

Abstract
APOBEC3G (A3G) is a single-stranded DNA cytosine deaminase that functions in innate immunity against retroviruses and retrotransposons. Although A3G can potently restrict Vif-deficient HIV-1 replication by catalyzing excessive levels of G-to-A hypermutation, sublethal levels of A3G-catalyzed mutation may contribute to the high level of HIV-1 fitness and its incurable prognosis. To chemically modulate A3G catalytic activity with the goal of reducing the HIV-1 genomic mutation rate, we synthesized and biochemically evaluated a class of 4-amino-1,2,4-triazole-3-thiol small molecule inhibitors that were identified by high-throughput screening. This class of compounds exhibits low micromolar (3.9 – 8.2 µm) inhibitory potency and remarkable specificity for A3G versus related deaminase APOBEC3A. Chemical modifications to inhibitors, A3G mutational screening, and thiol reactivity studies implicate C321, a residue proximal to the active site, as the critical A3G target for this class of molecules.
Keywords: Drug Discovery, APOBEC3G, Heterocycles, Hypomutation, Antiviral Agents
Introduction
Since the discovery of acquired immune deficiency syndrome (AIDS) and the identification of human immunodeficiency virus (HIV), the causative retrovirus of AIDS, a wealth of biochemical and immunological investigation has fueled the development of more than twenty-five antiretrovirals and the introduction of highly active antiretroviral therapy (HAART).[1,2] Though these therapies have slowed the global AIDS epidemic and drastically prolonged the life expectancy of HIV-1-positive patients, the imminent development of drug resistance and the toxic side effects associated with HAART prompt continued efforts towards the discovery of new therapeutics with unique mechanisms of action.[3]
Research over the past decade has elucidated a previously unknown mechanism of host-virus interaction between a family of retrovirus restriction factors found in human host cells, namely APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H, and the virally encoded virion infectivity factor (Vif).[4–6] APOBEC3G (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G; A3G), a single-stranded (ss)DNA cytosine to uracil (C-to-U) deaminase and the archetypal member of this family, is a potent restrictor of Vif-deficient HIV-1 replication. Wild-type virus, however, uses Vif to nucleate the formation of an E3 ubiquitin ligase complex that degrades A3G and enables virus replication in A3G-expressing cells. The APOBEC3-Vif interaction is therefore essential for HIV-1 infection and provides a novel, and minimally explored, target in HIV-1 drug discovery.
Current models hypothesize that A3G restricts HIV-1 replication by incorporating into budding viral particles by a RNA-Gag interaction, hijacking transport with the particle until a target cell is infected, and inhibiting viral cDNA synthesis via both deamination-independent[7] and deamination-dependent mechanisms.[4,5,8–10] Deamination-independent inhibition of HIV-1 replication is predicted to occur through direct A3G interaction with viral genomic RNA, causing physical blockage to HIV-1 reverse transcriptase progression. The prominent mechanism by which A3G restricts Vif-deficient HIV-1 replication, however, is through C-to-U deamination events on minus-strand viral DNA which become immortalized as G-to-A hypermutations in the plus (genomic) strand. Such extensive mutation ultimately renders the virus non-infective.
The potent anti-HIV-1 activity of A3G has recently sparked efforts to discover inhibitors of Vif, agonists of A3G and molecules that mask the A3G-Vif interaction surfaces.[11–17] Small molecules that antagonize Vif reinstate the innate antiviral activity of A3G by boosting G-to-A hypermutation, accomplishing lethal mutagenesis. Inhibitor design strategies to increase the HIV-1 mutation rate can be defined as “therapy by hypermutation”.[18]
Conversely, strong evidence exists to support the hypothesis that A3G is being exploited by HIV-1 through modulation by Vif. By taking advantage of the pro-mutational capabilities of A3G, HIV-1 can accomplish an advantageous level of sublethal mutation to enable viral fitness.[18–21] Sequenced genomes from HIV-1-positive patients support this model by exhibiting considerable A3G-dependant mutation patterns despite the ability of Vif to trigger A3G degradation.[22,23] Thus, genetic variation attributable to A3G likely contributes to the characteristically high mutation rate of HIV-1, its ability to evade immune clearance mechanisms, and its rapid evolution of resistance to antiretroviral therapies. Therefore, A3G-catalyzed mutation may be essential for HIV-1 to achieve its unprecedented level of viral fitness, and current therapies and vaccination strategies may benefit from dampening A3G activity as HIV-1 evolution would be predicted to slow. Our strategy to reduce HIV-1 mutation by inhibiting A3G can be classified as “therapy by hypomutation”.[18]
We recently described the development of a fluorescence-based, high-throughput screening (HTS) assay that was used to identify small molecule inhibitors of A3G.[24] In our previous study, screening of 1280 pharmacologically active compounds (LOPAC, Sigma) demonstrated the A3G-specific inhibitory capabilities of catechol-containing scaffolds, including our prototype A3G antagonist MN30 (IC50 = 9.1 µm, PubChem CID-5353329). Mutational screening, structural analysis, and mass spectrometry identified C321, located proximal to the active site, as the binding site for these catechol-based A3G inhibitors. To identify additional A3G-specific inhibitors with unique chemotypes, we screened over 325,000 compounds (Sanford-Burnham Medical Research Institute, NIH MLPCN collection) utilizing our fluorescence-based A3A/A3G DNA cytosine deamination assay.[25] A class of 4-amino-1,2,4-triazole-3-thiols and structurally related analogues were identified and developed as A3G inhibitors based on MLS-0036803, a HTS hit renamed MN256.0105 (1) in-house. Scaffolds identical or analogous to 1 have been previously investigated for fungicidal,[26] antioxidant,[27] cytotoxic,[28] antimicrobial[28] and anti-osteoarthritic[29] applications.
Results and Discussion
Synthesis of 4-amino-1,2,4-triazole-3-thiol analogues
To elucidate the mechanism of inhibition for the 4-amino-1,2,4-triazole-3-thiol class of screening hits and to reconfirm the original lead molecule, MN256.0105 (1) was synthesized (Scheme 1). 4-amino-1,2,4-triazole-3-thiol (4) was obtained in high yield by reaction of thiocarbohydrazide (2) in refluxing aqueous formic acid. Recrystallization from EtOH yielded pure 4 in 95% yield. Compound 1 was then obtained by condensation of 4 with 4-bromobenzaldehyde in excess AcOH in 82% yield. Analogue 8 was similarly synthesized in two steps, first by reaction of thiocarbohydrazide with AcOH under refluxing conditions to yield heterocycle 5. The highest yields were accomplished by introducing an empty Dean-Stark trap to remove by distillation excess AcOH upon conclusion of the reaction, as opposed to the previously reported method of concentration in vacuo.[30] Imine formation by reaction with p-anisaldehyde under acidic conditions delivered 8 (83%, two steps).
Scheme 1.
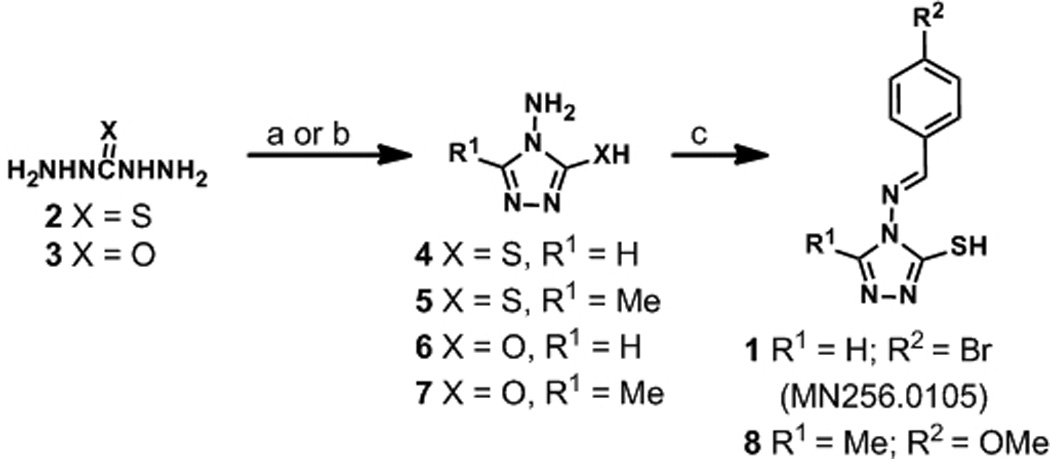
Reagents and conditions: (a) HCO2H (aq.) or AcOH, reflux (to dryness), 95%-quant. (for 4 and 5); (b) (EtO)3CH or (EtO)3CCH3, 60 °C - reflux, 34–69% (for 6 and 7); (c) p-MeOC6H4CHO or p-BrC6H4CHO, AcOH, reflux, 82–84%.
Chemical modification of the 4-amino-1,2,4-triazole-3-thiol heterocycle (4) was pursued to evaluate the necessity of the thiol. As shown in Scheme 1, the thiol moiety of congeners 4 and 5 was replaced by a hydroxyl group to deliver 6 and 7. These scaffolds were prepared by reacting carbohydrazide with excess triethylorthoformate (6) or triethylorthoacetate (7) to achieve the core 4-amino-1,2,4-triazole-3-ol heterocycles in 69% and 34% yields, respectively.
We were also interested in evaluating scaffolds similar to 1 that were not subject to imine hydrolysis to evaluate effects of the additional aromatic functionality on A3G inhibitory potency. The synthesis of non-hydrolysable amide 9 was accomplished in moderate yield through treatment of amine 5 with benzoyl chloride in refluxing dioxane (Scheme 2). We additionally pursued thiol protection methodologies to further gauge the necessity of a free sulfhydryl group for biological potency. For this investigation, we synthesized conjugated thiadiazole 10 and S-alkylated benzyl analogue 11. Compound 10 was prepared by treatment of 5 with p-methoxybenzonitrile and aqueous H3PO4 at elevated temperatures over 5 h (47% yield). Benzyl protection of 5 was carried out by reaction with benzyl bromide, in yields consistent with previous reports, to afford compound 11.[31]
Scheme 2.
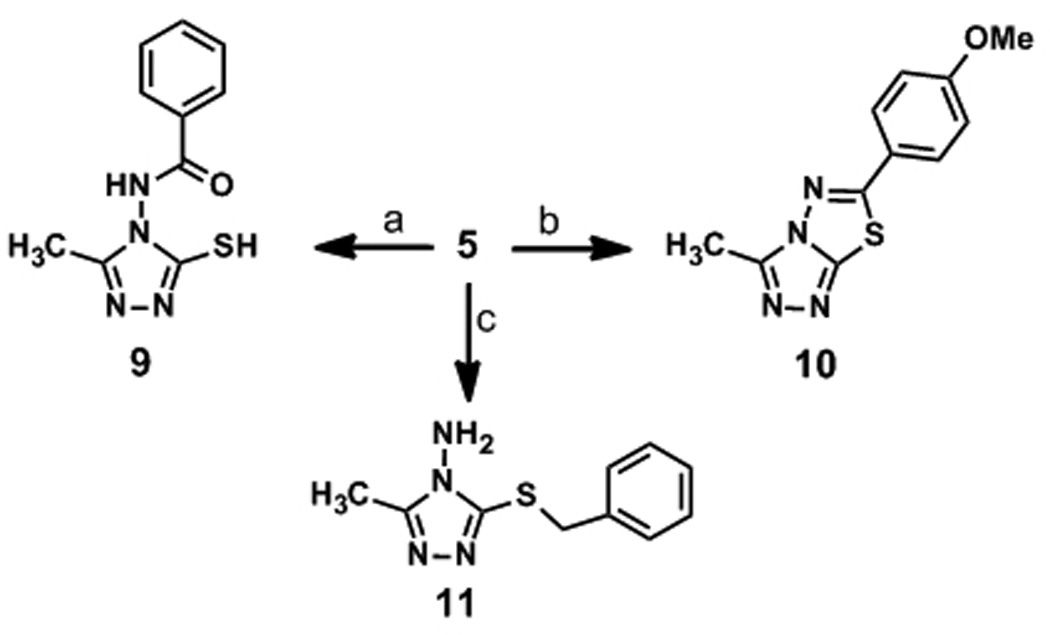
Reagents and conditions: (a) BzCl, dioxane, reflux, 44%; (b) p-methoxybenzonitrile, H3PO4 (aq.), reflux, 47%; (c) BnBr, NaH, DMF, rt, 14%.
Biological evaluation of synthetic analogues
Synthesized small molecules were evaluated for A3G inhibition using our fluorescence-based deamination assay.[24] Using full-length human A3G purified from HEK293T cells and a ssDNA oligonucleotide functionalized with a 5´ 6-FAM fluorophore and a 3´-TAMRA quenching molecule, deamination efficiency was quantified by measured fluorescence. In the absence of inhibitor, deamination of the target cytosine to uracil (C-to-U) is followed by uracil DNA glycosylase-catalyzed excision of U and NaOH-catalyzed phosphodiester backbone cleavage, which releases the 6-FAM fluorophore from the TAMRA quench. Dose-response curves were generated to determine IC50 values for synthesized analogues (Table 1).
Table 1.
APOBEC3G Inhibition (IC50) by 1 and 4–11.[a]
| Candidate Inhibitor | IC50 (µM)a |
|---|---|
| 1 | 4.3 ± 1.1 |
| 4 | No Activity |
| 5 | 6.1 ± 1.1 |
| 6 | No Activity |
| 7 | No Activity |
| 8 | 3.9 ± 1.2 |
| 9 | 8.2 ± 1.2 |
| 10 | No Activity |
| 11 | No Activity |
Performed in triplicate. Values shown are the mean +/− S.D.
Synthesized 1 exhibited equivalent A3G inhibitory activity to the HTS screened material, verifying results obtained from preliminary A3G inhibition (IC50 = 4.3 ± 1.1 µm versus IC50 = 2.0 µm, respectively). Structurally related analogue 8 (IC50 = 3.9 ± 1.2 µm) demonstrated equipotent anti-A3G activity to HTS hit 1 (Table 1). As a result, we hypothesized that small modifications to the phenyl and triazole rings have little effect on A3G inhibition.
Based on previous work with MN30,[24] we hypothesized that the thiol functionalities of 1 and 8 garnered the A3G inhibitory activity. Moreover, the suspected hydrolytic susceptibility of Schiff bases 1 and 8 under biological conditions prompted us to test the 4-amino-1,2,4-triazole-3-thiol precursors 4 and 5 as A3G inhibitors. Surprisingly, 4 did not display any inhibitory activity at 100 µm concentrations, whereas the related 5-methyl analogue 5 (IC50 = 6.1 ± 1.1 µm) was found to be nearly equipotent to its parent scaffold 8 (IC50 = 3.9 ± 1.2 µm). Incorporation of a non-hydrolysable linker, yielding 9, maintained inhibition (IC50 = 8.2 ± 1.2 µm) demonstrating that pendent aromatic functionalities on the exocyclic amine have little effect on the potency of A3G inhibition by molecules of this class.
To demonstrate the necessity of the thiol component of these scaffolds, hydroxyl-substituted analogues 6 and 7 were tested against purified A3G. No inhibitory activity was detected for either molecule (Table 1). Furthermore, evaluation of thiolalkylated scaffolds 10 and 11 revealed no A3G inhibition, further demonstrating the necessity of the free thiol.
In addition to screening against wild-type A3G, compounds 1 and 4–11 were tested for inhibitory activity against the related cytosine deaminase enzyme, APOBEC3A (A3A), to gauge inhibitor specificity. A3A is 65% identical and 73% similar in the A3G C-terminal catalytic domain at the amino acid level. Remarkably, analogues 1 and 4–11 all failed to inhibit A3A activity at doses up to 100 µm, in spite of the inherent reactivity of the thiol functionality. To begin querying if molecules of this class can exhibit activity in cells, we screened 1 and 4–11 for inhibitory activity against A3G in HEK293T cell lysates. Unfortunately, no measureable amounts of A3G inhibition were detected for any of the molecules screened. Consequently, the reactive thiol moiety may be unavailable for A3G inhibition in the complex environment of cell lysate.
Reactivity studies of APOBEC3G inhibitors with cysteine-like substrates
The known reactivity properties of thiols with cysteine residues prompted our evaluation of this scaffold to form disulfide bonds under biological conditions. Amide analogue 9, which contains a free thiol, was dissolved in DMSO, diluted with PBS buffer (pH 7.4) and treated with excess cysteamine. Reaction aliquots were removed at the following time points: 0 min, 30 min, 1 hr and 12 hr and were analyzed by reverse phase HPLC for the presence of new peaks. Complete disappearance of the starting material was observed within 30 min yielding one new product, which was characterized as disulfide 12 by mass spectrometry (Figure 2). Therefore, molecules of this class that contain free thiols are capable of intercepting other thiols and forming disulfide bonds, which may confer their primary mechanism of enzyme inhibition.
Figure 2.

(A) 4-Amino-1,2,4-triazole-3-thiol 9 and cysteamine were shaken in aqueous PBS (pH = 7.4) at 37 °C. Aliquots were analysed by reverse-phase HPLC at t = 0, 30, 60 mins and 12 hr. (B). Reaction at t = 0 min (top) and t = 12 hr (bottom). Peak 12 was isolated and characterized by mass spectrometry HRMS-ESI+ m/z [M+Na+] 332.0616 (calc’d); 332.0629 (found).
Mutation studies identify C321 as the inhibitor binding site
Since free thiols are required for A3G inhibition by molecules of this class, the surface cysteine residues of A3G were investigated as potential inhibitor binding sites. Previous work has demonstrated that cysteine 321 (C321) of the catalytic A3G domain is an inhibitor binding site for covalent modification by small molecules.[24] Employing this methodology, a triple mutant construct A3G-2K3A, which has three surface Cys-to-Ala substitutions (namely C243A, C321A and C356A), and a single mutant A3G-C321A were tested in parallel with wild type enzyme against thiols 1 and 8 utilizing the previously described deamination assay.[24] It is important to note that in the context of full-length A3G, these specific substitutions have no effect on localization, deamination, oligomerization or HIV-1 Vif-deficient restriction capabilities.[24,32,33]
We found that the mutant enzymes were only partially resistant to compounds 1 and 8 with recovered deamination efficiency between 19 – 46%. Interestingly, deaminase activity was not fully recovered in comparison to DMSO control or the previously reported catechol-based covalent inhibitor MN30.[24] Such results imply that these thiol-containing inhibitors may also function, at least in part, through a second inhibitory mechanism. Initial hypotheses derived from the field of transition metal catalysis suggest that the 4-amino-1,2,4-triazole-3-thiol scaffold chelates divalent metals.[34–37] A3G, which contains a catalytic zinc atom, may therefore suffer a decrease in deaminase efficiency in the presence of zinc chelating molecules; however, additional experiments are necessary to validate this possibility.
Conclusion
High-throughput screening and analogue synthesis have identified a class of 4-amino-1,2,4-triazole-3-thiols that inhibit the DNA cytosine deaminase A3G. Counter-screening of the small molecule analogues against related APOBEC3 family member A3A demonstrated marked specificity for A3G inhibition despite the reactivity of the inhibitors to sulphur nucleophiles. Replacement or protection of the thiol moeity of the inhibitors completely abrogated their inhibitory capabilities, providing evidence that a free thiol is a key structural feature of this class of inhibitors. A combination of mutagenesis studies and HPLC assays implicate the inhibitory activity of this class of molecules is accomplished through disulfide bond formation to C321, a residue adjacent to the A3G active site. These findings further support our hypothesis that covalent modification to C321 causes conformational change to the A3G protein, where Y315 shifts and sterically blocks substrate DNA cytosines from entering the A3G active site.[24]
Mutant screening potentially implicates a second mechanism of action based on the inability of the C321A mutant to fully recover deamination capabilities in comparision with DMSO control. We hypothesize that the 4-amino-1,2,4-triazole-3-thiol scaffold may engage the catalytic zinc atom in A3G confering a secondary mechanism of enzyme inhibiton, although more evidence is needed to support this theory.
Experimental Section
General Synthesis Information
Chemical reagents were purchased from Sigma-Aldrich, Alfa Aesar or Acros and used without additional purification. Bulk solvents were from Fisher Scientific and anhydrous solvents were purchased from Sigma-Aldrich. Reactions were performed under an atmosphere of dry N2 unless otherwise noted. Silica gel chromatography was performed on a Teledyne-Isco Combiflash Rf-200 instrument using Redisep Rf Gold High Performance silica gel columns (Teledyne-Isco) or self-packed columns with SiliaFlash 60Å silica gel (SiliCycle). HPLC analyses were performed on an Agilent 1200 series instrument equipped with a diode array detector and a Zorbax SB-C18 column (4.6 × 150 mm, 3.5 µm, Agilent Technologies) or a Zorbax SB-AQ column (4.6 × 150 mm, 3.5 µm, Agilent Technologies) for analytical-scale analysis. Compounds used in biological testing were no less than 98% pure as determined by two-wavelength HPLC analysis (254 and 215 nm), except for compounds 4 and 8, which were 92% and 93% pure, respectively. Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker Avance instrument operating at 400 MHz (for 1H) and 100 MHz (for 13C) at ambient temperature. Chemical shifts are reported in parts per million and normalized to internal solvent peaks or tetramethylsilane (0 ppm). Exchangeable protons (thiols, alcohols and amines) were verified by 1H NMR D2O exchange experiments. High-resolution mass spectrometry (HRMS) was recorded in positive-ion mode on a Bruker BioTOF II instrument.
4-amino-1,2,4-triazole-3-thiol (4)
Prepared as previously described.[38] 1H NMR ([D6]DMSO): δ= 13.63 (s, 1H, SH), 8.45 (s, 1H), 5.68 ppm (s, 2H, NH2); 13C NMR ([D6]DMSO): δ= 166.0, 142.3 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C2H4N4S: 139.0054, found: 139.0045.
4-((4-bromobenzylidene)amino)-1,2,4-triazole-3-thiol (1)
To a solution of 4-amino-1,2,4-triazole-3-thiol (4, 0.219 g, 1.89 mmol) in AcOH (8.0 mL) at room temperature was added 4-bromobenzaldehyde (0.349 g, 1.89 mmol). The reaction was then heated to reflux. After 2 h, the reaction was cooled and poured into ice water. The resulting precipitate was collected, washed with water and recrystallized from hot, absolute EtOH to yield compound 1 as a pale yellow solid (0.438 g, 82%): 1H NMR (CDCl3): δ= 10.47 (s, 1H), 8.08 (s, 1H), 7.73 (d, J = 2.0 Hz, 2H), 7.63 ppm (d, J = 2.0 Hz, 2H); 13C NMR (CDCl3): δ= 158.83, 158.78, 140.9, 132.4, 131.2, 130.0, 127.3 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C9H7BrN4S: 304.9472, found: 304.9468.
4-amino-5-methyl-1,2,4-triazole-3-thiol (5)
A mixture of thiocarbohydrazide (2, 2.60 g, 24.5 mmol) in AcOH (5.0 mL) in a 100 mL round bottom flask was heated to reflux into an empty Dean-Stark trap. Since product formation is kinetically fast, this process served to remove excess acid which enhanced reaction yields. The reaction was allowed to proceed until product formed as a white precipitate and all acid was removed. The residual solid was removed from the flask with water, filtered and recrystallized from hot, aqueous EtOH to yield compound 5 as a white powder (3.16 g, 99%): 1H NMR ([D6]DMSO): δ= 13.39 (s, 1H, SH), 5.51 (s, 2H, NH2), 2.11 ppm (s, 3H, CH3); 13C NMR ([D6]DMSO): δ= 165.37, 149.08, 10.37 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C3H6N4S: 153.0211, found: 153.0213.
4-amino-1,2,4-triazol-3-ol (6)
Prepared as previously described.[39] 1H NMR ([D6]DMSO): δ= 11.52 (s, 1H, OH), 7.80 (s, 1H), 5.26 ppm (2H, NH2); 13C NMR ([D6]DMSO): δ= 154.2, 139.3 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C2H4N4O: 123.0283, found: 123.0292.
4-amino-5-methyl-1,2,4-triazol-3-ol (7)
Carbohydrazide (3, 0.500 g, 5.550 mmol) was suspended in triethylorthoacetate (1 mL), heated from 60 – 90 °C over 45 min, and then refluxed. After 2 h, the reaction was cooled, concentrated in vacuo and the crude solid was recrystallized from hot, absolute EtOH to yield compound 7 as a white crystalline solid (0.214 g, 34%): 1H NMR ([D6]DMSO): δ= 11.23 (s, 1H, OH), 5.10 (s, 2H, NH2), 2.07 ppm (s, 3H); 13C NMR ([D6]DMSO): δ= 154.3, 145.4, 10.7 ppm; HRMS (ESI +) m/z [M+H]+ calc’d for C3H6N4O: 115.0614, found: 115.0615.
4-((4-methoxybenzylidene)amino)-5-methyl-1,2,4-triazole-3-thiol (8)
To a solution of 3 (0.100 g, 0.768 mmol) in AcOH (3.5 mL) at room temperature was added p-anisaldehyde (93 µL, 0.800 mmol). The reaction was heated to reflux for 2 h, then cooled and poured into ice water. The resulting precipitate was collected and washed with water to yield compound 8 as a pale yellow solid (0.160 g, 84%) without further purification: 1H NMR ([D6]DMSO): δ= 13.67 (s, 1H), 9.71 (s, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.10 (d, J = 8.0 Hz, 2H), 3.85 (s, 3H), 2.32 ppm (s, 3H); 13C NMR (CDCl3): δ= 163.3, 162.4, 161.5, 149.8, 130.8, 125.2, 114.6, 55.6, 11.3 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C11H12N4OS: 271.0630, found: 271.0617.
N-(3-mercapto-5-methyl-1,2,4-triazol-4-yl)benzamide (9)
A solution of 3 (0.100 g, 0.770 mmol) in dioxane (5 mL) was treated with benzoyl chloride (100 µL, 0.770 mmol) and heated to reflux for 24 h. The reaction mixture was cooled to rt and concentrated in vacuo. SiO2 purification (gradient 40–100% ethyl acetate in hexanes) gave compound 9 as a white solid (80.0 mg, 44%): 1H NMR ([D6]DMSO): δ= 13.73 (s, 1H, SH), 11.78 (s, 1H, NH), 8.01-7.98 (m, 2H), 7.71-7.57 (m, 3H), 2.20 ppm (s, 3H); 13C NMR ([D6]DMSO): δ= 167.0, 165.4, 149.9, 132.9, 130.9, 128.8, 127.9, 10.0 ppm; HRMS-ESI+ m/z [M+Na]+ calc’d for C11H11N3OS: 257.0473, found: 257.0288.
6-(4-methoxyphenyl)-3-methyl-1,2,4-triazolo[3,4-b][1,3,4]thiadiazole (10)
4-Methoxybenzonitrile (0.100 g, 0.75 mmol) and 5 (0.098 g, 0.75 mmol) were suspended in aqueous H3PO4 (5 mL) and heated to reflux. After 5 h, the mixture was diluted with an excess of H2O, neutralized with aqueous NaOH (40% w/v soln) to pH 7–8, and concentrated in vacuo to yield compound 10 as a pale yellow powder (0.087 g, 47%): 1H NMR (CDCl3): δ= 7.83 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 8.0 Hz, 2H), 3.90 (s, 3H), 2.76 ppm (s, 3H); 13C NMR (CDCl3): δ= 166.2, 163.2, 153.1, 144.9, 129.0, 122.1, 115.0, 55.8, 10.6 ppm; HRMS-ESI + m/z [M+Na]+ calc’d for C12H11N3OS: 269.0473, found: 269.0468.
3-(benzylthio)-5-methyl-1,2,4-triazol-4-amine (11)
Prepared as previously described.[31] 1H NMR (CDCl3): δ= 7.45-7.43 (m, 2H), 7.36-7.30 (m, 3H), 4.60 (s, 2H), 2.37 ppm (s, 3H); 13C NMR ([D6]DMSO): δ= 158.7, 152.0, 137.1, 128.7, 127.7, 127.1, 34.4, 10.8 ppm; HRMS-ESI+ m/z [M+H]+ calc’d for C10H12N4S: 221.0861, found: 221.0871.
Expression and Purification of APOBEC3A, APOBEC3G, APOBEC3G 2K3A and APOBEC3G C321A
A3G, A3A, A3G 2K3A and A3G C321A were expressed and purified as previously described.[24]
DNA Deaminase Assay
The DNA deaminase assay was performed as previously described with the ssDNA oligomer 5’–6-FAM-AAA-TAT-TCC-CTA-ATA-GAT-AAT-GTG-A-TAMRA-3’.[24] Deaminase assays with mutant A3G were performed with 50 µm compound, 0.0675 µm A3G, 0.33 µm ssDNA, and excess UDG. None of the synthesized compounds inhibited uracil DNA glycosylase in the context of the in vitro assay.
HPLC Assay for Disulfide Formation
A solution of 9 (10.0 mg, 0.040 mmol) in DMSO (100 µL) was diluted with 1× aqueous PBS (5 mL, pH 7.4) and treated with cysteamine (0.684 g, 4.03 mmol, 100 eq). The solution was then shaken at 37 °C. Aliquots of the reaction mixture were taken at the following time points: 0 min, 30 min, 60 min and 12 h. These reaction aliquots were analyzed by reverse phase analytical HPLC. The HPLC analytical method (Zorbax SB-C18 4.6 × 150 mm, 3.5 µm column, Agilent Technologies; flow rate = 1.0 mL/min) involved isocratic 10% CH3CN in 0.1% (v/v) aqueous CF3CO2H (0 to 2 min), followed by linear gradients of 10–85% CH3CN (2–24 min) and 85%–95% CH3CN (24–26 min). The major peak 12 (Figure 2) was isolated, concentrated in vacuo, and characterized by mass spectrometry. HRMS (ESI+) m/z calc’d for C12H15N5OS2 [M+Na]+ 332.0616; found 332.0629.
Supplementary Material
Figure 1.

Chemical structure of A3G inhibitor MN256.0105 (1) and the frequency that the 4-amino-1,2,4-triazole-3-thiol scaffold was observed in A3G hits from high-throughput screening.
Figure 3.
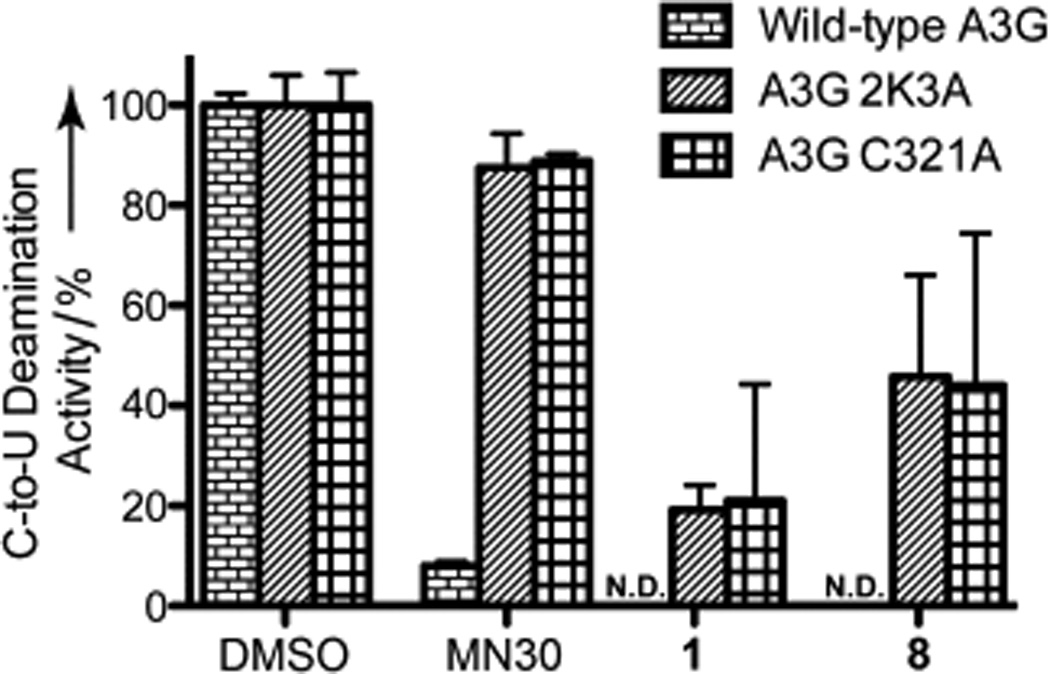
Potency of MN30 (Figure S1), 1 and 8 against wild-type A3G and mutants A3G-2K3A and A3G-C321A. The mean and SD of triplicate deaminase assays with 50 µm compound are shown relative to the DMSO only control. (N.D. = Not Detectable)
Acknowledgements
We acknowledge financial support from a University of Minnesota Innovation Grant (to D.A.H. and R.S.H.), the NIH (P01 GM091743 to R.S.H.), and start-up funds from the Department of Medicinal Chemistry and College of Pharmacy (to D.A.H.). M.E.O thanks the NIH for a Chemistry Biology Interface Predoctoral Traineeship (T32-GM08700) and Dr. Lyle and Sharon Bighley and the College of Pharmacy for a Bighley Graduate Fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org.
References
- 1.Mehellou Y, De Clercq E. J. Med. Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 2.Thompson MA, Aberg JA, Cahn P, Montaner JS, Rizzardini G, Telenti A, Gatell JM, Gunthard HF, Hammer SM, Hirsch MS, Jacobsen DM, Reiss P, Richman DD, Volberding PA, Yeni P, Schooley RT. JAMA. 2010;304:321–333. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- 3.Volberding PA, Deeks SG. Lancet. 2010;376:49–62. doi: 10.1016/S0140-6736(10)60676-9. [DOI] [PubMed] [Google Scholar]
- 4.Albin JS, Harris RS. Expert Rev. Mol. Med. 2010;12 doi: 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malim MH, Emerman M. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. J. Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop KN, Verma M, Kim E-Y, Wolinsky SM, Malim MH. PLoS Pathog. 2008;4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schumacher AJ, Hache G, Macduff DA, Brown WL, Harris RS. J. Virol. 2008;82:2652–2660. doi: 10.1128/JVI.02391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyagi E, Opi S, Takeuchi H, Khan M, Goila-Gaur R, Kao S, Strebel K. J. Virol. 2007;81:13346–13353. doi: 10.1128/JVI.01361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Browne EP, Allers C, Landau NR. Virology. 2009;387:313–321. doi: 10.1016/j.virol.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JL, Bu W, Burdick RC, Pathak VK. Trends Pharmacol. Sci. 2009;30:638–646. doi: 10.1016/j.tips.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu X-F. FASEB J. 2007;21:217–222. doi: 10.1096/fj.06-6773com. [DOI] [PubMed] [Google Scholar]
- 13.Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM. Nat. Biotechnol. 2008;26:1187–1192. doi: 10.1038/nbt.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cen S, Peng Z-G, Li X-Y, Li Z-R, Ma J, Wang Y-M, Fan B, You X-F, Wang Y-P, Liu F, Shao R-G, Zhao L-X, Yu L, Jiang J-D. J. Biol. Chem. 2010;285:16546–16552. doi: 10.1074/jbc.M109.085308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ejima T, Hirota M, Mizukami T, Otsuka M, Fujita M. Int. J. Mol. Med. 2011;28:613–616. doi: 10.3892/ijmm.2011.737. [DOI] [PubMed] [Google Scholar]
- 16.Ali A, Wang J, Nathans RS, Cao H, Sharova N, Stevenson M, Rana TM. ChemMedChem. 2012;7:1217–1229. doi: 10.1002/cmdc.201200079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohammed I, Parai MK, Jiang X, Sharova N, Singh G, Stevenson M, Rana TM. ACS Med. Chem. Lett. 2012;3:465–469. doi: 10.1021/ml300037k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris RS. Nat. Biotechnol. 2008;26:1089–1090. doi: 10.1038/nbt1008-1089. [DOI] [PubMed] [Google Scholar]
- 19.Haché G, Mansky LM, Harris RS. AIDS Rev. 2006;8:148–157. [PubMed] [Google Scholar]
- 20.Sadler HA, Stenglein MD, Harris RS, Mansky LM. J. Virol. 2010;84:7396–7404. doi: 10.1128/JVI.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulder LCF, Harari A, Simon V. Proc. Natl. Acad. Sci. USA. 2008;105:5501–5506. doi: 10.1073/pnas.0710190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, Mallal S. J. Virol. 2006;80:9259–9269. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, Daniels M, Ferrari G, Haynes BF, McMichael A, Shaw GM, Hahn BH, Korber B, Seoighe C. PLoS Pathog. 2009;5:e1000414. doi: 10.1371/journal.ppat.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, Harki DA, Solberg J, Hook DJ, Pandey KK, Parniak MA, Johnson JR, Krogan NJ, Somasundaran M, Ali A, Schiffer CA, Harris RS. ACS Chem. Biol. 2012;7:506–517. doi: 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M, Harris RS. Manuscript in Preparation [Google Scholar]
- 26.Sun X, Tao Y, Liu Y, Jia Y, Chen B. Acta Chim. Sinica. 2008;66:234–238. [Google Scholar]
- 27.Demchenko AM, Yanchenko VO, Smolskiy OS, Aheyev VO, Lozynskiy MO. Farmatsevtichnii Zhurnal (Kiev) 2004;68 [Google Scholar]
- 28.Badr SM, Barwa RM. Bioorg. Med. Chem. 2011;19:4506–4512. doi: 10.1016/j.bmc.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 29.Maingot L, Leroux F, Landry V, Dumont J, Nagase H, Villoutreix B, Sperandio O, Deprez-Poulain R, Deprez B. Bioorg. Med. Chem. Lett. 2010;20:6213–6216. doi: 10.1016/j.bmcl.2010.08.108. [DOI] [PubMed] [Google Scholar]
- 30.Eweiss NF, Bahajaj AA, Elsherbini EA. J. Heterocycl. Chem. 1986;23:1451–1458. [Google Scholar]
- 31.Cartwright DDJ, Clark BAJ, McNab H. J. Chem. Soc., Perkin Trans. 2001;1:424–428. [Google Scholar]
- 32.Harjes E, Gross PJ, Chen K-M, Lu Y, Shindo K, Nowarski R, Gross JD, Kotler M, Harris RS, Matsuo H. J. Mol. Biol. 2009;389:819–832. doi: 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shandilya SMD, Nalam MNL, Nalivaika EA, Gross PJ, Valesano JC, Shindo K, Li M, Munson M, Royer WE, Harjes E, Kono T, Matsuo H, Harris RS, Somasundaran M, Schiffer CA. Structure. 2010;18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh K, Singh DP, Barwa MS, Tyagi P, Mirza Y. J. Enzym. Inhib. Med. Ch. 2006;21:557–562. doi: 10.1080/14756360600642131. [DOI] [PubMed] [Google Scholar]
- 35.Singh K, Singh DP, Barwa MS, Tyagi P, Mirza Y. J. Enzym. Inhib. Med. Ch. 2006;21:749–755. doi: 10.1080/14756360600838648. [DOI] [PubMed] [Google Scholar]
- 36.Tabatabaee M, Ghassemzadeh M, Sadeghi A, Shahriary M, Neumüller B, Rothenberger A. Z. Anorg. Allg. Chem. 2009;635:120–124. [Google Scholar]
- 37.Ghassemzadeh M, Tabatabaee M, Soleimani S, Neumüller B. Z. Anorg. Allg. Chem. 2005;631:1871–1876. [Google Scholar]
- 38.McCarrick RM, Eltzroth MJ, Squattrito PJ. Inorg. Chim. Acta. 2000;311:95–105. [Google Scholar]
- 39.Zhang J, Zhang T, Yu K. Chem. Heterocycl. Compd. 2003;39:461–466. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.