

Abstract
It is now affordable to order clinically interpreted whole genome sequence reports from clinical laboratories. One major component of these reports is derived from the knowledge base of previously identified pathogenic variants, including research articles, locus specific and other databases. While over 150,000 such pathogenic variants have been identified, many of these were originally discovered in small cohort studies of affected individuals, so their applicability to asymptomatic populations is unclear. We analyzed the prevalence of a large set of pathogenic variants from the medical and scientific literature in a large set of asymptomatic individuals (N=1,092) and found 8.5% of these pathogenic variants in at least one individual. In the average individual in the 1000 Genomes Project, previously identified pathogenic variants occur on average 294 times (σ= 25.5) in homozygous form and 942 times (σ = 68.2) in heterozygous form. We also find that many of these pathogenic variants are frequently occurring: there are 3,744 variants with MAF >= 0.01 (4.6%) and 2,837 variants with MAF >= 0.05 (3.5%). This indicates that many of these variants may be erroneous findings or have lower penetrance than previously expected.
Keywords: whole genome sequencing, WGS, personalized medicine, incidental findings, incidentalome
Introduction
It is now possible to order clinically interpreted whole genome sequences (WGS) from clinical laboratories (genomeweb, 2011a; genomeweb, 2011b; Review, 2011) and direct-to-consumer groups . These data have the potential to improve medical care, but the methods to translate genomic variation into accurate clinical interpretations remain to be defined, particularly for asymptomatic individuals (Brunham and Hayden, 2012). WGS interpretation must address both novel variation that is likely to be pathogenic as well as over 150,000 variants with implied – but unconfirmed – disease associations that have been reported in the medical and scientific literature (Stenson, et al., 2012), locus specific databases (Vihinen, et al., 2012), researcher submissions (Yu, et al., 2008), clinical genetics practice (McKusick-Nathans Institute of Genetic Medicine; NCBI, 2012a), and genome-wide association studies (GWAS) (NHGRI).
Many of these variants were identified in symptomatic populations and may be erroneously associated with disease due to small cohort sizes, limited validation studies, and unmatched control populations, creating a mixture of well-established associations with unverified anecdotes (Homer, et al., 2008). The consequence of this case ascertainment bias is that the probability of observing a particular variant given the presence of a particular disease is not equivalent to the probability of developing the disease given the presence of each variant. Some variants may also be incompletely penetrant or potentially associated with variable expressivity. Furthermore, some of these associations, especially those reported before the completion of the Human Genome Project (HGP), are limited in applicability because of potential inconsistencies with our current standards for genomic coordinates, nomenclature and gene structure (Tong, et al., 2011). Consequently, it is difficult to translate these findings into estimates of disease risk for asymptomatic individuals, creating a major bottleneck in clinical application of WGS.
In a recent study, we estimated that 10.6% of variants, genome-wide, have sufficient clinical relevance and scientific validity for investigators to share them with research participants (Cassa, et al., 2012). This estimate specifies that there are over 12,000 variants that are appropriate to review and report, linked to both common and rare disease, and adverse drug response (Kohane, et al., 2012). But, without accurate risk estimates for each variant in patients lacking clinical suspicion, it is difficult to determine whether these are clinically relevant findings, or false indications that may frighten patients and cause needless diagnostic workups and costly screenings (Fabsitz, et al., 2010; Tong, et al., 2011).
Considering that many of these variants are more common in the population than their associated diseases are, many of these variants must be either false positives or incompletely penetrant, and should be filtered or annotated before they reach a clinical WGS report. But, just how many of these variants are sufficiently prevalent that we would be ill-advised to counsel an asymptomatic carrier about such an association?
To answer this question, we combined the data from the largest pathogenic variant database, the Human Gene Mutation Database (HGMD), with data from the largest publicly available source of WGS data from asymptomatic individuals, the 1000 Genomes Project (TGP). We analyzed the prevalence of HGMD variants by predicted impact and pathogenicity classifications in a large asymptomatic population.
Materials and Methods
Set of variants and whole genome sequence data
We included all single nucleotide substitution variants with genomic coordinate and reference/alternate allele information available in HGMD version 2012.2 (Stenson, et al., 2009) (N=81,432 variants). We used publicly available WGS call data from the TGP (N=1,092 individuals) (1000 Genomes Phase 1, Version 3, release date 4/30/12) (Consortium, 2012), as the set of genomic samples.
Whole genome analysis of asymptomatic individuals for variant allele frequency and count data
In each asymptomatic WGS, we checked for the presence of each HGMD pathogenic variant. Among the 81,432 variants, we found 6,917 variants present in at least one individual. We recorded whether each individual carried each variant in heterozygous or homozygous minor form, and calculated the maximum, minimum, and average number of variants present in each individual. For each variant, we calculate the minor allele frequency (MAF) using the maximum likelihood estimate from the observed variants in the TGP, which is the number of alternate alleles divided by the total number of alleles. For 717 variants, the allele frequency of the non-reference allele was above 50%; for these alleles, we treated the reference allele as the minor allele so all minor allele frequencies were below 50%.
Analysis of variant pathogenicity classifications and amino acid changes
We analyzed the pathogenicity classifications for each variant we observed in TGP. We reviewed the HGMD reported classification in Table 1, and those calculated by PolyPhen 2 (Adzhubei, et al., 2013). For each HGMD variant classification, we generated the population frequency distribution of observed variants in that class, using TGP data. For PolyPhen 2 scores, we plot each variant's score by variant minor allele frequency bin. We also grouped all variants in HGMD into four major categories of amino acid changes: synonymous, missense, nonsense, or none/other, where the associated variant is non-coding or no information is provided. For each amino acid category, we generated the population frequency distribution of observed variants in that class, using TGP data.
Table 1.
List of HGMD Classifications
| HGMD Classification | Classification Description |
|---|---|
| Disease-associated polymorphism (DP) | A polymorphism reported to be in significant association with a disease/phenotype (p<0.05) that is assumed to be functional (e.g. as a consequence of location, evolutionary conservation, replication studies etc), although there may as yet be no direct evidence (e.g. from an expression study) of function. |
| Disease-associated polymorphism with additional supporting functional evidence (DFP) | A polymorphism reported to be in significant association with disease (p<0.05) that has evidence of being of direct functional importance (e.g. as a consequence of altered expression, mRNA studies etc). |
| In vitro/laboratory or in vivo functional polymorphism (FP) | A polymorphism reported to affect the structure, function or expression of the gene (or gene product), but with no disease association reported as yet. |
| Frameshift or truncating variant (FTV) | A polymorphic or rare variant reported in the literature (e.g. detected in the process of whole genome/exome screening) that is predicted to truncate or otherwise alter the gene product (i.e. a nonsense or frameshift variant) but with no disease association reported as yet. Please note that any variant affecting the obligate donor/acceptor splice site of a gene will not be included in this category unless there is evidence for an effect on the splicing phenotype. Variants occurring in pseudogenes will also be excluded unless evidence for a functional effect is present for both the pseudogene itself (Balakirev ES & Ayala FJ, 2003) and the variant in question. |
| Disease causing mutation (DM) | Pathological mutation reported to be disease causing in the corresponding report (i.e. all other HGMD data). |
HGMD classifies variant reports in one of five major categories. Source: HGMD.
Results
We identified a total of 6,917 of these variants in at least one individual in the TGP (8.5% of HGMD variants in this study). The number of HGMD variants identified in each individual is graphed for both homozygous minor and heterozygous form in Figure 1. We found that individuals in the TGP had an average of 294 (σ= 25.5) variants in homozygous form and 942 (σ = 68.2) variants in heterozygous form (Table 2).
Figure 1.
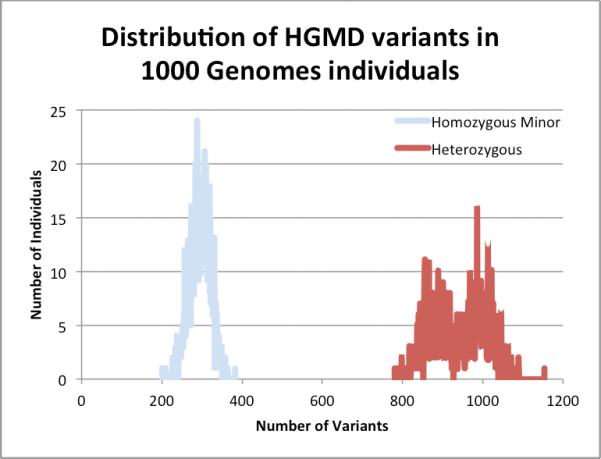
Frequency distribution of HGMD variants identified in 1000 genomes individuals. We identified a total of 6,917 HGMD variants in the 1,092 asymptomatic individuals with whole genome sequence data in the 1000 Genomes Project. The number of individuals is plotted for both homozygous variant genotypes as well as heterozygous genotypes.
Table 2.
Aggregate results from the whole genome interpretation of asymptomatic individuals from the 1000 Genomes Project
| Number of HGMD 2012.1 variants in each 1000 Genomes Project individual (N=1,092) | |||
|---|---|---|---|
| Homozygous Variant Genotypes | Heterozygous Genotypes | Homozygous Reference Genotypes | |
| Average (Std. Dev.) | 294.4 (σ=25.5) | 942.3 (σ=68.2) | 5,680.2 (σ=53.5) |
| [Minimum, Maximum] | [201, 383] | [780, 1154] | [5519, 5801] |
| For all HGMD variants identified in this study | |||
|---|---|---|---|
| Homozygous Variant Genotypes | Heterozygous Genotypes | Homozygous Reference Genotypes | |
| Average (Std. Dev.) | 43.9 (σ=76.5) | 148.8 (σ=181.5) | 899.3 (σ=250.6) |
| [Minimum, Maximum] | [0, 464] | [0, 1092] | [0, 1092] |
Whole genome sequence data of asymptomatic individuals in the 1000 Genomes Project were analyzed, using the substitution variants from HGMD. The total number of variants identified in each sequence is reported, along with the subset of those that are homozygous and heterozygous.
We found that many of these disease-associated variants are observed in asymptomatic individuals quite frequently, which suggests that there are opportunities to filter and prioritize variants that are very common and therefore unlikely to be strongly associated with disease. By population frequency, 3,744 variants have MAF >= 0.01 (54.1% of the 6,917 observed, 4.6% of all study variants) and 2,837 variants have MAF >= 0.05 (41.0% of the 6,917 observed, 3.5% of all study variants). We graph the distribution of observed HGMD variants by MAF in Figure 2. This indicates that many may be erroneous findings or be incompletely penetrant.
Figure 2.
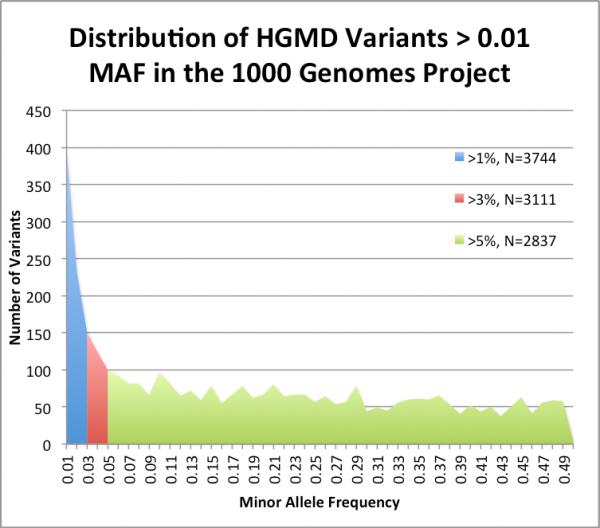
Distribution of HGMD variants with minor allele frequency (MAF) > 0.01 in the 1000 Genomes Project. We plot the number of HGMD variants by MAF in individuals from the 1000 Genomes Project. There are 3,744 HGMD variants with MAF > 0.01, 3,111 variants with MAF > 0.03, and 2,837 variants with MAF > 0.05.
All observed variants were grouped by their HGMD pathogenicity classification and predicted amino acid change. For each variant classification type, we graph the frequency distribution of observed variants in that class in Figure 3. As expected, there are many variants that are classified as polymorphic with MAF >= 0.01 in the asymptomatic population (DFP = 903, DP = 1,495, FP = 741). However there are also variants classified as polymorphic that are below a population frequency of 1% (DFP = 62, DP = 151, FP = 342), so these variants previously classified as polymorphic may be worthy of review for study or platform bias, or may be uncommon disease-associated variants. Unexpectedly, there are also many variants that are classified as disease causing mutations or disease-associated nonsense mutations (DM = 583, FTV = 32) that are present in asymptomatic individuals with MAF >= 0.01.
Figure 3.
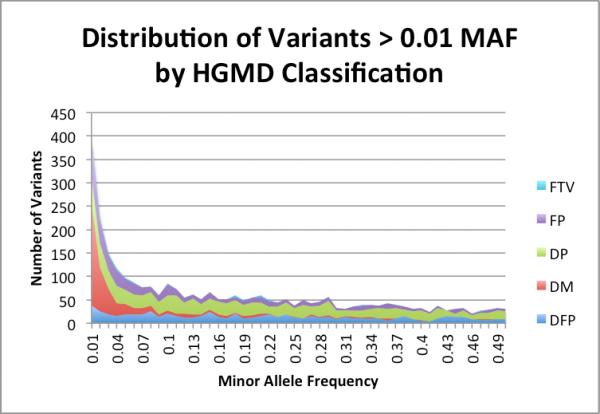
Distribution of variants with MAF > 0.01 from the 1000 Genomes Project by HGMD classification.
We graph the distribution of variants by four major categories of predicted amino acid changes in Figure 4. As expected, most HGMD variants identified in this set of asymptomatic individuals were synonymous (275), missense (4,440), or none/other (1,905). Of the remaining 226 identified nonsense variants, 31.4% (74) were present with MAF >= 0.01. Of the missense variants, 42.8% (1,900) were present with MAF >= 0.01.
Figure 4.
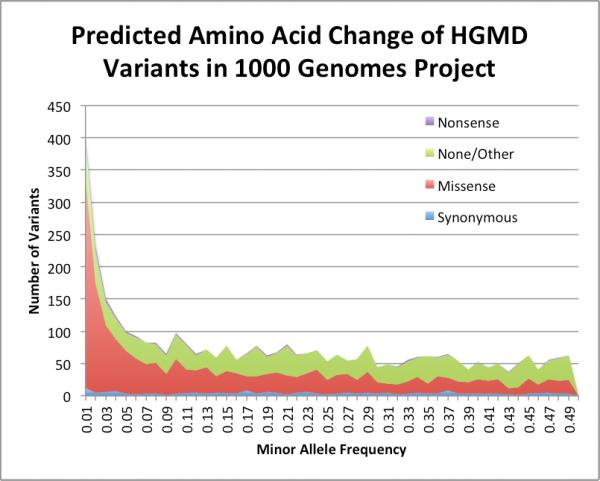
Distribution of variants with MAF > 0.01 from the 1000 Genomes Project by amino acid change.
We computed PolyPhen 2 scores for variants observed in TGP, and plotted these by minor allele frequency, in Figure 5. We observed the fraction of variants predicted as damaging by Polyphen 2 decreases as the variant MAF increases, so that the vast majority of high-frequency variants are predicted to be neutral. It is also worth noting that even at low frequencies, PolyPhen 2 predicts a substantial number of observed variants as benign (52% of the 4,431 total variants for which PolyPhen 2 predictions could be made, and 40% of 2,580 variants with MAF < 0.01).
Figure 5.
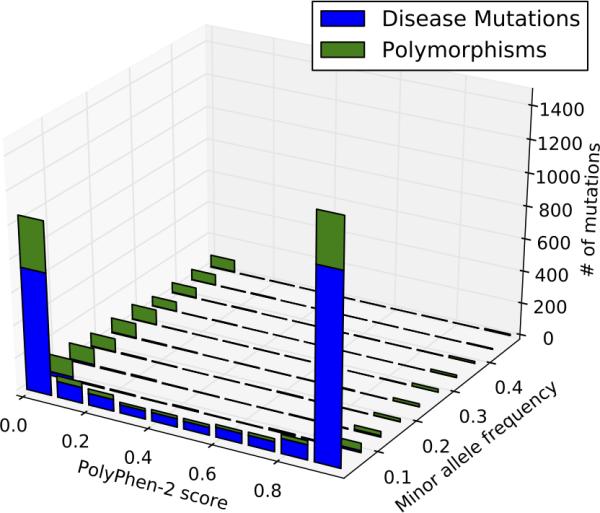
Distribution of variants PolyPhen 2 versus 1000 Genomes Project minor allele frequency. The PolyPhen score is bimodal, with most of the scores being found at the extremes: high numbers are pathogenic and low numbers are benign. Disease Mutations are mostly but not overwhelmingly predicted as pathogenic, while polymorphisms are mostly but not overwhelmingly predicted as benign. The number of pathogenic predictions decreases with increasing MAF, so that a variant with MAF > 0.3 is far more likely to be predicted benign than pathogenic.
Variants classified as Disease Mutations (DM) in HGMD are mostly, but not overwhelmingly predicted as pathogenic (58% of 2,555 DM variants for which predictions could be made). Polymorphisms are mostly, but not overwhelmingly predicted as benign (66% of 360 disease-associated polymorphisms with additional supporting evidence, 902 disease-associated polymorphisms, and 603 in vitro/in vivo functionally validated polymorphisms, for which PolyPhen 2 predictions could be made). The number of pathogenic predictions decreases with increasing MAF, so that a variant with MAF > 0.3 is far more likely to be predicted benign than pathogenic (84% of 357 variants observed with MAF > 0.3 for which PolyPhen 2 predictions could be made).
Discussion
In this study, we demonstrate that a large number of published disease-associated variants from HGMD are so common that they are likely to have limited predictive value for asymptomatic individuals. When one of these common variants is encountered in an asymptomatic individual, the significance is unclear when the prior probability of disease is low and there is no other confirmatory evidence such as familial segregation, validation studies, or case-control population data (Kohane, et al., 2006). We believe these findings are an important warning to those who use published disease-associated variants in the clinical interpretation of whole exome and genome sequence data.
While there is a substantial percentage (approximately 10.6%) of previously identified variants that are of sufficient clinical relevance and scientific validity to share with research participants (Cassa, et al., 2012), this study demonstrates that a similar percentage (8.5%) of these disease-associated variants are present in completely healthy individuals. We observe that 4.6% of these disease-associated variants are present with sufficient frequency (MAF >0.01), that they are unlikely to be highly penetrant Mendelian disease variants. Further, 40% of low frequency HGMD variants are predicted as benign by PolyPhen 2, suggesting that even many of these rare missense variants are not necessarily pathogenic.
We do not intend for this report to be a criticism of HGMD, but a commentary on the current state of the application of this knowledge base in WGS interpretation and personalized medicine. HGMD acknowledges that a substantial number of variants in the database are polymorphic and are included because of their association with disease (Stenson, et al., 2012). We also identified many variants that are classified as polymorphic, but that do not appear frequently in the asymptomatic population, and many others that are classified as disease causing mutations (DM) that are quite common. HGMD recently introduced a new category of mutation, “DM?” which updates a previously updated DM variant where the author of the report has indicated that there may be some degree of doubt, or subsequent evidence in the literature calls the deleterious nature of the variant into question (Biobase/HGMD, 2013).
Current approaches to variant filtering focus on the exclusion of common variation (Biesecker, 2010) or inclusion of variants with deleterious effects predicted using evolutionary and functional considerations (Adzhubei, et al., 2013; Kumar, et al., 2009; Thompson, et al., 2013). But the variant filtering process comes with complexities; simple frequency-based filters do not exclude all common, benign variation, and they do not maintain all pathogenic variation. Specific counterexamples of moderate frequency variants with clinical importance include hereditary hemochromatosis, Factor V Leiden deficiency, and numerous pharmacogenomics associations (Klein, et al., 2001). This filtering may be additionally informed by the population prevalence of disease (Biesecker, 2010; Park, et al., 2009) as well as validated case-control population data (EBI, 2012; NCBI, 2012a; NCBI, 2012b; NHLBI, 2012), and in silico predictive algorithms that assess variant pathogenicity using functional and evolutionary significance (Adzhubei, et al., 2013; Kumar, et al., 2009).
There are several limitations to this study. The variants studied only represent a subset of the total knowledge base, although this sample represents an easily accessible subset of variants with chromosome coordinate data that will likely be evaluated in most WGS pipelines. While these variants have been previously associated with disease in the scientific literature, they largely have been derived from small disease cohorts with limited control populations such that a reassessment of the evidence for pathogenicity is required.
We have likely produced an underestimate for the number of HGMD variants that are present in asymptomatic individuals generally. In this study, we have estimated the minor allele frequency values for disease-associated variants using the maximum likelihood estimate from low coverage data (2010). We expect that there is a great deal of rare variation that was not observed given the number of individuals and low coverage (Keinan and Clark, 2012; Nelson, et al., 2012). This means that there is likely additional rare variation that makes our present estimates an underestimate. In this preliminary study we did not make important exclusions for bias, such as for the sampling frequency of variants that cause early onset diseases.
Interpreted whole genome sequences are now available as clinically certified laboratory tests, making this data available for physicians in the clinical care of patients. Central to the clinical interpretation of these variants is the development of a standardized methodology to predict the pathogenicity of these variants, and to prioritize those that require expert review. Even if a patient is receiving an interpretation for one disease, the results will undoubtedly uncover risk variants for other diseases for which a patient has no strong prior probability, and is essentially “asymptomatic”. Furthermore, even variants with clear evidence for pathogenicity in a disease cohort may be incompletely penetrant, making an initial assessment of risk to a healthy individual difficult. This makes it essential to understand the limitations of the current knowledge base of genomic variants (Roberts, et al., 2012), as well as the clinical value to be derived from WGS for many rare Mendelian disorders (Kohane and Shendure, 2012).
Conclusion
Our findings demonstrate the limitations of using pathogenic variant databases in the WGS interpretation of asymptomatic individuals. These findings have substantial implications for those that leverage these genomic knowledge bases for use in clinical sequence interpretation. Microarrays and targeted sequencing are already used diagnostically, and it is anticipated that whole-genome sequencing will be integrated into clinical care. Issues to address in future research include decision support systems for prioritizing large numbers of identified variants, in conjunction with family history and/or clinical presentation.
Acknowledgments
Grant Numbers: This research was supported by NHGRI grant HG007229 (Dr. Cassa), and by NIGMS grant GM078598 (Daniel Jordan).
References
- Knome, Inc. Know thyself. Personal Human Genome Sequencing.
- A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013. Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed]
- Biesecker LG. Exome sequencing makes medical genomics a reality. Nat Genet. 2010;42(1):13–4. doi: 10.1038/ng0110-13. [DOI] [PubMed] [Google Scholar]
- Biobase/HGMD What's New at HGMD. 2013.
- Brunham LR, Hayden MR. Medicine. Whole-genome sequencing: the new standard of care? Science. 2012;336(6085):1112–3. doi: 10.1126/science.1220967. [DOI] [PubMed] [Google Scholar]
- Cassa CA, Savage SK, Taylor PL, Green RC, McGuire AL, Mandl KD. Disclosing pathogenic genetic variants to research participants: Quantifying an emerging ethical responsibility. Genome Res. 2012 doi: 10.1101/gr.127845.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GP. 1000 Genomes Project FTP Server. p 1000 Genomes Phase 1, Version 3, release date 4/30/12. 2012.
- EBI The European Genome-phenome Archive. 2012.
- Fabsitz RR, McGuire A, Sharp RR, Puggal M, Beskow LM, Biesecker LG, Bookman E, Burke W, Burchard EG, Church G. Ethical and practical guidelines for reporting genetic research results to study participants: updated guidelines from a National Heart, Lung, and Blood Institute working group. Circ Cardiovasc Genet. 2010;3(6):574–80. doi: 10.1161/CIRCGENETICS.110.958827. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- genomeweb Baylor Whole Genome Laboratory Launches Clinical Exome Sequencing Test. 2011a.
- genomeweb Partners HealthCare Center's LMM to Introduce Clinical Whole-Genome Sequencing Interpretation Service in 2012. 2011b.
- Homer N, Szelinger S, Redman M, Duggan D, Tembe W, Muehling J, Pearson JV, Stephan DA, Nelson SF, Craig DW. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet. 2008;4(8):e1000167. doi: 10.1371/journal.pgen.1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, Clark AG. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336(6082):740–3. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TE, Chang JT, Cho MK, Easton KL, Fergerson R, Hewett M, Lin Z, Liu Y, Liu S, Oliver DE. Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. Pharmacogenomics J. 2001;1(3):167–70. doi: 10.1038/sj.tpj.6500035. others. [DOI] [PubMed] [Google Scholar]
- Kohane IS, Hsing M, Kong SW. Taxonomizing, sizing, and overcoming the incidentalome. Genet Med. 2012 doi: 10.1038/gim.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohane IS, Masys DR, Altman RB. The incidentalome: a threat to genomic medicine. JAMA. 2006;296(2):212–5. doi: 10.1001/jama.296.2.212. [DOI] [PubMed] [Google Scholar]
- Kohane IS, Shendure J. What's a Genome Worth? Sci Transl Med. 2012;4(133):133fs13. doi: 10.1126/scitranslmed.3004208. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- McKusick-Nathans Institute of Genetic Medicine JHUB, MD Online Mendelian Inheritance in Man, OMIM®.
- NCBI ClinVar. 2012a.
- NCBI database of Genotypes and Phenotypes (dbGaP) 2012b. [DOI] [PMC free article] [PubMed]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337(6090):100–4. doi: 10.1126/science.1217876. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHGRI genome.gov, editor. Genome-Wide Association Studies.
- NHLBI NHLBI GO Exome Sequencing Project. 2012.
- Park J, Lee DS, Christakis NA, Barabasi AL. The impact of cellular networks on disease comorbidity. Mol Syst Biol. 2009;5:262. doi: 10.1038/msb.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Review T. Making Genome Sequencing Part of Clinical Care. 2011.
- Roberts NJ, Vogelstein JT, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. The predictive capacity of personal genome sequencing. Sci Transl Med. 2012;4(133):133ra58. doi: 10.1126/scitranslmed.3003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4(2):69–72. doi: 10.1186/1479-7364-4-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shaw K, Cooper DN. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinformatics. 2012. Chapter 1:Unit1 13. [DOI] [PubMed]
- Thompson BA, Greenblatt MS, Vallee MP, Herkert JC, Tessereau C, Young EL, Adzhubey IA, Li B, Bell R, Feng B. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2013;34(1):255–65. doi: 10.1002/humu.22214. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong MY, Cassa CA, Kohane IS. Automated validation of genetic variants from large databases: ensuring that variant references refer to the same genomic locations. Bioinformatics. 2011;27(6):891–3. doi: 10.1093/bioinformatics/btr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M, den Dunnen JT, Dalgleish R, Cotton RG. Guidelines for establishing locus specific databases. Hum Mutat. 2012;33(2):298–305. doi: 10.1002/humu.21646. [DOI] [PubMed] [Google Scholar]
- Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A navigator for human genome epidemiology. Nat Genet. 2008;40(2):124–5. doi: 10.1038/ng0208-124. [DOI] [PubMed] [Google Scholar]