

Abstract
The p53 protein is a sequence-specific DNA-binding factor that regulates inflammatory genes such as CCL2/MCP-1 that may play a role in various diseases. A recent study has indicated that the knockdown of human p53 leads to a strong negative regulation of CCL2 induction. We are therefore interested in how p53 regulates CCL2 gene expression. In the following study, our findings indicate that UV-induced p53 accumulation in mouse macrophages significantly decreases LPS-induced CCL2 production, and that p53 binds to CCL2 5’UTR in the region (16-35). We also found that a p53 domain (p53pep170) mimics full length p53 to down-regulate CCL2 promoter activity. Treatment of p53-deficient mouse primary macrophages with synthetic p53pep170 was found to decrease LPS-induced production of CCL2 without association with cellular endogenous p53. CCL2 production induced by lentiCLG in human monocytes or mouse primary macrophages was blocked in the presence of p53pep170. Overall, these results demonstrate that p53 or its derived peptide (p53pep170) is an important regulator of CCL2 gene expression via its binding activity, and acts as a novel model for future studies linking p53 and its short peptide to pave the way to possible pharmaceutical intervention of CCL2-mediated inflammatory and cancer diseases.
Keywords: p53, CCL2, transcriptional regulation, macrophage, infection
INTRODUCTION
It is well known that p53 regulates apoptotic signaling pathways. Isoforms of human p53 with alternative translation initiation sites have been studied [1-3]. Aberrant expression of these isoforms occurs in a variety of tumors [4-6]. A mouse expressing an N-terminal deletion mutant of p53 (Δ122p53) has been recently used to in vivo explore the functions of p53 isoform for tumor development and has been found to be involved in the increased serum concentrations of interleukin-6 and other proinflammatory cytokines and lymphocyte aggregates in the lung and liver, as well as other pathologies [7]. Besides its role as a tumor suppressor, deficiency or mutation of p53 has also been linked to autoimmune disorders [8, 9] as well as lung inflammation [10]. As certain cancers are caused by chronic inflammation, regulation of inflammation may ultimately lead to tumor suppression [11, 12]. However, the potential role of p53 in the regulation of inflammatory genes of chronic lung disease [10, 13, 14] needs to be investigated further.
It is well known that chemokines and chemokine receptors are involved in a variety of inflammatory disorders [15-17]. CCL2 (also known as MCP-1) has been suggested as a potential therapeutic target in inflammatory diseases due to the gene’s several-fold production increase in the peripheral blood, synovial fluid, and synovial tissue of patients [18]. CCL2 is also suggested to be a potent mediator of monocytes/macrophages as these cells have been shown to be directly involved in the induction and perpetuation of synovitis and subsequent joint destruction in rheumatoid arthritis [19, 20]. Macrophages/monocytes modulated by the CCL2/CCR2 axis have been recently found to be key players in the pathology of many human glomerular and tubulointerstitial diseases [21]. CCL2 has been found to be a regulated gene at the protein level and elevated in cancer cells [22]. Inhibition of CCL2 during tumor development resulted in decreased tumor volume in tumor-bearing mice [23]. Thus, the inhibition of elevated CCL2 production may provide a new therapeutic intervention in CCL2-induced inflammatory and cancer diseases [24]. Additionally, the transcriptional mechanisms of the CCL2 gene have been studied. The binding of transcription factors such as SP1 and NF B in the proximal region regulate basal CCL2 gene transcription in response to LPS stimulation [25-30].
We have previously shown that p53 is involved in inflammatory gene expression via regulation of its promoter activity [31, 32]. Additionally, a recent study has indicated that the knockdown of human p53 leads to a strong negative regulation of CCL2 induction [33]. Therefore, we are interested in the involvement of p53 in CCL2 gene expression.
In the present study, we found that UV-induced p53 accumulation in mouse macrophages significantly decreased LPS-induced CCL2 production, and that p53 naturally occurs in a cell by way of protein-DNA interaction with CCL2 5’UTR&promoter. In order to further investigate whether p53 is involved in CCL2 transcriptional activity, we analyzed CCL2 5’UTR& promoter, along with its derived DNA constructs. We found that the specific binding site for p53-CCL2 interaction is located in the region of CCL2 5’UTR& promoter (16-35), and that a p53 domain (aa 162-170: KSQHMTEVV, named p53pep170) mimics full length p53 to down-regulate CCL2 promoter activity. Treatment of mouse primary macrophages with synthetic p53pep170 was found to decrease LPS-induced production of CCL2 and cytokines such as TNF-α and IL-15. Using macrophages (p53-/-) from p53-deficient mice, we found that p53pep170 played an essential role in the inhibition of CCL2 production without association of cellular endogenous p53. In the presence of synthetic p53pep170, infection of lentiviral CCL2 (LentiCLG) was significantly blocked in human monocytes and mouse primary macrophages. These results suggest that p53, or its derived p53pep170 peptide, is an important regulator of CCL2 gene expression. This study may pave the way for future studies linking p53 and its short peptide to possible pharmaceutical intervention of inflammatory diseases and cancer mediated by CCL2.
MATERIALS AND METHODS
Cell Culture
All bacterial cloning constructs used Escherichia coli strain Top10 (Invitrogen). U2OS (Human osteosarcoma) cells from American Type Culture Collection (ATCC, Manassas, VA) and A549 (Human lung cancer cells from ATCC) were grown in DMEM with 10% fetal bovine serum (FBS) and maintained in a 37°C humidified atmosphere containing 5% CO2. Mouse primary macrophages elicited by i.p. thioglycollate were obtained from C57BL/6 mice (The Jackson Laboratory) or p53-deficient mice (bred by our lab) and purified by conventional methods [34]. The human monocytes were purchased from Advanced Biotechnologies Inc (ABI, Columbia, MD). Both mouse primary macrophages and human monocytes were grown in RPMI medium 1640 supplemented with 10% FBS and maintained in a humidified atmosphere of 5% CO2 at 37°C.
Breeding p53-Deficient Mice
Mice with p53 heterozygous type (129S2-Trp53tm1TYj/J, The Jackson Laboratory, Bar Harbor, ME) were bred to generate a number of wild-type as control, or homozygous p53-deficient mice following BU IACUC instructions (Protocol number: AN-15138).
Establishment of a Stable Cell Line
1x106 A549 cells were infected with MOI:2 of LentiCLG (as described below in Section 3, DNA constructs) for 3 days. A single cell containing CCL2 5’UTR&promoter/CCL2/luciferase/GFP was screened and transferred into a new plate until enough cells (About 10-100 cells) were grown. The stable lung cancer cell line with biological markers (luciferase and GFP, driven by CCL2 5’UTR&promoter enhance element) was confirmed by luciferase assay, microscopy-based observation, and PCR. Cells were collected and named A5CLG. Cells were grown in DMEM with 10% FBS and maintained in a 37°C humidified atmosphere containing 5% CO2.
DNA Constructs
Primer pairs used for PCR of DNA constructs are shown below (Table 1) and all cloned DNA were confirmed by DNA sequencing. 1. The clone mp53wt, which contains a full-length mouse p53 gene (aa 1-391) was provided by OpenBiosystems. A series of mouse p53 wild type or deletions was generated by PCR. For A-ALONE, B-ALONE or C-ALONE, they were added with one methionine (M) for initiation and one stop codon at the end. For mp53-dB, mp53-dC or mp53-muB, the first and second PCR-generated DNA fragments were diluted to 1ng per reaction and re-amplified by PCR with the primer pairs 5’-atgactgccatggaggagtc-3’ and 5’-tcagtctgagtcaggcccca-3’. A full-length mouse CCL2 gene (BC145867) was generated by PCR with 1ng of mouse cDNA (Provided by OpenBiosystems) as template. Finally, all PCR fragments above were inserted into pcDNA3HA [35]. 2. A series of truncated mouse CCL2 5’UTR&promoter DNAs (GQ917241) and IL-1β 5’UTR DNA fragment (X04964) was generated by PCR with mouse genomic DNA (Clontech) as template and appropriate primer pairs, as shown in the following table. All PCR products of reporter DNAs (CCL2 5’UTR&promoter or IL-1β 5’UTR) were purified and inserted into pGL3-basic vectors.
Table 1.
| DNA Clone | 1st PCR Amplification | 2d PCR Amplification | ||
|---|---|---|---|---|
| No | Name | Primer Pair | Primer Pair | |
| A series of mouse p53 DNA constructs | 1 | Mp53-d1 | 5’-atgactgccatggaggagtc-3’ + 5’-tcaatcttctggaggaagtagtt-3’ | |
| 2 | Mp53-d2 | 5’-atgactgccatggaggagtc-3’ + 5’-tcaagctcctgacactcggaggg-3’ | ||
| 3 | Mp53-d3 | 5’-atgactgccatggaggagtc-3’ + 5’-tcagtaagttttttgagaaggga-3’ | ||
| 4 | Mp53-d4 | 5’-atgactgccatggaggagtc-3’ + 5’-tcactggcagaatagcttattga-3’ | ||
| 5 | Mp53-d | 5’-atgactgccatggaggagtc-3’ + 5’-tcaagctggaggtgtggcgctga-3’ | ||
| 6 | Mp53-FA | 5’-atgactgccatggaggagtc-3’ + 5’-tcacttgtagatggccatggcgc-3’ | ||
| 7 | Mp53-FB | 5’-atgactgccatggaggagtc-3’ + 5’-tcacacgacctccgtcatgtgct-3’ | ||
| 8 | Mp53-FC | 5’-atgactgccatggaggagtc-3’ + 5’-tcaatcaccatcggagcagcgct-3’ | ||
| 9 | A-ALONE | 5’-atgcgcgccatggccatctacaagtaa-3’ + 5’-Ttacttgtagatggccatggcgcgcat-3’ | ||
| 10 | B-ALONE | 5’-atgaagtcacagcacatgacggaggtcgtgtaa-3’ + 5’-ttacacgacctccgtcatgtgctgtgacttcat-3’ | ||
| 11 | C-ALONE | 5’-atgagacgctgcccccaccatgagcgctgctccgatggtgattaa3’ + 5’-ttaatcaccatcggagcagcgctcatggtgggggcagcgtctcat-3’ | ||
| 12 | Mp53-dB | 5’-atgactgccatggaggagtc-3’ + 5’-cttgtagatggccatggcgc-3’ | 5’-gcgccatggccatctaCaagagacgctgcccccaccatgag-3’ + 5’-tcagtctgagtcaggcccca-3’ | |
| 13 | Mp53-dC | 5’-atgactgccatggaggagtc-3’ + 5’-cacgacctccgtcatgtgct-3’ | 5’-cagcacatgacGgaggtcgtgggcctggctcctccccagca-3’ + 5’-tcagtctgagtcaggcccca-3’ | |
| 14 | Mp53-muB | 5’-atgactgccatggaggagtc-3’ + 5’-ctcgtgcgtctgcaccatctttgagaccatcttgtagatggccatggc-3’ | 5’-gtctcaaagatggtgcagacgcaCgagagacgctgcccccaccat-3’ + 5’-tcagtctgagtcaggcccca-3’ | |
| 15 | mp53WT | 5’-atgactgccatggaggagtc-3’ + 5’-tcagtctgagtcaggcccca-3’ | ||
| mouse CCL2 5 ‘UTR&promoter DNA constructs | 16 | mcl2pwt | 5’-acgaaggaaacagggcagag-3¡̄ + 5’-ggtggtggaggaagagagag-3’ | |
| 17 | mcl2p715 | 5’-agtaacagcatctacttacc-3’ + 5’-ggtggtggaggaagagagag-3’ | ||
| 18 | mcl2p515 | 5’-ctgcaaaatatctggtaacc-3’ + 5’-ggtggtggaggaagagagag-3’ | ||
| 19 | mcl2p315 | 5’-gcagagccactccattcaca-3’ + 5’-ggtggtggaggaagagagag-3’ | ||
| 20 | mcl2p115 | 5’-caacttccactttccatcac-3’ + 5’-ggtggtggaggaagagagag-3’ | ||
| 21 | mCCL2wt | 5’- ctcgagccatgcaggtccctgtcatg -3’ + 5’- aagcttctagttcactgtcacactg -3’ | ||
| 22 | IL-1-β- p | 5’-aagtgcgtgtctctccag-3’ + 5’-agctgcttcagacacctg-3’ | ||
| 23 | mcl2p53m | 5’-caacttccactttccatcacttatccagggtgatgctactccttggcaccaagcaccctgcctgact ccacccccctggcttacaataaaaggctgcctc-3’ + 5’-ggtggtggaggaagagagagctggcttcagtgagagttggctggtgctggcgtctggctctctg cacttctggctgctctgaggcagccttttattgtaa-3’ | 5’-caacttccactttccatcac-3’ + 5’-ggtggtggaggaagagagag-3’ | |
3. Recombinant lentivirus. A mouse CCL2 5’UTR& promoter DNA (GQ917241) as template with the primer pairs: 5’-ggatcccaacttccactttccatcac-3’ and 5’-ctcgaggg tggtggaggaagagagag-3’, a mouse CCL2 in-frame DNA (BC145867) as template with primer pairs: 5’-CTCGAGATGCAGGTCCCTGTCAT-3’ and 5’-AAGCTTCTAGTTCACTGTCACACTG-3’, or a luciferase in-frame DNA (pGL3-basic plasmid, Promega) with primer pairs: 5’-AAGCTTATGGAAGACGCCAAAAAC-3’ and 5’-GGGCCCTTACACGGCGATCTTTCC-3’ (The underlined sequences are tagged enzymes: BamHI, XhoI, HindIII or ApaI) were generated by PCR. The PCR-amplified DNA fragments were digested with enzymes (BamHI, XhoI, HindIII, or ApaI) and ligated with T4 ligase. The ligated DNA fragment was recovered and inserted into pLenti6.3/V5-TOPO vectors (Invitrogen). The real cloned DNA with the right size and right orientation was screened and confirmed by DNA sequencing. The cloned DNA (CCL2 5’UTR&promoter/CCL2/luciferase/GFP) was co-transfected with ViraPower Packaging Mix (Invitrogen) into 293FT producer cells by using Lipofectamine 2000 (Invitrogen) and cultured at 37°C, 5% CO2 for 2-5 days. The viral pellet from its supernatant was harvested and suspended in an appropriate volume of PBS. The titer (1x108pfu) of viral particles (Named lentiCLG) was measured following manufacturer’s instructions. 4. Control lentivirus. pLenti6.3/V5-TOPO as backbone DNA without CCL2 insert fragment was co-transfected with ViraPower Packaging Mix into 293FT producer cells by using Lipofectamine 2000 and cultured at 37°C, 5% CO2 for 2-5 days. The viral pellet from its supernatant was harvested and suspended in an appropriate volume of PBS as described above. The titer (1x108pfu) of viral particles (named Lenti6.3virus) was measured following manufacturer’s instructions.
Luciferase Assay
Overnight precultured or freshly splitting cells were used for luciferase assay with a commercial kit (Cat#: E1500, luciferase reporter assay system, Promega) according to the protocol provided by the manufacturer.
ELISA
Medium from cells was analyzed by ELISA with the commercial kits (Cat#KMC0012 for IL-1β or Cat#KMC 1011 for CCL2, Invitrogen) following manufacturer’s instructions.
Bio-Plex Cytokine Assay
Mouse primary macrophages (1 x 106) were treated with 0.1 μg/ml E. coli LPS, 100 μg/ml SCpep, 10 μg/ml p53pep170, or 100 μg/ml p53pep170. Cells were then incubated overnight in RPMI 1640 supplemented with 10% FBS at 37°C and 5% CO2. Culture supernatants were harvested and centrifuged at 1,500 × g to remove cell debris. Concentrations of several cytokines and chemokine (CCL2) were measured for both the treated and untreated cell condition. The immunoreactivity was quantified by Bio-Plex 200 System (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions (MPXMCYTO-70K, Millipore) and then graphed.
IP-p53 Constructions
U2OS cells (1 x 106) were respectively transfected with individual p53 construction DNA using Lipofect-amine 2000 (Invitrogen) for 3 hrs, washed with PBS, and cultured overnight. The protein from treated cells was extracted with lysis buffer (Promega) and immuno-precipitated (IP) with HA plus IgG and a Protein A/G Plus-Agarose (sc-2003, Santa Cruz Biotechnology) following the manufacturer’s instructions. The IP protein from each concentration was used for EMSA.
Western Blot Analysis
Cell lysate from each experimental group was detected by Western blotting with antibodies against actin (C-11, Santa Cruz) or p53 (FL-393-G, Santa Cruz). Protein band intensity was analyzed using VersaDoc Imaging System model 4000MP with Quantity One Quantitation Software version 4.6.3 (Bio-Rad).
DNA Probes
A double stranded nucleotide of cl2/p53oligo (5’-ggaggaaggccagcccagca-3’) or SColigo, the control with scrambled DNA sequences (5’-gtctcaaagatggtgca gacgca cgag-3’) was synthesized and then radiolabeled with [γ32P]ATP using T4 polynucleotide kinase (Promega) following the manufacturer’s instructions. The labeled double-stranded DNA was purified using G-50 Sephadex columns (Boehringer, Ingelheim, Germany) and precipitated with ethanol. After centrifugation, the DNA was suspended in 10 μL water, measured for cpm/L and used as probes for electrophoretic mobility shift assay (EMSA), as described below.
EMSA
A commercial kit, Gel Shift Assay System (Promega), was used. IP-proteins used for EMSA were prepared as described above. The protein level of each IP-protein was confirmed by Western blot. A reaction mixture for EMSA contained 1 x 105 cpm/μL of radiolabeled double-stranded DNA probe (cl2/p53oligo or SColigo), 1mM DTT, 1 μg of each IP-protein (omitted from control), 2 μL of 5X binding buffer (Promega), and nuclease-free water to achieve a final volume of 10 μL. Mixtures were incubated at room temperature for 20 min, followed by electrophor-esis on nondenaturing 6% polyacrylamide gels in Tris-borate-EDTA buffer [90 mmol/L Tris-borate/2 mmol/L EDTA HEPES (pH 8)]. To evaluate the specificity of p53-CCL2 interactions, supershift was performed. 1μg anti-body (antiHA, sc-805, Santa Cruz) was mixed with IP-protein in binding buffer without probe for 30 min at room temperature, and then 32p-labeled probe was added with or without 1 pmol of cold competitor, and incubated for 30 min. The reaction was electrophoresed on 6% polyacryl-amide gels. The gel was dried and exposed to a SynGene BIO Imaging system (Frederick, MD). The signal was further improved by Photoshop (Adobe).
Isolation of Chromosomal DNA
DNA was isolated from A5CLG cells, or A549 cells as control, using a commercial kit (QuickGene DNA tissue kit, Fujifilm) based on the manufacturer’s instructions.
Chromatin Immunoprecipitation (ChIP)
Assay was performed with some modifications using a commercial kit (Cat#: 53009, ChIP-IT Express Enzymatic, Active Motif). 1x106 A5CLG cells or A549 cells as control were transfected with 1μg of p53 DNA overnight. The 10 μg nuclear extracts (NE) as input from the cross-linked cells were immunoprecipitated (IP) with 1μg p53 antibody (FL-393-G, Santa Cruz) or 1μg normal IgG as control at 4ºC for 4 hrs. DNA from each experimental group (Input or IP) was isolated by elution, reverse cross-linking and Proteinase K treatment according to the manufacturer’s instructions. The DNA was then used as a template to perform PCR with primer pairs, primerA+primerB (Fig. 4A) for amplification of a 210bp DNA fragment of CCL2 5’UTR&promoter or primerA+primerD for amplification of a 410bp DNA fragment of CCL2 5’UTR&promoter+ cDNA or GAPDH primer pairs (Invitrogen) as control.
Fig. (4).
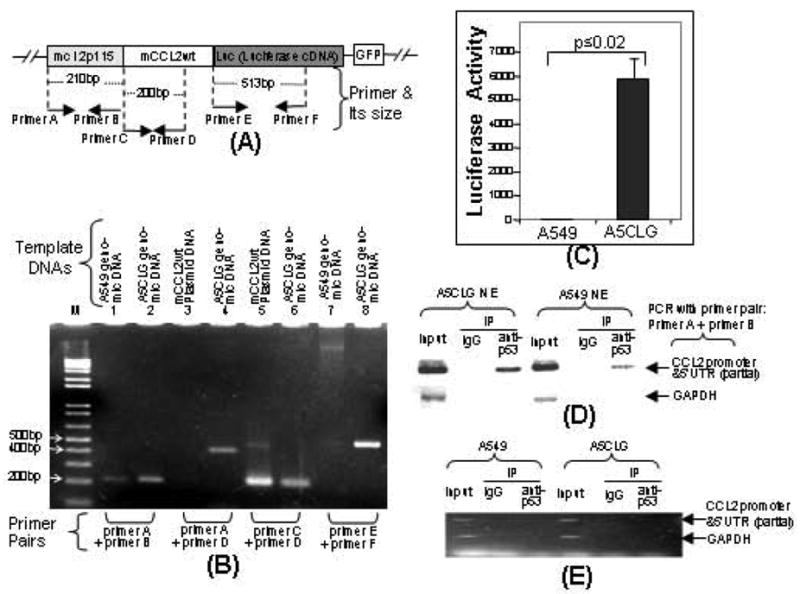
Confirmation of a stable cell line integrated with a cloned DNA (mcl2p115/mCCL2wt/Luc). A. Schematic diagram of the cloned DNA (mcl2p115/mCCL2wt/Luc) integrated into chromosome of A549 cells (Named A5CLG). The arrows indicate the location and size of primers (Named primers A-F) used in PCR amplification of coding regions. These primer pairs were used for PCR and ChIP as described (Panels B&D). B. Detection of the cloned DNA by PCR. Chromosomal DNA isolated from A5CLG cells or A549 cells and mCCL2wt plasmid DNA as positive control were used as template for PCR with primer pairs (Bottom). The PCR-amplified specific DNAs with molecular weight of about 210bp, 200bp, 397bp or 513bp were run in a 1.5% agarose gel and imaged. C. Luciferase assay. Cell lysate from 1x106 A5CLG cells or A549 cells as control was assessed by luciferase assay (n = 3). Values were normalized with respect to protein concentrations. D. ChIP-detection of protein-DNA interaction between p53 and CCL2 5’UTR&promoter. The PCR-amplified specific DNA fragments were indicated with arrows. E. Analysis of protein-mRNA interaction between p53 and CCL2 by RNA-IP. cDNA of A5CLG cells or A549 cells and mCCL2wt plasmid DNA as positive control were used as template for PCR with primer pairs (attached to bottom). RT-PCR-amplified DNA fragments are indicated with arrows.
RNA-Immunoprecipitation (RNA–IP)
Assay was performed with some modifications. 1x106 A5CLG cells or A549 cells as control were exposed to UV radiation (15mJ/cm2) and harvested 2 hrs after UV exposure. To avoid potential DNA contamination, the lysate as input from the cross-linked cells was treated with RNase-free DNase for 30min, washed three times with PBS, and then immunoprecipitated with 1μg p53 antibody (FL-393-G, Santa Cruz) or 1μg normal IgG as control at 4ºC for 4 hrs. RNAs from each experimental group (input or IP) were isolated by elution, reverse cross-linked, treated with Proteinase K, and reverse transcribed into cDNA using iScript (Bio-Rad). The cDNA was then used as a template to perform PCR with a primer pair (5’-ccttggcaccaagcac-3’ + 5’-ctggtgctggtgctgg-3’) for amplification of a 120bp DNA fragment of CCL2 5’UTR& promoter and a GAPDH primer pair (Invitrogen) for a control.
Peptides
Synthetic peptides were supplied by Biosynthesis, Inc. (Lewisville, TX). The non-FITC-labeled or FITC labeled peptide, named p53pep170 or FITC-p53pep170, consisted of the p53 sequence KSQHMTEVV and located in the region (aa 162-170), was synthesized. The synthetic peptide, named SCpep, consisted of a randomly scrambled sequence VSKMVQTHE (Medusa Random Sample Generator Software, Randombots.com). The synthetic peptide, named C-motif, consisted of the p53 sequence RRCPHHERCSDGD and located in the region from aa 171 to 183. The synthetic peptide, named A-motif, consisted of the p53 sequence RAMAIYK and served as a negative control peptide and located in the region (aa 155-161). For luciferase assay, the synthetic peptides were solubilized in DMSO and respectively added into U2OS cell cultures (5-100ug peptide/3x105 cells/ml) without a reagent after transfection with CCL2 reporter DNA by Lipofectamine 2000 for 3 hrs. Cells were incubated overnight and their pellets were collected and assessed. For ELISA, 1x105 macrophage cells were treated with LPS (0.1ug/ml) for 2 hrs, washed and added fresh medium, then the synthetic peptides (5-100ug peptide/ml) were added without a reagent and cells were incubated overnight. The supernatant from each treated cell group was collected and measured by CCL2/MCP-1 ELISA kit (Invitrogen) following the manufacturer’s instruction.
Statistical Analysis
All experiments were repeated at least three times. All the data were normally distributed. In case of multiple mean comparisons, data were analyzed by analysis of variance (ANOVA). In case of single mean comparison, data were analyzed by Student’s t-test. In case of time-course study, data were analyzed by two-way repeated measure ANOVA, and P values less than 0.05 were regarded as significant.
RESULTS
Involvement of p53 in CCL2 Production
In order to investigate whether endogenous p53 affects CCL2 gene expression, LPS-treated mouse primary macrophages were exposed to ultraviolet (UV) radiation according to the conventional method [36]. As shown in Fig. (1), UV-induced p53 accumulation in wild-type macrophages was not involved in LPS-induced IL-1-β gene expression (Fig. 1A, dotted bars) compared to the control (Fig. 1A), downward diagonal striped bars)). However, UV-induced p53 accumulation in the same cells from time point 0.5 to 2 hrs significantly decreased LPS-induced CCL2 production (Fig. 1B, dotted bars) to 42% at 0.5 hr, 44% at 1hr, and 47% at 2hrs compared to the control (Fig. 1B, downward diagonal striped bars). The analysis of protein from each experimental group of wild-type macrophages by Western blot (top panel) indicated that cells harvested 0.5 to 2 hours after the UV exposure exhibited about 3-fold increase in accumulation of endogenous p53 compared to untreated control cells. To examine if the cellular endogenous p53 protein specifically regulates CCL2 gene expression, the same dosage exam was performed using p53-deficient macrophages. As shown in Fig. (1C), treatment of p53-deficient cells with UV radiation (dotted bars) did not decrease LPS-induced CCL2 production compared to the control (downward diagonal striped bars). We therefore hypothesized that CCL2 is regulated via p53 binding activity. To address our hypothesis, U2OS cells were co-transfected overnight with IL-1-β-p (mouse IL-1-β promoter DNA) as negative control or with mcl2pwt (mouse CCL2 5’UTR&promoter DNA) and/or different concentration of mp53WT or mp53-muB as control (mp53-muB is a p53 mutant that has been shown not to interact with CCL2 5’UTR in this study, Figs. 3, 5D). mcl2pwt proteins and controls were then analyzed by luciferase assay. As shown in Fig. (1D), transfection of increasing concentrations of mp53WT from 0.1 to 1 μg caused a concomitant decrease in mcl2pwt-mediated luciferase activity compared to controls, suggesting that overexpression of p53 down-regulates CCL2 promoter activity.
Fig. (1).
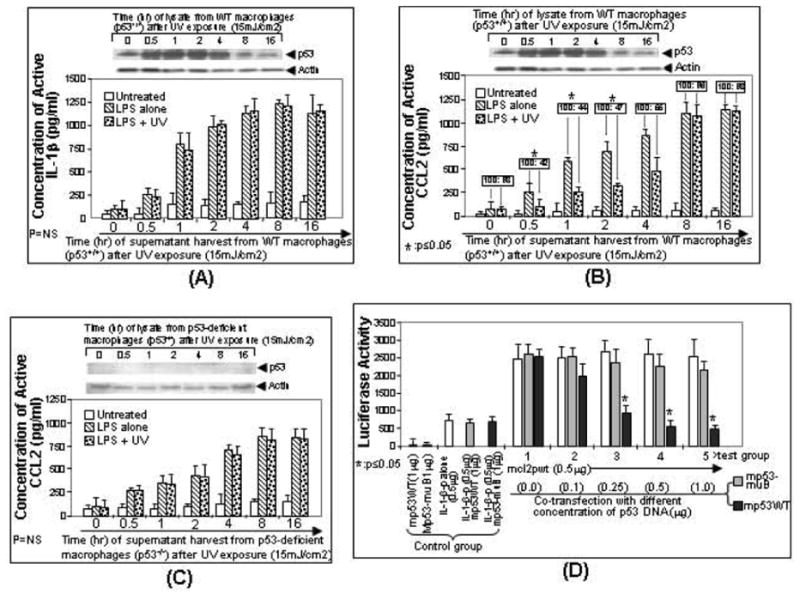
Regulation of LPS-induced CCL2 production by the accumulation of endogenous p53. Mouse primary macrophage cells from wild-type mice (A, B) or p53-deficient mice (C) were untreated as control (white bar) or treated with LPS (downward diagonal striped bar) or LPS plus UV radiation (dotted bar). Cells were then cultured. The medium from each group of treated cells were harvested at different times (0, 0.5, 1, 2, 4, 8 or 16 hrs) and assessed for IL-1-β production as control (A) or CCL2 production (B, C) by ELISA (n = 3). The concentration of CCL2 secreted from LPS-treated cells was assigned a value of 100% as the baseline, and the relative CCL2 concentration from co-treated cells by LPS plus UV was calculated (boxed ratio). Values were normalized with respect to protein concentrations. Data are presented as mean ± SEM. In order to detect p53 accumulation, the lysate from cells at each time point was purified and analyzed by Western blot with the antibody against p53 or actin as control (Top panel of A-C). D. Regulation of CCL2 promoter activity by overexpression of p53. U2OS cells were transfected with reporter DNA (mcl2pwt alone or mouse IL-1β promoter alone, white bar, control group) or co-transfected with mcl2pwt DNA and different concentrations (0, 0.1, 0.25, 0.5, or 1μg) of p53 DNA (grey bar for mp53WT or black bar for mp53-muB as control, experimental group 1-5). Cells were cultured overnight. The lysate from cells at each group was assessed by luciferase assay (n = 3). Data are presented as mean ± SEM.
Fig. (3).
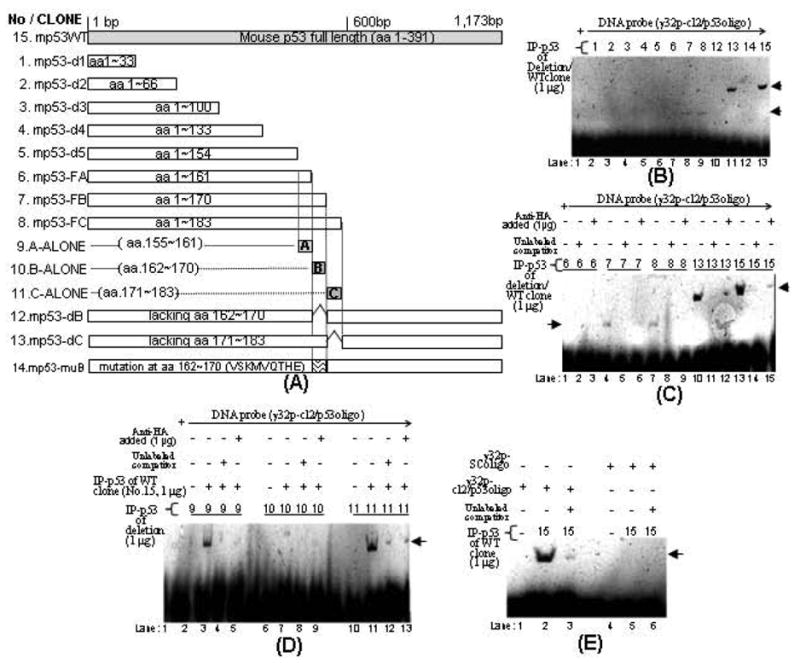
Diagram of mouse p53 deletions and their effects on the CCL2 promoter activity. A. Different lengths of p53 DNA were truncated and inserted into pcDNA3HA vector. Gray box: full length of p53. White boxes: deletions. The region representing amino acids of each cloned DNA was shown in boxes. Their DNA deletions were confirmed by sequencing. B. EMSA. A probe consisting of [32p]ATP-labeled cl2/p53oligo was added to each reaction buffer. Probe was alone (lane 1) or mixed with 1 μg of anti-HA immunoprecipitation (IP-p53 fusion protein of deletion 1, 2, 3, 4, 5, 6, 7, 8, 12, 13, 14 or wild-type clone 15, lane 2-13). The band shifts are indicated by arrows. C. Supershift assay 1. 32p-cl2/p53oligo probe was added to each reaction buffer and mixed with 1 μg of IP-p53 fusion protein (deletion 6, 7, 8, 13 or wild-type clone 15 in lane 1, 4, 7, 10, or 13) plus 1 pmol unlabeled competitor (lane 2, 5, 8, 11, or 14) or plus 1μg of HA antibody (lane 3, 6, 9, 12, or 15). The band shifts are indicated by arrows. D. Supershift assay 2. 32p-cl2/p53oligo probe was added to reaction buffer including 1 μg of IP-p53 fusion protein of deletion 9 (lanes 2-5), deletion 10 (lanes 6-9), or deletion 11 (lanes 10-13). The reaction buffer was then added with 1 μg of IP-p53 fusion protein of wild-type clone 15 (lanes 3-5, 7-9, and 11-13). The mixture was further added with 1 pmol unlabeled competitor (lane 4, 8, and 12) or 1μg HA antibody (lane 5, 9, and 13). The band shifts are indicated by arrows. E. EMSA. 32p-cl2/p53oligo probe (lanes 1-3) or 32p-SColigo probe as control (lanes 4-6) was added to each reaction buffer and mixed with 1 μg of IP-p53 fusion protein of wild-type clone 15 (lanes 2, 3, 5, or 6) plus 1 pmol unlabeled competitor (lane 3 or 6). The shifted bands are indicated by arrows.
Fig. (5).
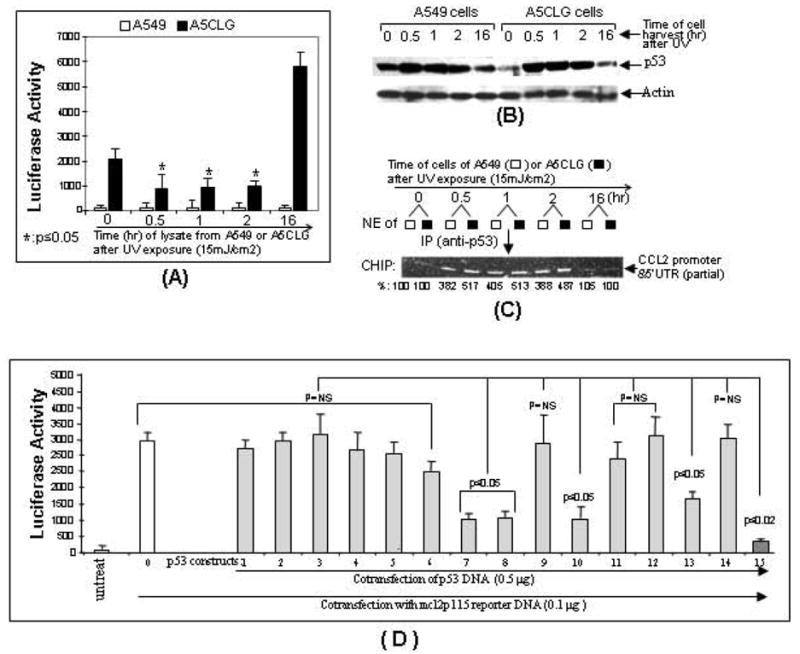
The effect of p53 accumulation on luciferase gene expression via regulation of CCL2 promoter activity. The freshly splitting A5CLG cells or A549 cells as control were exposed to UV radiation and cells were harvested at different times (0 hr, 0.5 hr, 1 hr, 2 hr or 16 hr). The proteins and nuclear extracts from each experimental group were respectively purified and assessed (n = 3) for luciferase assay (A), Western blot (B), or ChIP (C). Values of luciferase assay were normalized with the relative ratio of ChIP assay. Western blot was performed with antibodies against p53 and actin as control and their values were normalized with respect to the corresponding actin bands. For ChIP assay, the nuclear extracts were further immunoprecipitated with p53 antibody and used as template for PCR with primer pairs (primerA&B). The PCR-amplified DNA fragments are indicated with arrows. Intensity of DNA band detected at 0 hr of UV exposure was assigned to a base value (100%). Intensity of other bands was calculated relative to this base value and their ratio is attached under the corresponding DNA band. D. Proteins from each group of cells co-transfected with mcl2p115 (No. 0-15) and p53 constructs (No. 1-15) or untreated as control were measured by luciferase assay (n = 3). Data are presented as mean ± SEM.
Determination of p53-CCL2 Binding Site and Binding Domain
To determine which region of CCL2 5’UTR&promoter acts as a binding site for p53, mcl2pwt (full length 5’UTR&promoter shown in Fig. (2A) and its derived deletions (mcl2p715, mcl2p515, mcl2p315, mcl2p115 or mcl2p53m) were subcloned into pGL3-basic vector (Fig. 2B). These cloned DNAs were transiently transfected into U2OS cells to confirm their enhancement of luciferase production. The luciferase activity for mcl2pwt was assigned a value of 100% as the baseline, and the relative luciferase activities for the others were: 87.5% for mcl2p715, 93.1% for mcl2p515, 71.5% for mcl2p315, 81.8% for mcl2p115, and 69% for mcl2p53m. Next, U2OS cells were co-transfected with these clones plus mp53WT or pcDNA3 (control) overnight and their proteins were assessed by luciferase assay. As shown in Fig. (2C), the site of CCL2 5’UTR&promoter for p53 binding activity was located in the region from -115 to 85 because luciferase activity induced by mcl2p115 was significantly down-regulated by p53 overexpression. In addition to the data above, we located the putative p53 binding site in the region of 16-35 of CCL2 5’UTR (Fig. 2A). In order to confirm this binding site, U2OS cells were transfected with mcl2p53m (site mutation) alone and/or with mp53WT. As shown in Fig. (2C), no suppression of mcl2p53m-induced luciferase activity was observed in cells treated with mp53WT compared to the control, suggesting that this region is specific to p53 binding. To further determine the binding domain on p53, a series of mouse p53 constructs were created as shown (Fig. 3A) and respectively transfected into U2OS cells. The fusion protein from each group of cells was immunoprecipitated and used for EMSA as shown in Fig. (3B-E). The [32P]ATP-labeled double-stranded nucleotide (cl2/p53oligo) was treated with IP-p53 fusion proteins to test their specific binding activities. The shifted DNA bands as indicated by arrows (Fig. 3B), p53 deletion 7, 8, 13 or wild-type clone 15) showed that cl2/p53oligo binds to IP-mp53-FB, IP-mp53-FC, IP-mp53-dC or IP-mp53WT. In contrast, mixtures of cl2/p53oligo with IP-mp53-d1∼d5, IP-mp53-FA, IP-mp53-dB or IP-mp53-muB (deletion 1, 2, 3, 4, 5, 6, 12, or 14) did not generate a shifted band, suggesting the lack of a region (aa 162-170). The specificity of binding was further confirmed by supershift assay. As shown in Fig. (3C), the signal intensity of the band shift induced by DNA-protein complex due to the interaction of cl2/p53oligo with IP-mp53-FB, IP-mp53-FC, IP-mp53-dC or IP-mp53WT from deletion 7, 8, 13, or wild-type clone 15 was strongly reduced by adding unlabeled competitor or antibody, suggesting that the addition of an excess amount of cold probe and antibody supershifts or blocks CCL2-p53 complex. Additionally, no interaction of DNA probe with IP-A-, B-, or C-ALONE (Fig. 3D), deletion 9, 10, 11, lane 2, 6, or 10)) could be observed, perhaps due to the difficulty of detecting protein-DNA interaction with such small molecule fusion proteins by regular assay. Thus, a modified supershift assay was employed using them as a competitor of IP-mp53WT as shown (Fig. 3D). The band shift was still observed after a mixture of IP-mp53WT and IP-A-ALONE (deletion 9, lane 3) or IP-C-ALONE (deletion 11, lane 11). However, the mixture of IP-B-ALONE (deletion 10, lane 7) with IP-mp53WT significantly reduced the band shift. This suggests that IP-B-ALONE indeed binds to the DNA probe and competes IP-mp53WT away from protein-DNA interaction because IP-B-ALONE is a small molecule and about 40 fold of IP-mp53WT in assay mixture when they are the same concentration (1 g fusion protein). To further test the specificity of cl2/p53oligo, EMSA was performed with IP-p53 fusion protein from wild-type clone 15 plus SColigo DNA probe or cl2/p53oligo probe as the positive control. No band shift was observed (Fig. 3E, lane 5) compared to the positive control (lane 2). These findings above suggest that, 1) the site 16-35 of CCL2 5’UTR&promoter is specific to p53 interaction and 2) p53 protein region (aa 162-170) is an important binding domain for p53 binding activity.
Fig. (2).
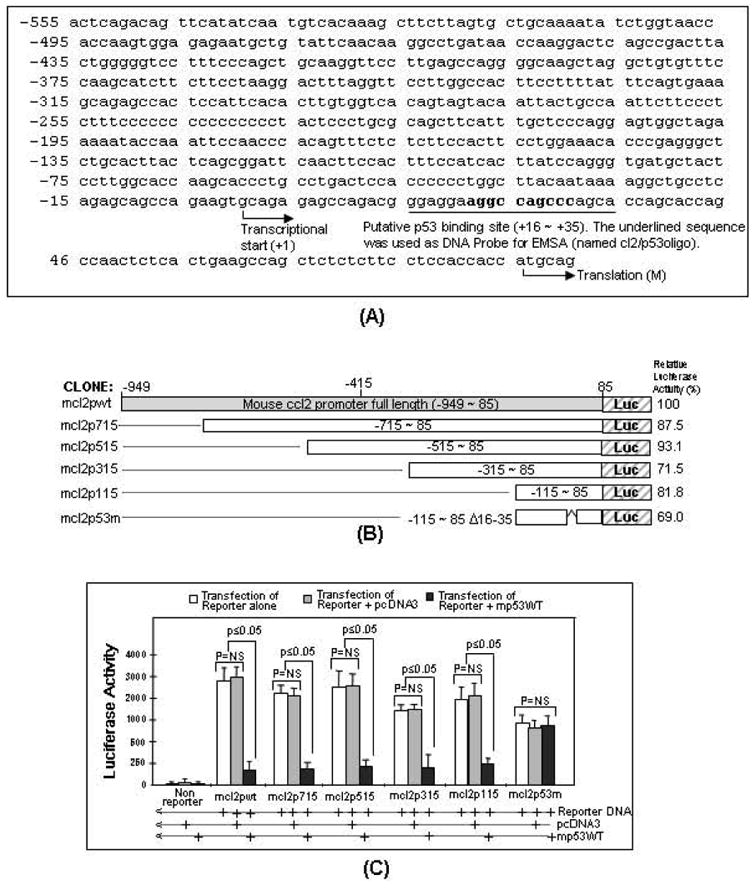
Analysis of p53 binding site in CCL2 5’UTR&promoter. A. Mouse CCL2 5’UTR&promoter sequence from -949 to +85. BLAST-search of the sequence showed that it was identical to mouse genomic DNA (AL713839) and it ended before the start codon of CCL2. The putative binding sites for p53 in CCL2 5’UTR&promoter were analyzed by PROMO version 3.0.2 (UPC) and bolded. The binding sites for other potential transcription factors have not been analyzed. The oligonucleotide including p53 binding site (Underlined sequence, named cl2/p53oligo) and its complementary oligo were synthesized. Both synthetic oligonucleotides were annealed and labeled with [32P]ATP as a double strand DNA probe for EMSA. B. Diagram of mouse CCL2 5’UTR&promoter DNA constructs and its promoter activity. Different lengths of CCL2 5’UTR&promoter DNA were truncated and inserted into the pGL3-basic vector. Gray box: region representing full length of CCL2 5’UTR&promoter (mcl2pwt). White boxes: region representing CCL2 5’UTR&promoter deletions from 85 to -715 (mcl2p715), 85 to -515 (mcl2p515), 85 to -315 (mcl2p315), 85 to -115 (mcl2p115) or 85 to -115 with deletion of 16-35 (mcl2p53m). Their DNA deletions were confirmed by sequencing. The relative promoter activity from each construct was shown. C. Determination of p53 binding site in CCL2 5’UTR&promoter. U2OS cells were transfected with pcDNA3 alone or mp53WT alone without reporter gene as controls, or with various reporter genes alone (white bars): mcl2pwt, mcl2p715, mcl2p515, mcl2p315, mcl2p115 and mcl2p53m, or co-transfected with reporter genes plus 0.5μg pcDNA3 (gray bars) or plus 0.5μg mp53WT (black bars) overnight. The lysate from each experimental group was assessed by luciferase assay (n = 3). Values were normalized with respect to protein concentrations. Data are presented as mean ± SEM.
Analysis of Interaction of p53 and CCL2 5’UTR& promoter in Cells
To further analyze DNA-protein interaction of CCL2 5’UTR&promoter and p53 in cells, we have established a stable cell line (named A5CLG) containing the CCL2 promoter-enhanced cDNAs of a full length CCL2, a full length luciferase and GFP in its chromosome (Fig. 4A). These integrated DNAs were confirmed by PCR as shown in Fig. (4B). A CCL2 5’UTR&promoter DNA fragment (210bp, No. 2), a CCL2 DNA fragment (200bp, No. 6), CCL2 5’UTR&promoter plus its cDNA (397bp, No. 4), or a partial-length of luciferase cDNA (513bp, No. 8) was detected compared to the controls (Nos.1, 3, 5 or 7). A5CLG or control cells (A549) were cultured under normal conditions without any stimulation overnight. Their cell lysate was then assessed by luciferase assay. As shown in Fig. (4C), a high concentration of luciferase production in A5CLG cells was detected compared to control cells, suggesting that A5CLG cells contain the integrated gene in chromosomes and these artificial genes can be strongly enhanced by CCL2 promoter. Thus, A5CLG cells were used to analyze DNA-protein interaction of CCL2 promoter and p53 using chromatin immunoprecipitation (ChIP). As shown in Fig. (4D), the PCR-amplified DNA fragment of CCL2 5’UTR&promoter (210bp) was detected in both A5CLG and control cells, suggesting that p53 occurs in a cell through interaction with CCL2 promoter. To further determine whether there is any possibility that p53 binds to transcribed CCL2 mRNA, RNA–IP was performed. As shown in Fig. (4E), no reverse transcription (RT)-PCR-amplified DNA fragment (120bp) was detected in immunoprecipitation of A5CLG or control cells, suggesting that p53 does not interact with CCL2 mRNA.
Analysis of Endogenous p53-CCL2 Binding Activity
In order to assess the effect of endogenous p53 on regulation of CCL2 promoter activity seen above, A5CLG and control cells were exposed to ultraviolet (UV) radiation. The lysate was collected at different times and used for luciferase assay, Western blot and ChIP. As shown in Fig. (5A), the luciferase activity of cells harvested immediately after UV exposure was detected as the control and assigned to the baseline of 100%, the luciferase activity from A5CLG cells at each time after UV exposure was decreased to 42.8% at 0.5hr, 45.2% at 1hr, or 47.6% at 2 hrs. The analysis of protein from each UV exposed group of cells by Western blot (Fig. 5B) indicated that the cells harvested 0.5 to 2 hours after the UV exposure exhibited about a 3-fold increase in accumulation of endogenous p53 compared to the baseline. The further ChIP assay showed that UV-induced endogenous p53 occurs in both A5CLG and control cells through 4- to 5-fold increased interaction with CCL2 promoter (Fig. 5C) compared to the control. Taken together, the data suggests that increasing accumulation of p53 decreases CCL2 promoter activity. To further confirm p53 binding domain and its function, U2OS cells were co-transfected with DNAs of mcl2p115 (as a CCL2 reporter system), and various p53 constructs and extract from each experimental group were assessed by luciferase assay (Fig. 5D). The promoter activity of mcl2p115 alone (No. 0) was assigned the value of 100% as the positive control. The CCL2 promoter activity was nearly maintained by the overexpression of mp53-d1, mp53-d2, mp53-d3, md53-d4, mp53-d5, and mp53-FA (Nos. 1-6) because their regions are upstream of aa 161. After the transient transfection of mp53-FB, mp53-FC, mp53-dC or mp53WT, CCL2 promoter activity was reduced to 30% (No. 7), 32% (No. 8), 48% (No. 13), or 10.8% (No. 15) since they all contain a region with aa 162-170. Furthermore, B-ALONE containing only 9 amino acids in the region (aa 162-170) still reduced the value of promoter activity to 34% (No. 10). However, A-ALONE (No. 9), C-ALONE (No. 11), mp53-dB (No. 12) and mp53-muB (No. 14) were unable to inhibit CCL2 promoter activity because the amino acids 162-170 were either lacking or mutated (Fig. 3A). This suggests that aa 162-170 plays an important role in both the binding activity, as well as the inhibition of CCL2 promoter activity. Thus, a short peptide (Named p53pep170 corresponding to the region aa 162-170 of mp53WT) and a control peptide (SCpep consisting of randomly scrambled sequence of p53pep170) were synthesized (Bio Synthesis, Inc.), and the effects of peptide treatments in human or mouse monocytes on CCL2 production were analyzed.
Analysis of p53pep170-Mediated Cytokine and Chemokine Gene Expression in Response to LPS Stimulation
To examine whether p53pep170 treatment is involved in LPS-induced cytokine or chemokine production, Bio-Plex cytokine assay (BioRad) was performed. As shown in Fig. (6A, B), LPS-induced cytokines (PDGF, IL-15, IL-10, and TNF-α) and chemokine (CCL2) were significantly reduced by p53pep170 administration (No. 8) relative to the LPS alone as the positive control (No. 2). This regulation of cytokines or CCL2 was specific since no effect of SCpep on LPS-induced gene expression (No. 6) could be observed. Additionally, no evidence was found on toxicity of peptide treatment in macrophage cells since there was no significant effect on cell death induced by peptide treatment compared to controls (Fig. 6A). To examine if the p53pep170 is associated with cellular endogenous p53 protein to regulation of CCL2 gene expression, a dose course test was performed. As shown in Fig. (6C), the treatment of p53pep170 in mouse primary macrophages from wild-type (White bars) or p53-deficient mice (Grey bars) caused a decrease (61% for wild-type mice or 54.5% for p53-deficient mice) in LPS-induced CCL2 production as compared to the controls, which cements our hypothesis that the p53pep170 is not associated with cellular endogenous p53 to play an essential role in the inhibition of CCL2 production. To further examine whether p53pep170 treatment is involved in suppression of CCL2 gene expression via down-regulation of its promoter activity, human monocytes (Fig. 6D, panels A-H) or mouse macrophages (Panels I-P) were infected with lentiCLG (Panels A-P). The analysis of fluorescent signals showed that lentiCLG-induced GFP production was significantly inhibited with addition of 100μg/ml p53pep170 (Panels H&P) compared to the controls (Panels E, F, M, N). In order to test the effect of p53pep170 on general lentiviral infection, human monocytes (Panels Q&R) or mouse macrophages (Panels S&T) were treated with Lenti6.3virus plus 100μg/ml p53pep170. As no reduction in GFP signal was observed, this indicates that p53pep170 does not interfere with it.
Fig. (6).
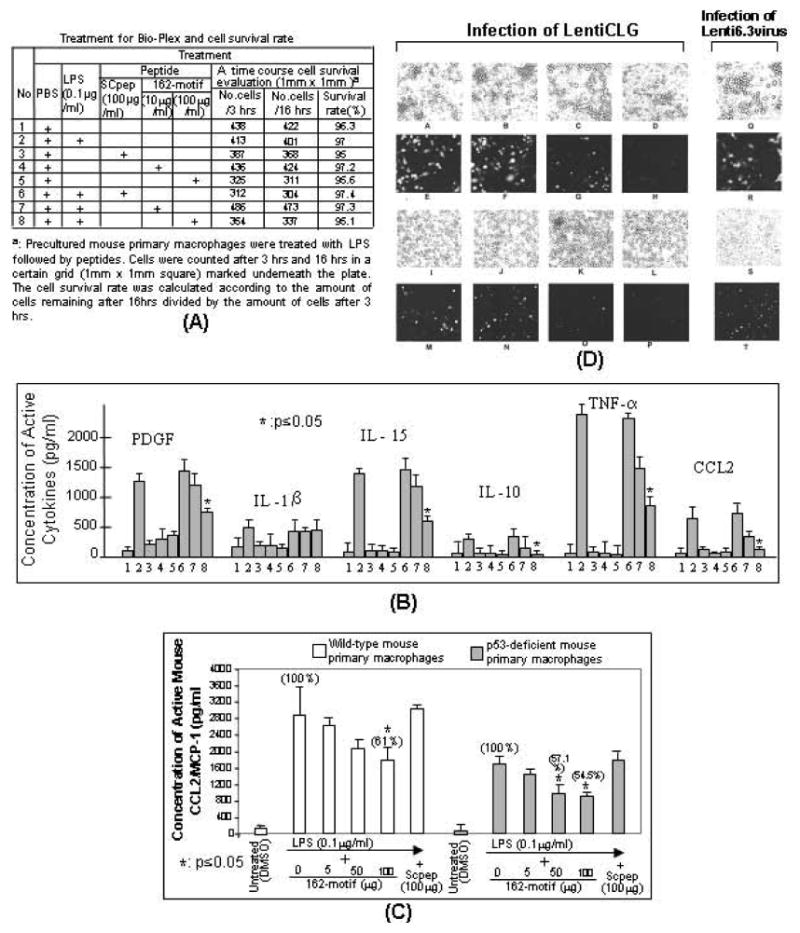
Effect of p53pep170 on gene expression in cells. A. Treatment for Bio-Plex and cell survival rate is described in the table. B. Bio-Plex assay. The cultured medium from each treated mouse primary macrophage cells group (table) was used and the major measurements were quantified (n=3) by Bio-Plex 200 System (Bio-Rad). Data are presented as mean ± SEM. Values were normalized with respect to the cell survival rate and protein concentrations. C. ELISA assay. Mouse primary macrophages (1 x 106) from wild-type (white bars) or p53-deficient mice (grey bars) were stimulated with 0.1 μg/ml E. coli LPS alone, LPS plus different dosages of p53pep170 (5 μg/ml, 50 μg/ml or 100 μg/ml), and LPS with SCpep (100 μg/ml) as control. Their cultured media were used for ELISA (n=3). Data are presented as mean ± SEM. Values were normalized with respect to the cell survival rate and protein concentrations. D. Down-regulation of lentiCLG-induced CCL2 production by p53pep170 in both human primary monocytes and mouse primary macrophages. The human monocytes (Panels A-H) and mouse macrophages (I-P) were infected with LentiCLG (MOI=2) plus DMSO alone (A&E or I&M) or 10ug/ml p53pep170 (C&G or K&O) or 100ug/ml p53pep170 (D&H or L&P) or 100ug/ml Scpep (B&F or J&N) as the control. To test whether p53pep170 has an effect on suppression in lentivirus infection, human monocytes (Q&R) and mouse macrophages (S&T) were infected with Lenti6.3virus (MOI=2) plus 100ug/ml p53pep170. Cells were incubated for 37°C, 5% CO2 for 5 days. The phase contrast panels (A&E, B&F, C&G, D&H, I&M, J&N, K&O, L&P, Q&R, or S&T) were the pair of sections. The treated cells were exposed to visible light (A-D, I-L, Q&S) and to fluorescent light (E-H, M-P, R&T) using an Olympus BX40 microscope at 1000x magnification. The GFP-induced fluorescent signal in human primary monocytes (E-H, R) or in mouse primary macrophages (M-P, T) was observed. The images were taken with MicroFIRE camera under uniform exposure time: 30 msec for visible light (A-D, I-L, Q&S) and 1 sec for fluorescent light (E-H, M-P, R&T). The data analysis was processed by the program Image-Pro plus 5.0. Multiple tests have been done with similar results.
DISCUSSION
CCL2 is implicated in a wide range of diseases, playing an important role in inflammatory responses as well as cancer and HIV-related disorders. Patients with autoimmune conditions [37] and rheumatoid arthritis [38] are found to have elevated levels of CCL2, and studies have demonstrated that CCL2 is involved in atherosclerotic plaque formation [39], pulmonary hypertension [40], insulin resistance [16, 17, 41] and allergic asthma [42]. Furthermore, CCL2 is linked to the development of carcinomas such as in prostate, colorectal and breast cancer, as well as metastasis and tumor recurrence [43-46]. In addition, CCL2 has been found to be involved in HIV-mediated diseases [47, 48]. As CCL2 is associated with a variety of disorders, several regulatory factors have been found to affect its production, such as enhancers AP-1, AP-2beta, STAT6, NFκB [49-51], TNF-α [52], Tat [53], LPS [54], S1P [55], and Nef [56]. There are also a numbers of suppressors, such as RS102895 [57], p38 or SB203580 [58], HDL [38], and Smad3 [59]. In one study, p53 was found to be involved in the regulation of CCL2 production in HPV-positive cells [60]. However, there are several areas that need further investigation, such as how p53 regulates CCL2, what domain of p53 is involved in this regulation, and whether this function is valid in human monocytes or mouse primary macrophages.
We are interested in the mechanism by which these specific domains regulate the targeted gene transcription via binding activity. We report that the specific protein/peptide in the region of p53 aa 162-170, expressed by DNA clones (Fig. 3A, B-ALONE), specifically interacts with CCL2 5’UTR and causes a significant reduction of its promoter activity (Fig. 5C, No. 10). All other p53 deletions either lacking or containing mutations in amino acid sequence 162-170 do not affect CCL2 promoter activity. Synthetic peptide for this sequence, named p53pep170, is able to reduce CCL2 production in mouse macrophages while peptides derived from upstream and downstream sequences do not affect CCL2. It is known that tetramerization of p53 protein contributes to its activation and high binding affinity to DNA and protein [61-63]. Studies have shown that the tetramerization domain on p53 C-terminal is important in protein-protein interaction [64, 65]. However, it was found that p53 mutants lacking the C-terminal still bound to DNA and maintained significant transactivation and growth suppressor activity [66-69]. In this study, we have observed a similar result that, without its C-terminal, a short peptide of p53 alone is able to suppress the CCL2 gene expression via its binding activity. These results bring forth the possibility of future investigations looking to uncover the mechanism behind the observed binding and suppression.
In addition, the comparison of human p53 peptide (p53pep164 [31], with the mouse p53 peptide (p53pep170) in this study indicates that both peptides are located in the same region (Sequence-specific DNA-binding site) and are almost homologous in their amino acid sequences: KQSQHMT for human and KSQHMTVV for mouse. It suggests that this peptide from either human or mouse p53 plays a major role in the regulation of inflammatory genes. This finding provides the potential for utilizing an animal model to confirm the in vivo effect of p53pep170 prior to its application in a clinical trial.
The various agents used for delivery of small molecules or compounds into cells have been widely studied [70-72]. But most agents used for molecule delivery into cells or animals will consequently induce some toxicity or side effects. Thus, delivery of molecule without agents is an expected method. We have most recently found that the synthetic p53 peptide is easily taken up by both human THP-1 cells in vitro and mouse organs in vivo without any transfection agents even though the mechanism of peptide delivery into the plasma and nuclear membranes of cells is under investigation [32].
In summary, we found that UV-induced p53 accumulation in mouse macrophages significantly decreased LPS-induced CCL2 production. We in vitro have confirmed the role of p53 as the regulator to promoter activities of CCL2. We have also identified the nine amino acid residue p53pep170 to be the active site for this regulation. As p53pep170 is found to reduce LPS-induced CCL2 production in mouse primary macrophages of p53-deficient mice, it suggests that p53 or its derived peptide (p53pep170) is an important regulator of CCL2 gene expression.
Acknowledgments
This work was supported by NIH grants R01DE014079 (SA).
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
References
- 1.Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987–97. doi: 10.1128/MCB.24.18.7987-7997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohaly G, Chemnitz J, Dehde S, et al. A novel human p53 isoform is an essential element of the ATR-intra-S phase checkpoint. Cell. 2005;122:21–32. doi: 10.1016/j.cell.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 3.Virginie M, Staphane P, Mustapha A, et al. Delta160p53 is a novel N-terminal p53 isoform encoded by delta133p53 transcript. FEBS Lett. 2010;584:4463–8. doi: 10.1016/j.febslet.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Avery-Kiejda KA, Zhang XD, Adams LJ, et al. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin Cancer Res. 2008;14:1659–68. doi: 10.1158/1078-0432.CCR-07-1422. [DOI] [PubMed] [Google Scholar]
- 5.Hofstetter G, Berger A, Fiegl H, et al. Aoternative splicing of p53 and p73: the novel p53 splice variant p53 delta is an independent prognostic marker in ovarian cancer. Oncogene. 2010;29:1997–2004. doi: 10.1038/onc.2009.482. [DOI] [PubMed] [Google Scholar]
- 6.Anensen N, Oyan AM, Bourdon JC, Kalland KH, Bruserud O, Gjertsen BT. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin Cancer Res. 2006;12:3985–92. doi: 10.1158/1078-0432.CCR-05-1970. [DOI] [PubMed] [Google Scholar]
- 7.Slatter TL, Hung H, Campbell H, et al. Hyperproliferation, cancer, and inflammation in mice expressing a D133p53-like isoform. blood. 2011;117:5166–77. doi: 10.1182/blood-2010-11-321851. [DOI] [PubMed] [Google Scholar]
- 8.Okuda Y, Okuda M, Bernard CC. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2003;135:29–37. doi: 10.1016/s0165-5728(02)00428-9. [DOI] [PubMed] [Google Scholar]
- 9.Yamanishi Y, Boyle DL, Pinkoski MJ, et al. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am J Pathol. 2002;160:123–30. doi: 10.1016/S0002-9440(10)64356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S, Mendoza T, Ortiz LA, et al. Bleomycin sensitivity of mice expressing dominant-negative p53 in the lung epithelium. Am J Respir Crit Care Med. 2002;166:890–7. doi: 10.1164/rccm.2109094. [DOI] [PubMed] [Google Scholar]
- 11.Komarova EA, Krivokrysenko V, Wang K, et al. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005;19:1030–2. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- 12.Staib F, Robles AI, Varticovski L, et al. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005;65:10255–64. doi: 10.1158/0008-5472.CAN-05-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 14.Baum N, Schiene-Fischer C, Frost M, et al. The prolyl cis/trans isomerase cyclophilin 18 interacts with the tumor suppressor p53 and modifies its functions in cell cycle regulation and apoptosis. Oncogene. 2009;28:3915–25. doi: 10.1038/onc.2009.248. [DOI] [PubMed] [Google Scholar]
- 15.Schilling M, Strecker JK, Schabitz WR, Ringelstein EB, Kiefer R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience. 2009;161:806–12. doi: 10.1016/j.neuroscience.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Yang SJ, IglayReger HB, Kadouh HC, Bodary PF. Inhibition of the chemokine (C-C motif)ligand 2/chemokine (C-C motif) receptor 2 pathway attenuates hyperglycaemia and inflammation in a mouse model of hepatic steatosis and lipoatrophy. Diabetologia. 2009;52:972–81. doi: 10.1007/s00125-009-1309-8. [DOI] [PubMed] [Google Scholar]
- 17.Kondo M, Maegawa H, Obata T, et al. Transcription factor activating protein-2beta: a positive regulator of monocyte chemoattractant protein-1 gene expression. Endocrinology. 2009;150:1654–61. doi: 10.1210/en.2008-1361. [DOI] [PubMed] [Google Scholar]
- 18.Haringman J, Gerlag DM, Smeets TJM, et al. A randomized controlled trial with an anti-CCL2 monoclonal antibody in patients with Rheumatoid Arthritis. Arihritis&Rheumatism. 2006;54:2387–92. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 19.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Monocyte chemotactic proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J. 1994;8:1055–60. doi: 10.1096/fasebj.8.13.7926371. [DOI] [PubMed] [Google Scholar]
- 20.Tak PP, Brersnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum. 2000;43:2619–32. doi: 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 21.Mclntosh LM, Barnes JL, Barnes VL, McDonald JR. Selective CCR2-targeted macrophage depletion ameliorates experimental mesangioproliferative glomerulonephritis. Clin Exp Immunol. 2009;155:295–303. doi: 10.1111/j.1365-2249.2008.03819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102:522–8. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romeo S, Abangan J, Christopher RW, et al. MCP1 directs trafficking of hematopoietic stem cell-derived fibroblast precursors in solid tumor. Am J Pathol. 2010;176:1914–26. doi: 10.2353/ajpath.2010.080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ajuebor MN, Swain MG, Perretti M. Chemokines as novel therapeutic targets in inflammatory diseases. Biochem Pharmacol. 2002;63:1191–6. doi: 10.1016/s0006-2952(02)00854-7. [DOI] [PubMed] [Google Scholar]
- 25.Ueda A, Okuda K, Ohno S, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;53:2052–63. [PubMed] [Google Scholar]
- 26.Dwarakanath RS, Sahar S, Reddy MA, Castanotto D, Rossi JJ, Natarajan R. Regulation of monocyte cdhemoattractant protein-1 by the oxidized lipid, 13-hydroperoxyoctadecadienoic acid, in vascular smooth muscle cells via nuclear factor-kappa B (NF-kB) J Mol Cell Cardiol. 2004;36:585–95. doi: 10.1016/j.yjmcc.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence DMP, Seth P, Durham L, et al. Astrocyte differentiation selectively upregulates CCL2/monocyte chemoattractant protein-1 in cultured human brain-derived progenitor cells. GLIA. 2006;53:81–91. doi: 10.1002/glia.20261. [DOI] [PubMed] [Google Scholar]
- 28.Kondo M, Maeqawa H, Obata T, et al. Transcription factor activating protein-2beta: a positive regulator of monocyte chemoattractant protein-1 gene expression. Endocrinology. 2009;150:1654–61. doi: 10.1210/en.2008-1361. [DOI] [PubMed] [Google Scholar]
- 29.Chen LI, Nishinaka T, Kwan K, et al. The retinoblastoma gene product RB stimulates Sp1-mediated transcription by liberating Sp1 from a negative regulator. Mol Cell Biol. 1994;14:4380–9. doi: 10.1128/mcb.14.7.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabath DE, Koehler KM, Yang WQ, Phan V, Wilson Joel. DNA-Protein Interactions in the Proximal-Globin Promoter: Identification of Novel CCACCC- and CCAAT-binding Proteins. Blood Cells, Molecules, and Diseases. 1998;15:183–98. doi: 10.1006/bcmd.1998.0185. [DOI] [PubMed] [Google Scholar]
- 31.Tang X, Molina M, Amar S. p53 short peptide (p53pep164) regulates lipopolysaccharide-induced tumor necrosis factor-alpha factor/cytokine expression. Cancer Res. 2007;67:1308–16. doi: 10.1158/0008-5472.CAN-06-1600. [DOI] [PubMed] [Google Scholar]
- 32.Tang X, O’Reilly A, Asano M, Merrill JC, Yokoyama KK, Amar S. p53 peptide prevents LITAF-induced TNF-alpha-mediated mouse lung lesions and endotoxic shock. Curr Mol Med. 2011;11:439–52. doi: 10.2174/156652411796268731. [DOI] [PubMed] [Google Scholar]
- 33.Katrin H, Bladimiro RO, Gilles B, et al. Regulation of MCP-1 chemokine transcription by p53. Mol Cancer. 2010;9:82. doi: 10.1186/1476-4598-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimomura H, Matsuura M, Saito S, Hirai Y, Isshiki Y, Kawahara K. Lipopolysaccharide of Burkholderia cepacia and its unique character to stimulate murine macrophages with relative lack of interleukin--beta-inducing ability. Infect Immun. 2001;69:3663–9. doi: 10.1128/IAI.69.6.3663-3669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang X, Marciano DL, Leeman SE, Amar S. LPS induces the interaction of a transcription factor, LPS-induced TNF-alpha factor, and STAT6(B) with effects on multiple cytokines. Proc Natl Acad Sci USA. 2005;102:5132–7. doi: 10.1073/pnas.0501159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng H, You H, Zhou XZ, et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–53. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 37.Goser S, Ottl R, Brodner A, et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation. 2005;112:3400–7. doi: 10.1161/CIRCULATIONAHA.105.572396. [DOI] [PubMed] [Google Scholar]
- 38.Scanu A, Oliviero F, Gruaz L, et al. High-density lipoproteins downregulate production in human fibroblast-like synoviocytes stimulated by urate crystals. Arthritis Res Ther. 2010;12:R23. doi: 10.1186/ar2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Combadiere C, Potteaux S, Rodero M, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda Y, Yonemitsu Y, Kataoka C, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2002;283:2021–8. doi: 10.1152/ajpheart.00919.2001. [DOI] [PubMed] [Google Scholar]
- 41.Bernatoniene J, Zhang Q, Dogan S, Mitchell TJ, Paton JC, Finn A. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J Infect Dis. 2008;198:1823–33. doi: 10.1086/593177. [DOI] [PubMed] [Google Scholar]
- 42.Ip WK, Wong CK, Lam CW. Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2. but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin Exp Immunol. 2006;145:162–72. doi: 10.1111/j.1365-2249.2006.03085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hembruff SL, Jokar I, Yang L, Cheng N. Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms. Neoplasia. 2010;12:425–33. doi: 10.1593/neo.10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu H, Sun L, Guo C, et al. Tumor cell-microenvironment interaction models coupled with clinical validation reveal CCL2 and SNCG as two predictors of colorectal cancer hepatic metastasis. Clin Cancer Res. 2009;15:5485–93. doi: 10.1158/1078-0432.CCR-08-2491. [DOI] [PubMed] [Google Scholar]
- 45.Lu X, Kang Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem. 2009;284:29087–96. doi: 10.1074/jbc.M109.035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka K, Kurebayashi J, Sohda M, et al. The expression of monocyte chemotactic protein-1 in papillary thyroid carcinoma is correlated with lymph node metastasis and tumor recurrence. Thyroid. 2009;19:21–5. doi: 10.1089/thy.2008.0237. [DOI] [PubMed] [Google Scholar]
- 47.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–06. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez E, Rovin BH, Sen L, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci USA. 2002;99:13795–800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levina V, Su Y, Nolen B, et al. Chemotherapeutic drugs and human tumor cells cytokine network. Int J Cancer. 2008;123:2031–40. doi: 10.1002/ijc.23732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen YM, Chiang WC, Lin SL, Wu KD, Tsai TJ, Hsieh BS. Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: modulation by type III phosphodiesterase inhibition. J Pharmacol Exp Ther. 2004;309:978–86. doi: 10.1124/jpet.103.062620. [DOI] [PubMed] [Google Scholar]
- 51.Fulkerson PC, Zimmermann N, Hassman LM, Finkelman FD, Rothenberg ME. Pulmonary chemokine expression is coordinately regulated by STAT1, STAT6, and IFN-gamma. J Immunol. 2004;173:7565–74. doi: 10.4049/jimmunol.173.12.7565. [DOI] [PubMed] [Google Scholar]
- 52.Ho AW, Wong CK, Lam CW. Tumor necrosis factor-alpha up-regulates the expression of CCL2 and adhesion molecules of human proximal tubular epithelial cells through MAPK signaling pathways. Immunobiology. 2008;213:533–44. doi: 10.1016/j.imbio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 53.Khiati A, Chaloin O, Muller S, Tardieu M, Horellou P. Induction of monocyte chemoattractant protein-1 (MCP-1/CCL2) gene expression by human immunodeficiency virus1 Tat in human astrocytes is CDK9 dependent. J Neurovirol. 2010;16:150–67. doi: 10.3109/13550281003735691. [DOI] [PubMed] [Google Scholar]
- 54.Leonard EJ, Skeel A, Yoshimura T, Rankin J. Secretion of monocyte chemoattractant protein-1 (MCP-1) by human mononuclear phagocytes. Adv Exp Med Biol. 1993;351:55–64. doi: 10.1007/978-1-4615-2952-1_7. [DOI] [PubMed] [Google Scholar]
- 55.Chen XL, Grey JY, Thomas S, et al. Sphingosine kinase-1 mediates TNF-alpha-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol. 2004;287:H1452–8. doi: 10.1152/ajpheart.01101.2003. [DOI] [PubMed] [Google Scholar]
- 56.Lehmann MH, Masanetz S, Kramer S, Erfle V. HIV-1 Nef upregulates CCL2/MCP-1 expression in astrocytes in a myristoylation- and calmodulin-dependent manner. J Cell Sci. 2006;119:4520–30. doi: 10.1242/jcs.03231. [DOI] [PubMed] [Google Scholar]
- 57.Yao H, Peng F, Dhillon N, et al. Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neurosci. 2009;29:1657–69. doi: 10.1523/JNEUROSCI.2781-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marra F, Delogu W, Petrai I, et al. Differential requirement of members of the MAPK family for CCL2 expression by hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G18–26. doi: 10.1152/ajpgi.00336.2003. [DOI] [PubMed] [Google Scholar]
- 59.Eldeen MB, Deshmane SL, Simbiri K, Khalili K, Amini S, Sawaya BE. MH2 domain of Smad3 reduces HIV-1 Tat-induction of cytokine secretion. J Neuroimmunol. 2006;176:174–80. doi: 10.1016/j.jneuroim.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Hacke K, Rincon-Orozco B, Buchwalter G, et al. Regulation of MCP-1 chemokine transcription by p53. Mol Cancer. 2010;9:82–93. doi: 10.1186/1476-4598-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Joerger AC, Rajagopalan S, Natan E, Veprintsev DB, Robinson CV, Fersht AR. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci USA. 2009;106:17705–10. doi: 10.1073/pnas.0905867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 63.Chene P. The role of tetramerization in p53 function. Oncogene. 2001;20:2611–7. doi: 10.1038/sj.onc.1204373. [DOI] [PubMed] [Google Scholar]
- 64.Kaustov L, Lukin J, Lemak A, et al. The conserved CPH domains of Cul7 and PARC are protein-protein interaction modules that bind the tetramerization domain of p53. J Biol Chem. 2007;282:11300–7. doi: 10.1074/jbc.M611297200. [DOI] [PubMed] [Google Scholar]
- 65.Maki CG. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J Biol Chem. 1999;274:16531–5. doi: 10.1074/jbc.274.23.16531. [DOI] [PubMed] [Google Scholar]
- 66.Shaulian E, Zauberman A, Milner J, Davies EA, Oren M. Tight DNA binding and oligomerization are dispensable for the ability of p53 to transactivate target genes and suppress transformation. EMBO J. 1993;12:2789–97. doi: 10.1002/j.1460-2075.1993.tb05940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slingerland JM, Jenkins JR, Benchimol S. The transforming and suppressor functions of p53 alleles: effects of mutations that disrupt phosphorylation, oligomerization and nuclear translocation. EMBO J. 1993;12:1029–37. doi: 10.1002/j.1460-2075.1993.tb05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Unger T, Mietz JA, Scheffner M, Yee CL, Howley PM. Functional domains of wild-type and mutant p53 proteins involved in transcriptional regulation, transdominant inhibition, and transformation suppression. Mol Cell Biol. 1993;13:5186–94. doi: 10.1128/mcb.13.9.5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Reed M, Wang P, et al. p53 domains: identification and characterization of two autonomous DNA-binding regions. Genes Dev. 1993;7:2575–86. doi: 10.1101/gad.7.12b.2575. [DOI] [PubMed] [Google Scholar]
- 70.Juliano RL, Alam R, Dixit V, Kang HM. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:324–35. doi: 10.1002/wnan.4. [DOI] [PubMed] [Google Scholar]
- 71.Demers A, McNicoll N, Febbraio M, et al. Identification of the growth hormone-releasing peptide binding site in CD36: a photoaffinity cross-linking study. Biochem J. 2004;382:417–24. doi: 10.1042/BJ20040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tam SP, Ancsin JB, Tan R, Kisilevsky R. Epub 2005 Aug 1. Peptides derived from serum amyloid A prevent, and reverse, aortic lipid lesions in apoE-/- mice. J Lipid Res. 2005;46:2091–101. doi: 10.1194/jlr.M500191-JLR200. [DOI] [PubMed] [Google Scholar]