prevents actin polymerization), papuamide B (disrupts plasma membrane integrity) and caspofungin (disturbs cell wall integrity) [56]. Other genes connect mitochondria to polarity: members of the Mgm1/Ugo1/Fzo1 complex, which is required for mitochondrial morphology and genome maintenance, were repeatedly identified in the cell polarity screens analysed here (see electronic supplementary material, table S2). Direct links have also been reported between mitochondria and F-actin: disruption of actin cables results in altered mitochondrial morphology, and mitochondria use actin cables to facilitate migration to the daughter cell during mitosis [123]. One possible model for the involvement of mitochondrial maintenance genes in polarity suggests that mitochondrial dysfunction inhibits cellular polarization through the action of the mitochondrial retrograde signalling pathway which relays alterations in mitochondrial function to the nucleus, leading to specific adaptive changes in nuclear gene expression [82].
Our analysis of published cell polarity screens also highlighted genes of unknown function that appeared in several screens and may represent previously unappreciated regulators of cell polarity. Of particular note is AIM44, whose product localizes to the bud neck [35]; a strain deleted for AIM44 is highly sensitive to numerous chemicals affecting polarity processes, including papuamide B, latrunculin, caspofungin and auroebasidin B among others [56]. The genetic profile of AIM44 is highly correlated with several genes that are involved in regulating the onset of cell polarization, including SWI5, a cell-cycle regulated transcription factor required for M/G1 and G1-specific gene expression, RGL1, a regulator of Rho1 signalling which is localized to the bud neck, and PKH1, which regulates endocytosis and the maintenance of cell wall integrity [47]. Together, these data implicate AIM44 in cell polarity and suggest that further characterization of its function should be fruitful. Indeed, recent work by Meitinger et al. [124] suggests that AIM44 plays a role in regulating Cdc42 and Rho1 to establish a novel cell polarity cue at the cell division site. We provide the integrated cell polarity dataset in full in the electronic supplementary material, table S2, and anticipate that further amalgamated analyses using these and other data will provide a useful resource for cell polarity research.
4. Perspectives and future directions
The study of cell polarity in yeast has been dramatically enhanced by the application of high-throughput approaches, which can provide an unbiased view of conserved biological processes. So far, systematic genetic screens have largely focused on the analysis of single and double deletion mutants using simple phenotypic read-outs, for example colony size, which has provided biologically rich information. In the next few years, cell polarity research is likely to be advanced on several fronts. First, yeast researchers are now poised to shift towards large-scale analysis of higher order combinations of genetic lesions, including triple mutant analysis, and combinations of genetic and chemical perturbations. Higher order multiplexing of mutant strains is now technically feasible owing to recent adaptations of pooled competitive growth assays [58]. Also, most genome-wide screens to date have used deletion alleles of non-essential genes and mechanistically uncharacterized conditional alleles of essential genes [31–34]. Many cell polarity components are essential or have multiple roles, and their analysis demands a more sophisticated approach. High-throughput methods for generation of gene mutations [31,125–127] mean it is now feasible to construct strain libraries carrying point mutations in all yeast genes, as opposed to deletion or other loss-of-function alleles. It is clear that SGA-based screens with strains carrying different mutations in the same gene provide unique biological information [32,62,128,129]. Thus, GI analysis with strains carrying specific mutations in cell polarity genes promises to lead researchers into uncharted genetic territory.
Second, advances in automated image acquisition and analysis for assessment of cell polarity phenotypes in sensitized genetic or conditional backgrounds mean that image-based screens should move to the forefront of cell polarity research in yeast [130]. To date, image-based screens using the yeast deletion and other arrayed collections have collected static images in a single focal plane [53,90–98]. However, cell polarity is highly dynamic, and future analyses will involve the high-throughput acquisition of three- and four-dimensional spatial and temporal image sequences, using multiple fluorescent markers to highlight numerous compartments simultaneously. Until recently, the huge amount of data generated by high-dimensional screens has made analysis time-consuming and labour-intensive. However, new algorithms for data extraction and expansions of readily accessible software are making this type of screening more practical for yeast laboratories [131–133], and automated image-based analysis of cell polarity phenotypes should become routine. This is important because visual analysis of complex phenotypes is highly subjective and qualitative, while computational extraction of cellular features and subsequent statistical analysis provides an essential foundation for quantitative biology. Finally, proteomics approaches are also being expanded to take into account the dynamic nature of cellular processes, defining abundance, localization, protein–protein interactions and post-translational modification for the yeast proteome, as well as changes in the proteome in response to different growth conditions [116,117,134]. In the future, using techniques that allow quantification of absolute protein levels at the proteome level, for example stable isotope labelling by amino acids in cell culture (SILAC) [135,136], researchers will be able to accurately assign protein abundance and stoichiometry of cell polarity complexes in specific conditions, for instance the phases of the cell cycle.
Computational and mathematical modelling of the cell polarity machinery also provides new perspectives in understanding cell polarity. Some models are fairly linear and are based on positive feedback and global inhibition [137,138], whereas others are more complex and aim to directly address molecular interactions underlying cellular polarization [139,140]. Generally, these models rely on the application and interpretation of our current knowledge of specific targeting sequences and domains of proteins and have already been successfully applied to identify candidate genes, whose roles in polarity have then been assessed experimentally [141]. For example, recent work revealed that multiple clusters of polarity landmarks are formed in budding yeast cells and that the bud site is selected via competition between these sites followed by negative feedback to dismantle all clusters but one [142]. Interestingly, this observation predicts oscillatory polarization, as the positive feedback loop created by the polarity clusters is rapidly antagonized by the negative feedback loop, and hints that this system could be adapted to polarize several axes to produce the complex morphologies observed in higher eukaryotes. The integration of genome-wide data with the predictive power of computational modelling provides a unique way to survey previously unexplored areas of the complete cellular network. Very few groups have ventured into the field of mathematical modelling at a genome-wide level, but the recent success of Chau et al. [141] in modelling all possible feedback loops capable of polarizing the cell highlights the potential for future research in this area.
In summary, the budding yeast continues to provide a robust experimental platform to study gene function and malfunction, and a guide for studying and interpreting biological pathways in more complicated systems. Indeed, hypotheses generated in modern applications of yeast systems biology have contributed numerous valuable insights to the broad range of cellular activities that underlie the core biological process of polarity. More than 20 years ago, studies in S. cerevisiae pioneered the identification of the first major cell polarity regulators that are responsible for orienting cell growth. More recent work using high-throughput functional genomics approaches has begun to make sense of how molecular interactions between key polarity determinants are organized within cells and has provided new insight into how polarity is generated and maintained. Ultimately, these types of studies aim to approach the goal of a complete description of the functions and interactions of all the molecular components of a basic eukaryotic cell.
References
- 1.Botstein D, Fink GR. 2011. Yeast: an experimental organism for 21st century biology. Genetics 189, 695–704 (doi:10.1534/genetics.111.130765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bi E, Park HO. 2012. Cell polarization and cytokinesis in budding yeast. Genetics 191, 347–387 (doi:10.1534/genetics.111.132886) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullen PJ, Sprague GF. 2012. The regulation of filamentous growth in yeast. Genetics 190, 23–49 (doi:10.1534/genetics.111.127456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drubin DG. 1991. Development of cell polarity in budding yeast. Cell 65, 1093–1096 (doi:10.1016/0092-8674(91)90001-F) [DOI] [PubMed] [Google Scholar]
- 5.Pringle JR, Bi E, Harkins HA, Zahner JE, De Virgilio C, Chant J, Corrado K, Fares H. 1995. Establishment of cell polarity in yeast. Cold Spring Harb. Symp. Quant. Biol. 60, 729–744 (doi:10.1101/SQB.1995.060.01.079) [DOI] [PubMed] [Google Scholar]
- 6.Herskowitz I, Park HO, Sanders S, Valtz N, Peter M. 1995. Programming of cell polarity in budding yeast by endogenous and exogenous signals. Cold Spring Harb. Symp. Quant. Biol. 60, 717–727 (doi:10.1101/SQB.1995.060.01.078) [DOI] [PubMed] [Google Scholar]
- 7.Park HO, Bi E, Pringle JR, Herskowitz I. 1997. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc. Natl Acad. Sci. USA 94, 4463–4468 (doi:10.1073/pnas.94.9.4463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng Y, Hart MJ, Shinjo K, Evans T, Bender A, Cerione RA. 1993. Biochemical comparisons of the Saccharomyces cerevisiae Bem2 and Bem3 proteins. Delineation of a limit Cdc42 GTPase-activating protein domain. J. Biol. Chem. 268, 24 629–24 634 [PubMed] [Google Scholar]
- 9.Chant J. 1999. Cell polarity in yeast. Annu. Rev. Cell Dev. Biol. 15, 365–391 (doi:10.1146/annurev.cellbio.15.1.365) [DOI] [PubMed] [Google Scholar]
- 10.Pruyne D, Bretscher A. 2000. Polarization of cell growth in yeast. J. Cell Sci. 113, 571–585 [DOI] [PubMed] [Google Scholar]
- 11.Govindan B, Novick P. 1995. Development of cell polarity in budding yeast. J. Exp. Zool. 273, 401–424 (doi:10.1002/jez.1402730505) [DOI] [PubMed] [Google Scholar]
- 12.Schott D, Ho J, Pruyne D, Bretscher A. 1999. The COOH-terminal domain of Myo2p, a yeast myosin V, has a direct role in secretory vesicle targeting. J. Cell Biol. 147, 791–808 (doi:10.1083/jcb.147.4.791) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill KL, Catlett NL, Weisman LS. 1996. Actin and myosin function in directed vacuole movement during cell division in Saccharomyces cerevisiae. J. Cell Biol. 135, 1535–1549 (doi:10.1083/jcb.135.6.1535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoepfner D, van den Berg M, Philippsen P, Tabak HF, Hettema EH. 2001. A role for Vps1p, actin, and the Myo2p motor in peroxisome abundance and inheritance in Saccharomyces cerevisiae. J. Cell Biol. 155, 979–990 (doi:10.1083/jcb.200107028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossanese OW, Reinke CA, Bevis BJ, Hammond AT, Sears IB, O'Connor J, Glick BS. 2001. A role for actin, Cdc1p, and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J. Cell Biol. 153, 47–62 (doi:10.1083/jcb.153.1.47) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beach DL, Thibodeaux J, Maddox P, Yeh E, Bloom K. 2000. The role of the proteins Kar9 and Myo2 in orienting the mitotic spindle of budding yeast. Curr. Biol. 10, 1497–1506 (doi:10.1016/S0960-9822(00)00837-X) [DOI] [PubMed] [Google Scholar]
- 17.Tcheperegine SE, Gao X-D, Bi E. 2005. Regulation of cell polarity by interactions of Msb3 and Msb4 with Cdc42 and polarisome components. Mol. Cell Biol. 25, 8567–8580 (doi:10.1128/MCB.25.19.8567-8580.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moseley JB, Goode BL. 2006. The yeast actin cytoskeleton, from cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70, 605–645 (doi:10.1128/MMBR.00013-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. 1990. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J. Cell Biol. 111, 131–142 (doi:10.1083/jcb.111.1.131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engqvist-Goldstein AEY, Drubin DG. 2003. Actin assembly and endocytosis: from yeast to mammals. Annu. Rev. Cell Dev. Biol. 19, 287–332 (doi:10.1146/annurev.cellbio.19.111401.093127) [DOI] [PubMed] [Google Scholar]
- 21.Casamayor A, Snyder M. 2003. Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization, and function. Mol. Cell Biol. 23, 2762–2777 (doi:10.1128/MCB.23.8.2762-2777.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asano S, et al. 2006. Direct phosphorylation and activation of a Nim1-related kinase Gin4 by Elm1 in budding yeast. J. Biol. Chem. 281, 27 090–27 098 (doi:10.1074/jbc.M601483200) [DOI] [PubMed] [Google Scholar]
- 23.Lippincott J, Shannon KB, Shou W, Deshaies RJ, Li R. 2001. The Tem1 small GTPase controls actomyosin and septin dynamics during cytokinesis. J. Cell Sci. 114, 1379–1386 [DOI] [PubMed] [Google Scholar]
- 24.Caviston JP, Longtine M, Pringle JR, Bi E. 2003. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol. Biol. Cell 14, 4051–4066 (doi:10.1091/mbc.E03-04-0247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arkowitz RA. 2009. Chemical gradients and chemotropism in yeast. Cold Spring Harb. Perspect. Biol. 1, a001958 (doi:10.1101/cshperspect.a001958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito H. 2010. Regulation of cross-talk in yeast MAPK signaling pathways. Curr. Opin. Microbiol. 13, 677–683 (doi:10.1016/j.mib.2010.09.001) [DOI] [PubMed] [Google Scholar]
- 27.Waltermann C, Klipp E. 2010. Signal integration in budding yeast. Biochem. Soc. Trans. 38, 1257–1264 (doi:10.1042/BST0381257) [DOI] [PubMed] [Google Scholar]
- 28.Dickinson JR. 2008. Filament formation in Saccharomyces cerevisiae: a review. Folia Microbiol. (Praha) 53, 3–14 (doi:10.1007/s12223-008-0001-6) [DOI] [PubMed] [Google Scholar]
- 29.Winzeler EA, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 (doi:10.1126/science.285.5429.901) [DOI] [PubMed] [Google Scholar]
- 30.Giaever G, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (doi:10.1038/nature00935) [DOI] [PubMed] [Google Scholar]
- 31.Ben-Aroya S, Coombes C, Kwok T, O'Donnell KA, Boeke JD, Hieter P. 2008. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell. 30, 248–258 (doi:10.1016/j.molcel.2008.02.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, et al. 2011. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 29, 361–367 (doi:10.1038/nbt.1832) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mnaimneh S, et al. 2004. Exploration of essential gene functions via titratable promoter alleles. Cell 118, 31–44 (doi:10.1016/j.cell.2004.06.013) [DOI] [PubMed] [Google Scholar]
- 34.Breslow DK, et al. 2008. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 5, 711–718 (doi:10.1038/nmeth.1234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, Oshea EK. 2003. Global analysis of protein localization in budding yeast. Nature 425, 686–691 (doi:10.1038/nature02026) [DOI] [PubMed] [Google Scholar]
- 36.Breker M, Gymrek M, Schuldiner M. 2013. A novel single-cell screening platform reveals proteome plasticity during yeast stress responses. J. Cell Biol. 200, 839–850 (doi:10.1083/jcb.201301120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghaemmaghami S, Huh W, Bower K, Howson R. 2003. Global analysis of protein expression in yeast. Nature 425, 737–741 (doi:10.1038/nature02046) [DOI] [PubMed] [Google Scholar]
- 38.Sopko R, et al. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell 21, 319–330 (doi:10.1016/j.molcel.2005.12.011) [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, et al. 2007. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 17, 536–543 (doi:10.1101/gr.6037607) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelperin DM, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 19, 2816–2826 (doi:10.1101/gad.1362105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho CH, et al. 2009. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotechnol. 27, 369–377 (doi:10.1038/nbt.1534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 5, 239–241 (doi:10.1038/nmeth.1181) [DOI] [PubMed] [Google Scholar]
- 43.Magtanong L, et al. 2011. Dosage suppression genetic interaction networks enhance functional wiring diagrams of the cell. Nat. Biotechnol. 29, 505–511 (doi:10.1038/nbt.1855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson DI, Pringle JR. 1990. Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity. J. Cell Biol. 111, 143–152 (doi:10.1083/jcb.111.1.143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amy Hin Yan T, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (doi:10.1126/science.1065810) [DOI] [PubMed] [Google Scholar]
- 46.Tong AHY, et al. 2004. Global mapping of the yeast genetic interaction network. Science 303, 808–813 (doi:10.1126/science.1091317) [DOI] [PubMed] [Google Scholar]
- 47.Costanzo M, et al. 2010. The genetic landscape of a cell. Science 327, 425–431 (doi:10.1126/science.1180823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguilar PS, et al. 2010. A plasma-membrane E-MAP reveals links of the eisosome with sphingolipid metabolism and endosomal trafficking. Nat. Struct. Mol. Biol. 17, 901–908 (doi:10.1038/nsmb.1829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goehring AS, Mitchell DA, Tong AHY, Keniry ME, Boone C, Sprague GF. 2003. Synthetic lethal analysis implicates Ste20p, a p21-activated protein kinase, in polarisome activation. Mol. Biol. Cell 14, 1501–1516 (doi:10.1091/mbc.E02-06-0348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuldiner M, et al. 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123, 507–519 (doi:10.1016/j.cell.2005.08.031) [DOI] [PubMed] [Google Scholar]
- 51.Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, Boone C, Hall MN, Emr SD. 2004. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J. 23, 3747–3757 (doi:10.1038/sj.emboj.7600384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou J, Friesen H, Larson J, Huang D, Cox M, Tatchell K, Andrews B. 2009. Regulation of cell polarity through phosphorylation of Bni4 by Pho85 G1 cyclin-dependent kinases in Saccharomyces cerevisiae. Mol. Biol. Cell 20, 3239–3250 (doi:10.1091/mbc.E08-12-1255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burston HE, Maldonado-Baez L, Davey M, Montpetit B, Schluter C, Wendland B, Conibear E. 2009. Regulators of yeast endocytosis identified by systematic quantitative analysis. J. Cell Biol. 185, 1097–1110 (doi:10.1083/jcb.200811116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walhout M, Vidal M, Dekker J. 2012. Handbook of systems biology: concepts and insights. London, UK: Academic Press. [Google Scholar]
- 55.Baryshnikova A, et al. 2010. Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat. Methods 7, 1017–1024 (doi:10.1038/nmeth.1534) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hillenmeyer ME, et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 (doi:10.1126/science.1150021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hillenmeyer ME, Ericson E, Davis RW, Nislow C, Koller D, Giaever G. 2010. Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol. 11, R30 (doi:10.1186/gb-2010-11-3-r30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith AM, et al. 2010. Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 38, e142 (doi:10.1093/nar/gkq368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Onge RPS, Mani R, Oh J, Proctor M, Fung E, Davis RW, Nislow C, Roth FP, Giaever G. 2007. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat. Genet. 39, 199–206 (doi:10.1038/ng1948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Narayanaswamy R, Niu W, Scouras AD, Hart GT, Davies J, Ellington AD, Iyer VR, Marcotte EM. 2006. Systematic profiling of cellular phenotypes with spotted cell microarrays reveals mating-pheromone response genes. Genome Biol. 7, R6 (doi:10.1186/gb-2006-7-1-r6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14, 450–456 (doi:10.1038/ng1296-450) [DOI] [PubMed] [Google Scholar]
- 62.Haarer B, Viggiano S, Hibbs MA, Troyanskaya OG, Amberg DC. 2007. Modeling complex genetic interactions in a simple eukaryotic genome: actin displays a rich spectrum of complex haploinsufficiencies. Genes Dev. 21, 148–159 (doi:10.1101/gad.1477507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haarer B, Aggeli D, Viggiano S, Burke DJ, Amberg DC. 2011. Novel interactions between actin and the proteasome revealed by complex haploinsufficiency. PLoS Genet. 7, e1002288 (doi:10.1371/journal.pgen.1002288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sopko R, Huang D, Smith JC, Figeys D, Andrews BJ. 2007. Activation of the Cdc42p GTPase by cyclin-dependent protein kinases in budding yeast. EMBO J. 26, 4487–4500 (doi:10.1038/sj.emboj.7601847) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prosser DC, Drivas TG, Maldonado-Báez L, Wendland B. 2011. Existence of a novel clathrin-independent endocytic pathway in yeast that depends on Rho1 and formin. J. Cell Biol. 195, 657–671 (doi:10.1083/jcb.201104045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biver S, Portetelle D, Vandenbol M. 2011. Multicopy suppression screen in a Saccharomyces cerevisiae strain lacking the Rab GTPase-activating protein Msb3p. Biotechnol. Lett. 33, 123–129 (doi:10.1007/s10529-010-0407-5) [DOI] [PubMed] [Google Scholar]
- 67.Boettner DR, D'Agostino JL, Torres OT, Daugherty-Clarke K, Uygur A, Reider A, Wendland B, Lemmon SK, Goode BL. 2009. The F-BAR protein Syp1 negatively regulates WASp-Arp2/3 complex activity during endocytic patch formation. Curr. Biol. 19, 1979–1987 (doi:10.1016/j.cub.2009.10.062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Höfken T, Schiebel E. 2002. A role for cell polarity proteins in mitotic exit. EMBO J. 21, 4851–4862 (doi:10.1093/emboj/cdf481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll SY, Stirling PC, Stimpson HEM, Giesselmann E, Schmitt MJ, Drubin DG. 2009. A yeast killer toxin screen provides insights into A/B toxin entry, trafficking, and killing mechanisms. Dev. Cell. 17, 552–560 (doi:10.1016/j.devcel.2009.08.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pagé N, et al. 2003. A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 163, 875–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sambade M, Alba M, Smardon AM, West RW, Kane PM. 2005. A genomic screen for yeast vacuolar membrane ATPase mutants. Genetics 170, 1539–1551 (doi:10.1534/genetics.105.042812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lesage G, Sdicu A-M, Menard P, Shapiro J, Hussein S, Bussey H. 2004. Analysis of beta-1,3-glucan assembly in Saccharomyces cerevisiae using a synthetic interaction network and altered sensitivity to caspofungin. Genetics 167, 35–49 (doi:10.1534/genetics.167.1.35) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang L, et al. 2011. A high-throughput screen for chemical inhibitors of exocytic transport in yeast. ChemBioChem 11, 1291–1301 (doi:10.1002/cbic.200900681) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi Y, Stefan CJ, Rue SM, Teis D, Emr SD. 2011. Two novel WD40 domain-containing proteins, Ere1 and Ere2, function in the retromer-mediated endosomal recycling pathway. Mol. Biol. Cell 22, 4093–4107 (doi:10.1091/mbc.E11-05-0440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zewail A, Xie MW, Xing Y, Lin L, Zhang PF, Zou W, Saxe JP, Huang J. 2003. Novel functions of the phosphatidylinositol metabolic pathway discovered by a chemical genomics screen with wortmannin. Proc. Natl Acad. Sci. USA 100, 3345–3350 (doi:10.1073/pnas.0530118100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chasse SA, Flanary P, Parnell SC, Hao N, Cha JY, Siderovski DP, Dohlman HG. 2006. Genome-scale analysis reveals Sst2 as the principal regulator of mating pheromone signaling in the yeast Saccharomyces cerevisiae. Eukaryot. Cell 5, 330–346 (doi:10.1128/EC.5.2.330-346.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slessareva JE, Routt SM, Temple B, Bankaitis VA, Dohlman HG. 2006. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein alpha subunit at the endosome. Cell 126, 191–203 (doi:10.1016/j.cell.2006.04.045) [DOI] [PubMed] [Google Scholar]
- 78.Schluter C, Lam KKY, Brumm J, Wu BW, Saunders M, Stevens TH, Bryan J, Conibear E. 2008. Global analysis of yeast endosomal transport identifies the vps55/68 sorting complex. Mol. Biol. Cell 19, 1282–1294 (doi:10.1091/mbc.E07-07-0659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bonangelino CJ, Chavez EM, Bonifacino JS. 2002. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol. Biol. Cell 13, 2486–2501 (doi:10.1091/mbc.02-01-0005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lorenz MC, Heitman J. 1998. Regulators of pseudohyphal differentiation in Saccharomyces cerevisiae identified through multicopy suppressor analysis in ammonium permease mutant strains. Genetics 150, 1443–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang CM, Jiang YW. 2005. Genome-wide survey of non-essential genes required for slowed DNA synthesis-induced filamentous growth in yeast. Yeast 22, 79–90 (doi:10.1002/yea.1195) [DOI] [PubMed] [Google Scholar]
- 82.Jin R, Dobry CJ, McCown PJ, Kumar A. 2008. Large-scale analysis of yeast filamentous growth by systematic gene disruption and overexpression. Mol. Biol. Cell 19, 284–296 (doi:10.1091/mbc.E07-05-0519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pitoniak A, Birkaya B, Dionne HM, Vadaie N, Cullen PJ. 2009. The signaling mucins Msb2 and Hkr1 differentially regulate the filamentation mitogen-activated protein kinase pathway and contribute to a multimodal response. Mol. Biol. Cell 20, 3101–3114 (doi:10.1091/mbc.E08-07-0760) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mösch HU, Fink GR. 1997. Dissection of filamentous growth by transposon mutagenesis in Saccharomyces cerevisiae. Genetics 145, 671–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shively CA, Eckwahl MJ, Dobry CJ, Mellacheruvu D, Nesvizhskii A, Kumar A. 2013. Genetic networks inducing invasive growth in Saccharomyces cerevisiae identified through systematic genome-wide overexpression. Genetics 193, 1297–1310 (doi:10.1534/genetics.112.147876) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roberts RL, Fink GR. 1994. Elements of a single MAP kinase cascade in Saccharomyces cerevisiae mediate two developmental programs in the same cell type: mating and invasive growth. Genes Dev. 8, 2974–2985 (doi:10.1101/gad.8.24.2974) [DOI] [PubMed] [Google Scholar]
- 87.Ryan O, et al. 2012. Global gene deletion analysis exploring yeast filamentous growth. Science 337, 1353–1356 (doi:10.1126/science.1224339) [DOI] [PubMed] [Google Scholar]
- 88.Voordeckers K, De Maeyer D, van der Zande E, Vinces MD, Meert W, Cloots L, Ryan O, Marchai K, Verstrepen KJ. 2012. Identification of a complex genetic network underlying Saccharomyces cerevisiae colony morphology. Mol. Microbiol. 86, 225–239 (doi:10.1111/j.1365-2958.2012.08192.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Narayanaswamy R, Moradi EK, Niu W, Hart GT, Davis M, McGary KL, Ellington AD. 2009. Systematic definition of protein constituents along the major polarization axis reveals an adaptive reuse of the polarization machinery in pheromone-treated budding yeast. J. Proteome Res. 8, 6–19 (doi:10.1021/pr800524g) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ohya Y, et al. 2005. High-dimensional and large-scale phenotyping of yeast mutants. Proc. Natl Acad. Sci. USA 102, 19 015–19 020 (doi:10.1073/pnas.0509436102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ni L, Snyder M. 2001. A genomic study of the bipolar bud site selection pattern in Saccharomyces cerevisiae. Mol. Biol. Cell 12, 2147–2170 (doi:10.1091/mbc.12.7.2147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arlt H, Perz A, Ungermann C. 2011. An overexpression screen in Saccharomyces cerevisiae identifies novel genes that affect endocytic protein trafficking. Traffic 12, 1592–1603 (doi:10.1111/j.1600-0854.2011.01252.x) [DOI] [PubMed] [Google Scholar]
- 93.Bircham PW, Maass DR, Roberts CA, Kiew PY, Low YS, Yegambaram M, Matthews J, Jack CA, Atkinson PH. 2011. Secretory pathway genes assessed by high-throughput microscopy and synthetic genetic array analysis. Mol. BioSyst. 7, 2589–2598 (doi:10.1039/c1mb05175j) [DOI] [PubMed] [Google Scholar]
- 94.Singh J, Tyers M. 2009. A Rab escort protein integrates the secretion system with TOR signaling and ribosome biogenesis. Genes Dev. 23, 1944–1958 (doi:10.1101/gad.1804409) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Watanabe M, Watanabe D, Nogami S, Morishita S, Ohya Y. 2009. Comprehensive and quantitative analysis of yeast deletion mutants defective in apical and isotropic bud growth. Curr. Genet. 55, 365–380 (doi:10.1007/s00294-009-0251-0) [DOI] [PubMed] [Google Scholar]
- 96.Proszynski TJ, et al. 2005. A genome-wide visual screen reveals a role for sphingolipids and ergosterol in cell surface delivery in yeast. Proc. Natl Acad. Sci. USA 102, 17 981–17 986 (doi:10.1073/pnas.0509107102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frohlich F, Moreira K, Aguilar PS, Hubner NC, Mann M, Walter P, Walther TC. 2009. A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J. Cell Biol. 185, 1227–1242 (doi:10.1083/jcb.200811081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grossmann G, et al. 2008. Plasma membrane microdomains regulate turnover of transport proteins in yeast. J Cell Biol. 183, 1075–1088 (doi:10.1083/jcb.200806035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. 2012. Segregation of molecules at cell division reveals native protein localization. Nat. Methods 9, 480–482 (doi:10.1038/nmeth.1955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA 98, 4569–4574 (doi:10.1073/pnas.061034498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yu H, et al. 2008. High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110 (doi:10.1126/science.1158684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fields S, Song O. 1989. A novel genetic system to detect protein–protein interactions. Nature 340, 245–246 (doi:10.1038/340245a0) [DOI] [PubMed] [Google Scholar]
- 103.Drees BL, et al. 2001. A protein interaction map for cell polarity development. J. Cell Biol. 154, 549–571 (doi:10.1083/jcb.200104057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roumanie O, Weinachter C, Larrieu I, Crouzet M, Doignon F. 2001. Functional characterization of the Bag7, Lrg1 and Rgd2 RhoGAP proteins from Saccharomyces cerevisiae. FEBS Lett. 506, 149–156 (doi:10.1016/S0014-5793(01)02906-4) [DOI] [PubMed] [Google Scholar]
- 105.Stagljar I, Korostensky C, Johnsson N, Heesen te S. 1998. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl Acad. Sci. USA 95, 5187–5192 (doi:10.1073/pnas.95.9.5187) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wittke S, Lewke N, Müller S, Johnsson N. 1999. Probing the molecular environment of membrane proteins in vivo. Mol. Biol. Cell 10, 2519–2530 (doi:10.1091/mbc.10.8.2519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Remy I, Ghaddar G, Michnick SW. 2007. Using the beta-lactamase protein-fragment complementation assay to probe dynamic protein–protein interactions. Nat. Protoc. 2, 2302–2306 (doi:10.1038/nprot.2007.356) [DOI] [PubMed] [Google Scholar]
- 108.Tarassov K, et al. 2008. An in vivo map of the yeast protein interactome. Science 320, 1465–1470 (doi:10.1126/science.1153878) [DOI] [PubMed] [Google Scholar]
- 109.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17, 1030–1032 (doi:10.1038/13732) [DOI] [PubMed] [Google Scholar]
- 110.Gavin A-C, et al. 2006. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 (doi:10.1038/nature04532) [DOI] [PubMed] [Google Scholar]
- 111.Krogan NJ, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 (doi:10.1038/nature04670) [DOI] [PubMed] [Google Scholar]
- 112.Wiederhold E, Gandhi T, Permentier HP, Breitling R, Poolman B, Slotboom DJ. 2008. The yeast vacuolar membrane proteome. Mol. Cell. Proteomics 8, 380–392 (doi:10.1074/mcp.M800372-MCP200) [DOI] [PubMed] [Google Scholar]
- 113.Yin QY. 2005. Comprehensive proteomic analysis of Saccharomyces cerevisiae cell walls identification of proteins covalently attached via glycosylphosphatidylinositol remnants or mild alkali-sensitive linkages. J. Biol. Chem. 280, 20 894–20 901 (doi:10.1074/jbc.M500334200) [DOI] [PubMed] [Google Scholar]
- 114.Navarre C, Degand H, Bennett KL, Crawford JS, Mørtz E, Boutry M. 2002. Subproteomics: identification of plasma membrane proteins from the yeast Saccharomyces cerevisiae. Proteomics 2, 1706–1714 (doi:10.1002/1615-9861(200212)2:12<1706::AID-PROT1706>3.0.CO;2-K) [DOI] [PubMed] [Google Scholar]
- 115.Gruhler A. 2005. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 (doi:10.1074/mcp.M400219-MCP200) [DOI] [PubMed] [Google Scholar]
- 116.Babu M, et al. 2012. Interaction landscape of membrane–protein complexes in Saccharomyces cerevisiae. Nature 489, 585–589 (doi:10.1038/nature11354) [DOI] [PubMed] [Google Scholar]
- 117.Picotti P, et al. 2013. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494, 266–270 (doi:10.1038/nature11835) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ashburner M, et al. 2000. Gene ontology, tool for the unification of biology. Nat. Genet. 25, 25–29 (doi:10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Endo M, Takesako K, Kato I, Yamaguchi H. 1997. Fungicidal action of aureobasidin A, a cyclic depsipeptide antifungal antibiotic, against Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 41, 672–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yan A, Ahmed E, Yan Q, Lennarz WJ. 2003. New findings on interactions among the yeast oligosaccharyl transferase subunits using a chemical cross-linker. J. Biol. Chem. 278, 33 078–33 087 (doi:10.1074/jbc.M305337200) [DOI] [PubMed] [Google Scholar]
- 121.Knauer R, Lehle L. 1999. The oligosaccharyltransferase complex from Saccharomyces cerevisiae. Isolation of the OST6 gene, its synthetic interaction with OST3, and analysis of the native complex. J. Biol. Chem. 274, 17 249–17 256 (doi:10.1074/jbc.274.24.17249) [DOI] [PubMed] [Google Scholar]
- 122.Levy SF, Siegal ML. 2008. Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol. 6, e264 (doi:10.1371/journal.pbio.0060264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Aguilaniu H, Gustafsson L, Rigoulet M, Nyström T. 2003. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 (doi:10.1126/science.1080418) [DOI] [PubMed] [Google Scholar]
- 124.Meitinger F, Richter H, Heisel S, Hub B, Seufert W, Pereira G. 2013. A safeguard mechanism regulates Rho GTPases to coordinate cytokinesis with the establishment of cell polarity. PLoS Biol. 11, e1001495 (doi:10.1371/journal.pbio.1001495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu J, Cropp TA. 2012. A method for multi-codon scanning mutagenesis of proteins based on asymmetric transposons. Protein Eng. Des. Sel. 25, 67–72 (doi:10.1093/protein/gzr060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ben-Aroya S, Pan X, Boeke JD, Hieter P. 2010. Making temperature-sensitive mutants. Methods Enzymol. 470, 181–204 (doi:10.1016/S0076-6879(10)70008-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Araya CL, Fowler DM. 2011. Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 29, 435–442 (doi:10.1016/j.tibtech.2011.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Measday V, et al. 2005. Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc. Natl Acad. Sci. USA 102, 13 956–13 961 (doi:10.1073/pnas.0503504102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Boettner DR, Friesen H, Andrews B, Lemmon SK. 2011. Clathrin light chain directs endocytosis by influencing the binding of the yeast Hip1R homologue, Sla2, to F-actin. Mol. Biol. Cell 22, 3699–3714 (doi:10.1091/mbc.E11-07-0628) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vizeacoumar FJ, Chong Y, Boone C, Andrews BJ. 2009. A picture is worth a thousand words: genomics to phenomics in the yeast Saccharomyces cerevisiae. FEBS Lett. 583, 1656–1661 (doi:10.1016/j.febslet.2009.03.068) [DOI] [PubMed] [Google Scholar]
- 131.Carpenter AE, et al. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 (doi:10.1186/gb-2006-7-10-r100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lamprecht MR, Sabatini DM, Carpenter AE. 2007. CellProfiler: free, versatile software for automated biological image analysis. BioTechniques 42, 71–75 (doi:10.2144/000112257) [DOI] [PubMed] [Google Scholar]
- 133.Abramoff MD, Magalhães PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int. 11, 36–42 [Google Scholar]
- 134.Nagaraj N, et al. 2012. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteomics 11, M111.013722 (doi:10.1074/mcp.M111.013722) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rees J, Lilley K. 2011. Enabling technologies for yeast proteome analysis. Methods Mol. Biol. 759, 149–178 (doi:10.1007/978-1-61779-173-4_10) [DOI] [PubMed] [Google Scholar]
- 136.de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Fröhlich F, Walther TC, Mann M. 2008. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 (doi:10.1038/nature07341) [DOI] [PubMed] [Google Scholar]
- 137.Turing AM. 1952. The chemical basis of morphogenesis. Phil. Trans. R. Soc. Lond. B 237, 37–72 (doi:10.1098/rstb.1952.0012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Parent CA, Devreotes PN. 1999. A cell's sense of direction. Science 284, 765–770 (doi:10.1126/science.284.5415.765) [DOI] [PubMed] [Google Scholar]
- 139.Hodgkin AL, Huxley AF. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Layton AT, Savage NS, Howell AS, Carroll SY, Drubin DG, Lew DJ. 2011. Modeling vesicle traffic reveals unexpected consequences for Cdc42p-mediated polarity establishment. Curr. Biol. 21, 184–194 (doi:10.1016/j.cub.2011.01.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chau AH, Walter JM, Gerardin J, Tang C, Lim WA. 2012. Designing synthetic regulatory networks capable of self-organizing cell polarization. Cell 151, 320–332 (doi:10.1016/j.cell.2012.08.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Howell AS, Jin M, Wu C-F, Zyla TR, Elston TC, Lew DJ. 2012. Negative feedback enhances robustness in the yeast polarity establishment circuit. Cell 149, 322–333 (doi:10.1016/j.cell.2012.03.012) [DOI] [PMC free article] [PubMed] [Google Scholar]