

Abstract
Immediate Early Genes (IEGs) are expressed upon re-entry of quiescent cells into the cell cycle following serum stimulation. These genes are involved in growth control and differentiation and hence their expression is tightly controlled. Many IEGs are regulated through Serum Response Elements (SREs) in their promoters, which bind Serum Response Factor (SRF). However, many other IEGs do not have SREs in their promoters and their serum regulation is poorly understood. We have identified SRF-independent IEGs in SRF-depleted fibroblasts. One of these, Id1, was examined more closely. We mapped a serum responsive element in the Id1 promoter and find that it is identical to a BMP Responsive Element (BRE). The Id1 BRE is necessary and sufficient for the serum regulation of Id1. Inhibition of the BMP pathway by siRNA depletion of Smad 4, treatment with the BMP antagonist noggin, or the BMP receptor inhibitor dorsomorphin blocked serum induction of Id1. Further, BMP2 is sufficient to induce Id1 expression. Given reports that SRC inhibitors can block Id1 expression, we tested the SRC inhibitor, AZD0530, and found that it inhibits the serum activation of Id1. Surprisingly, this inhibition is independent of SRC or its family members. Rather, we show that AZD0530 directly inhibits the BMP type I receptors. Serum induction of the Id1 related gene Id3 also required the BMP pathway. Given these and other findings we conclude that the Id family of IEGs is regulated by BMPs in serum through similar BREs. This represents a second pathway for serum regulation of IEGs.
Keywords: Immediate Early Gene; BMP Response Element; Id1, Inhibitor of DNA binding/differentiation 1; Serum Response Element; AZD0530, Saracatinib
Introduction
When growth factors are removed from NIH3T3 mouse fibroblasts they become quiescent. Upon re-stimulation with serum or specific growth factors, these cells re-enter the cell cycle. A class of genes, Immediate Early Genes (IEGs), is expressed rapidly and transiently upon treatment with serum or growth factors [1–5]. The expression of these genes is independent of new protein synthesis, suggesting that is it is a relatively direct response to serum induction. IEGs are involved in a plethora of processes including growth [6], differentiation, lineage determination, learning and memory [7]. c-fos, c-jun and c-myc are among the earliest and most well studied IEGs. The regulation of these IEGs is tightly controlled and misregulation can lead to many diseases including cancer, neurological disorders [8], bone remodeling disorders [9], and other chronic diseases. As reviewed in Dunn et al. [10], IEG expression in many cancers is found to be sustained and abnormally high. Understanding the transcriptional regulation of IEGs is an important step in understanding how their deregulation results in disease and finding better therapies to counter them.
Hundreds of experimentally validated or hypothesized genes have a Serum Response Element (SRE) or CArG box in their promoter region [11–14]. The MADS box family member, Serum Response Factor (SRF), binds to the SREs of these genes [8, 15–17]. SRF is constitutively present at the promoters of the genes it regulates [18]. When quiescent cells are stimulated with the growth factors in serum, two pathways sufficient for SRE activation are activated, the mitogen activated protein kinase (MAPK; ERK1/2) and the RhoA GTPase pathways. The MAPK pathway, through a cascade of factors, leads to the phosphorylation and activation of SRF co-factors, the ternary complex factors (TCFs) Elk1, Sap1 and Net [19, 20]. SRF is also activated by the small GTPase RhoA via another group of SRF co-transcriptional activators, the myocardin related factors, Megakaryoblastic Leukemia 1/2 (MKL1/2) [21–24]. RhoA activation leads to changes in the actin cytoskeleton, which directly results in changes in the nuclear localization and activation of MKL1/2 and therefore activation of SRF target gene expression [25–29].
Some IEGs do not have clear SREs in their promoters. Their induction may be due to cryptic or distant SREs or entirely different pathways. As described here, some IEGs do not require SRF for their serum induction. It would be interesting to find out whether there is another common sequence element or pathway through which these SRE-lacking, SRF-independent IEGs are regulated. Inhibitor of DNA binding/differentiation 1 (Id1) is a member of this group.
Id1 is a member of the Helix Loop Helix (HLH) family of transcription factors [30, 31], which form heterodimers with other members of the HLH family. The Id1 protein lacks a basic DNA-binding domain but is still able to form heterodimers with other HLH proteins that contain basic domains (bHLH proteins) [32]. These heterodimers are unable to bind DNA, thereby inhibiting the transcriptional activity of the bHLH proteins.
Id1 is ubiquitously expressed [33] and is regulated by the TGF-β super-family of transcription factors. Id1 expression is increased by prolonged exposure to TGF-β1 in human epithelial cells [34]. Smad3 and ATF binding elements in the Id1 promoter mediate this regulation. Id1 is also activated by TGF-β1 in the human mammary gland cell line, MCF10A [35]. Smad3 also mediates this regulation. Several groups have shown that Id1 expression is also increased in response to BMP signaling [36–39]. Sequences in the Id1 promoter responsible for BMP activation were mapped to two close but distinct regions [13, 40, 41]. Subsequently, common BMP responsive sequences were found for the Id family of genes in Xenopus (TGGCGCCAG-N3-GTCTG) and these elements were conserved in mammals [42]. The element mutated by Korchynskyi et al. partially matches this consensus [13]. We refer to this sequence at −1067 to −1050 in the mouse Id1 promoter as the BMP responsive element (BRE). Overall expression of Id1 was shown to be regulated by an Egr-1 binding site upstream of the BRE [43]. Expression of Id1 in cells grown continually in serum-containing media vs. low serum media was reduced by mutations (m16 and m17) in the BRE region, however it was not clear what factors or pathways activated through this element [43]. It was also not clear whether rapid serum induction acted through this region.
BMP is a member of the TGFβ family of transcription factors. BMPs bind to transmembrane type I and type II receptors [44]. These receptors encode serine/threonine kinases. The activated type 1 receptor phosphorylates the receptor-regulated R-Smads, Smad1, Smad5 and Smad8 in the cytoplasm. These phospho-R-Smads then complex with the common Smad, Smad4. This R-Smad/Smad 4 complex moves to the nucleus where it binds to regulatory regions of target genes [45]. Here we show that a previously identified element [13, 43] in the Id1 promoter is a BMP responsive element and is also necessary and sufficient for serum regulation of the Id1 promoter in NIH3T3 cells.
Id genes are required for G1 progression and regulate cellular senescence [46]. Deregulation of Id1 expression is observed in many kinds of cancer including ovarian [47, 48], colon [49], breast [50, 51], and thyroid [52, 53] cancers. Id1 promotes migration and proliferation of cancer cells in vitro [54, 55], confers angiogenic properties on fully differentiated endothelial cells, contributes to therapeutic angiogenesis [56] and is required for angiogenesis [57], neurogenesis and vascularization of tumor xenographs [58]. Further, it has been demonstrated that Id1 promotes lung cancer growth in a BMP2, Smad1/5 dependent manner [59]. Id1 has been proposed as a molecular target in breast and ovarian cancers [60, 61]. Here we demonstrate that the src kinase inhibitor, saracatinib (AZD0530), directly inhibits the BMP type 1 receptors and is able to reduce the phosphorylation of Smad1/5/8 and the expression of Id1 in the colon cancer cell line HCT116. Therefore, this and other drugs that inhibit the BMP receptors may have therapeutic potential in cancers which have perturbed BMP signaling and increased Id1 expression.
Materials and Methods
Cell culture
SRF shRNA and control cell lines were generated by stable infection of NIH3T3 with lentiviral vectors (pLKO.1) containing short hairpin RNA (shRNA) directed to SRF; shSRF-1, 5′-GCCAGCAUUCACAGUCACCAAC- 3' and shSRF-2, 5'–GAUGGAGUUCAUCGACAACAA -3'; a non-targeting shRNA was used to generate the control cell line. The lentiviruses were made in Phoenix-ECO cells (ATCC CRL3214). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% New Born Calf serum (NCS) and 10 μg/ml puromycin (Invivogen). NIH3T3 cells were grown in 5% CO2 in DMEM supplemented with 10% NCS. HCT116 and SYF cells were grown in 5% CO2 in DMEM supplemented with 10% Fetal Bovine Serum (FBS).
Luciferase assays
NIH3T3 (4 × 104 cells/well) cells were plated on 24 well plates. The following day the media was changed to fresh 10% NCS in DMEM. The cells were transfected with Polyjet reagent (Signagen), 100 ng of pRLSV40P (SV40 promoter driving the Renilla luciferase gene) and 100 ng of pGL3 Id1 or c-fos reporter genes. For transfection with constitutively active BMP receptor genes, caALKs, 25 ng of each ALK was also transfected. The next day the media was changed to DMEM containing 0.2 % NCS (starvation media). For serum/BMP2 induction, the next day the cells were induced with 20% NCS or 20 ng/ml recombinant human BMP2 (Sino Biological Inc.) for 4 hours. The cells were harvested and luciferase activity assayed utilizing the Dual-Luciferase Reporter System (Promega). The Renilla luciferase activity was measured as an internal control and firefly luciferase values were normalized to the corresponding Renilla luciferase levels. For AZD0530 treatment, the cells were treated with 100 nM AZD0530 (Selleck Chemicals) for 1 hour before the addition of serum. For noggin, 10 ng/ml noggin (STEMGENT) was added overnight while the cells were being starved in 0.2% NCS.
siRNA treatment
siRNA duplexes for mSmad4 were synthesized by Integrated DNA Technologies. Two duplexes were used to deplete Smad4. Smad4-1 contains forward, 5' - GCAAUUGAGAGUUUGGUAAAGAAGC-3' and reverse 3' – CUCGUUAACUCUCAAACCAUUUCUUCG - 5'. Smad4-2 forward 5'- CAAAUACACCAACAAGUAACGAUGC and reverse 3' – UUGUUUAUGUGGUUGUUCCAUUGCUACG – 3'. NIH3T3 cells (2 × 104 cells/well) were plated on 6 well plates. The next day, fresh media was added to each well. The cells were transfected with 50 pmoles siRNA using 3 μl Powerfect (Signagen) as per the company's protocol. The next day the media was changed to 0.2% NCS in DMEM. The cells were harvested in Tri Reagent (Molecular Research Center, Inc.) and mRNA levels determined as described below.
Plasmids
The Id1 promoter fragments were cloned into the pGL3-basic plasmid utilizing restriction enzymes. Mouse Id1 −1577 to +54 and −1050 to +54 were amplified from mouse genomic DNA (Bioline). All other deletion mutants were made utilizing Id1 −1577 to +54 as a template. Point mutations were made utilizing the QuikChange Multi Site-Directed Mutagenesis Kit from Agilent in the mId1 −1150 background. The c-fos WT and pFos-pm18 luciferase constructs were as previously described [62]. The expression vector for constitutively active ALK1 pcDNA-ALK1 was a gift from Dr. Kristina Bostrom [63]. Expression vectors for constitutively active HA-ALK 2–5, 7 were gifts from Dr. Peter Ten Dijke [13]. The expression vector for C3 transferase was as described [62].
Gene expression
mRNA expression levels were assayed by plating NIH3T3 cells on 6 well plates; the next day the media was changed to 0.2% serum. The following day serum/BMP2 was added for the time indicated to a final concentration of 20% and 20 ng/ml, respectively. For LY294002, PD0325901, AZD0530 and Dorsomorphin treatment, the drugs were added one hour before addition of serum. 10 μm LY294002 (ChemieTek), 5 μm PD0325901 (ChemieTek), 100 nM AZD0530 (Selleck Chemicals), 1 μm Dorsomorphin (Chemdea), or 50 μg/ml cycloheximide (Sigma) were used. RNA was isolated using Tri Reagent (Molecular Research Center, Inc.) following the manufacturer's instructions. cDNA was made from 1 μg total RNA using ImProm-II reverse transcriptase (Promega) and random hexamer primers. Individual gene expression was quantified with sybr green present in the qPCR master reaction mix (Thermo Scientific) and the Step One Plus machine (Applied Biosystems) was used for real time PCR quantification of gene expression. 18s rRNA expression was measured for normalization of all samples. Supplemental table S2 shows the sequences of all the gene primers.
Immunoblotting
Cells grown as described in preceding methods were rinsed twice in ice cold Phosphate Buffered Saline (PBS) and lysed in 1× passive lysis buffer from Promega. Lysates were kept on ice for 20 minutes, then centrifuged at 11,000 × g for 20 minutes at 4°C. The samples were diluted with one half volume 3X protein sample buffer (188 mM Tris-Cl (pH 6.8), 3% SDS, 30% glycerol, 0.01% bromophenol blue, 15% β-mercaptoethanol) and boiled for 5 minutes. Proteins were separated by SDS-PAGE on 10% or 12% polyacrylamide gels. The proteins were transferred to a nitrocellulose membrane and the membrane was blocked in 6% nonfat dry milk for 1 hour, washed 3X in 1X Tris Buffered Saline (TBS) and incubated with primary antibody (1:1000) in TBS overnight at 4°C while shaking. The next day the membrane was washed 3X with TBS-tween (TBS, 0.1% tween-20). The membrane was then incubated with fluorescently labeled secondary antibody (1:10,000) (IRDye goat anti rabbit 800 or goat anti rabbit 680 or goat anti mouse 680; LI-COR Biosciences) for one hour at room temperature, washed twice with TBS-tween and once with TBS. The proteins were visualized using the Li-Cor Odyssey Infrared imaging system. Primary antibodies against the proteins were as follows: Phospho-src Family (Tyr416) Antibody, rabbit, Cell Signaling; Total ERK (p44/42(ERK1/2) antibody, rabbit, Cell Signaling), Actin (1–19) Antibody, rabbit, Santa Cruz Biotechnology; SRF, rabbit, described in [64]; Phospho-Smad1/5/8 (S463/S465/Smad8(S426/S428)) Antibody, rabbit, Cell Signaling; Smad1/5/8 Antibody, (N-18)-R, sc-6031-R, rabbit, Santa Cruz Biotechnology; Phospho (ERK) p44/42 MAP Kinase, rabbit, Cell Signaling; Phospho AKT (Thr 308) Antibody, rabbit, Cell Signaling.
In vitro kinase reactions
The in vitro kinase reasction were performed by the Reaction Biology Corporation using the “HotSpot” assay platform. Briefly, purified recombinant kinase/substrate pairs were incubated with the indicated concentrations of AZD0530 (Selleck Chemicals) or Staurosporine and with a mixture of ATP (Sigma) and 33P-ATP to a final concentration of 10 μM in reaction buffer (20 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO) for 120 minutes at 25 °C. The reactions were spotted onto P81 ion exchange filter paper (Whatman) and free phosphate was removed by washing of filters in 0.75% phosphoric acid. The kinase activity data were expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using Prism (GraphPad Software).
Results
Serum induction of Id1 is independent of SRF
In order to identify IEGs whose expression is independent of SRF we made stable cell lines expressing shRNA lentiviral vectors that target SRF in NIH3T3 mouse fibroblasts. The expression of SRF was significantly decreased in two clones (shSRF-1 and shSRF-2), decreasing SRF mRNA expression by at least 70% at all time points of serum induction (Fig. 1B). The decrease in expression was stronger at the protein level (Fig. 1A) with stronger reduction of SRF in shSRF-2. NIH3T3 cells containing shRNA that does not target any known mouse gene was used as the control cell line. We analyzed the serum regulation of many IEGs in NIH3T3 cells induced with serum for 0 to 2 hours following overnight serum starvation. The transcript levels of 19 IEGs that were previously identified in NIH3T3 cells [14] were measured by quantitative RT-PCR (qPCR). Not surprisingly, the serum regulation of many IEGs such as CTGF, egr-2, and nur77 were SRF dependant while others such as Id1, Id3 and mig6 were SRF independent (Fig. 1B and supplemental figure 1). Surprisingly, the expression of c-fos, the longest studied SRF target gene, was only slightly affected. This may be because residual SRF was sufficient to facilitate c-fos expression or that there is another mechanism by which c-fos can be serum regulated (see Discussion). cyr61 induction also appeared to be SRF-independent, despite previous mapping of SREs in its promoter [65]. As with c-fos, it may only require low levels of SRF or use a different mechanism. Other genes with previously reported SREs, such as vcl, nur77, and egr2 were in-fact SRF dependant (Figs. 1 and S1 and supplemental table 1.)
Figure 1. Effect of SRF depletion on the serum regulation of immediate early genes.

A) SRF levels in NIH3T3 cells stably expressing control shRNA or shRNA targeting SRF were measured by immunoblotting with anti-SRF sera. Anti-ERK1/2 antibodies were used as a loading control. B) Control and SRF shRNA cells were starved in 0.2% serum overnight and then serum induced with 20% NCS for the indicated times. The levels of the indicated genes were measured using quantitative real time-PCR (qPCR) with 18s rRNA levels used to normalize the samples. The values are the means of three experiments +/− standard deviations.
While, the level of SRF expression is significantly reduced by use of shRNAs targeting SRF, the residual SRF mRNA is still serum inducible (Fig. 1B). This is not surprising as SRF itself is an IEG. In SRF depleted cells, however, many of the IEGs analyzed appeared SRF-independent. (Fig. 1, Fig. S1 and supplemental table 1). Of these, the serum induction of Id1 actually increased upon depletion of SRF at the 30 and 60 minute time points (Fig. 1B). This and the lack of putative SREs in the Id1 gene suggest that Id1 induction is SRF-independent. Consequently, we proceeded to identify sequence elements and factors responsible for the serum regulation of the Id1 IEG.
Serum induction of Id1 does not require the PI3K or the MAPK pathway
SRF target genes are regulated through the RhoA or MAPK pathways. We studied whether these or other known pathways are involved in serum regulation of Id1 by utilizing pathway inhibitors. The serum induction of Id1 was assayed in the presence and absence of the phosphoinositide-3-kinase (PI3K) inhibitor LY294002 and the MAPK pathway MEK1 inhibitor PD0325901. NIH3T3 cells were pretreated for 1 hour with either LY294002 or PD0325901 followed by serum induction for 0 to 120 minutes. The potency of the drugs was confirmed by immunoblotting for phospho-AKT and phospho-ERK1/2, respectively (Fig. 2). The RNA levels of Id1 were unchanged when treated with the inhibitors (Figs. 2A and B). The need for ERK1/2 in c-fos serum regulation is well documented, at least partially due to the phosphorylation of the ELK1 family members [5]. Not surprising, c-fos expression was dramatically decreased when NIH3T3 cells were treated with PD0325901, while treating the cells with LY294002 had little effect on c-fos expression (Fig. 2). To study the RhoA pathway we utilized the RhoA inhibitor, C3 transferase [66], using the Id1 reporter gene as described below. There was only a small effect of C3 transferase on Id1 promoter activation by serum while c-fos promoter activation was inhibited strongly in the presence of the C3 transferase expression vector (supplementary Figure S2). The c-fos report used (pFos-pm18) lacks the TCF (ternary complex factor; Elk1 family) site such that it is activated by the RhoA-MKL pathway [22]. Its inhibition by C3 transferase shows that the RhoA pathway is effectively inhibited. These results suggest that Id1 serum regulation does not require the ERK1/2, RhoA or PI3K pathways.
Figure 2. Inhibition of MAPK and PI3K signaling does not affect Id1 induction.

A) NIH3T3 cells were grown overnight in 0.2% NCS, treated with the PI3K inhibitor LY294002 (10 μM) for 1 hour and then for 1 hour or the indicated times with 20% serum. Gene expression was analyzed by qPCR, as in figure 1. Phospho-ERK1/2 and phopho-AKT levels were measured by immunoblotting with phospho-specific antibodies. B) Cells were treated for 1 hour with the MEK1 inhibitor, PD0325901, (5 μM for the RNA experiments and 1 or 5 μM, as indicated, for the immunoblots). The cells were then serum-stimulated, as described above, and measured by qPCR for mRNA expression and immunoblotting for ERK1/2 phosphorylation. Actin levels were measured by immunoblotting as a loading control.
Mapping of sequence elements responsible for serum induction of Id1
In order to decipher the factor(s) and pathway(s) involved in the serum regulation of Id1, promoter mapping of the mouse Id1 promoter was undertaken in NIH3T3 cells. We found that a construct containing −1577 to +54 of the mouse Id1 gene was sufficient to mediate nearly a 5 fold serum-induced increase in luciferase expression (Fig. 3A, B). A series of deletion constructs were made between −1577 and −1050 of the mId1 promoter (Fig. 3A). This allowed us to map a region required for serum activation to a 100 bp region between −1150 and −1050 (Fig. 3B). This region of the promoter is particularly conserved as shown by the alignment of the mouse, human and chicken Id1 promoter sequences (Fig. 3C). Critical elements have previously been identified in this region for expression in cells grown in serum containing medium compared to cells starved for serum [43]. These and other studies identified binding sites for YY1, Egr1, CREB/ATF and a Smad binding element (SBE). In addition, systematic point mutation identified the M8 and M16/M17 regions as being required for expression [43]. In order to identify the sequence element(s) important for serum regulation of the Id1 promoter, a series of mutations was made in this 100 bp region of Id1 (Fig. 4A, B). Mutation of most of the known sites had some effect, but also retained some serum induction (Fig. 4D). The most striking difference was observed with mutation of the M16/M17 region (mBRE; Fig. 4D). This region was previously identified as a Smad binding element [13] and is similar to regions in the Id2 and Id3 promoters required for BMP induction [67, 68] (Fig. 4C). We will therefore refer to it as a BMP Response Element (BRE). We made finer mutations across the BRE and found that each reduced expression, except for mutations near the 3' end of the BRE (mBRE-R-b; Fig. 4B and E). These mutations are in the spacer region of the consensus sequence (Fig. 4C). Other mutations in the element (BRE-L and BRE-R) showed that the consensus region is needed for optimal Id1 serum induction (Fig. 4E).
Figure 3. Mapping the Serum regulation element of the mId1 promoter.

A) Map of mouse Id1 promoter deletion mutants. B) NIH3T3 cells were transfected with variants of the Id1 promoter along with pRLSV40P as an internal control. The next day transfected cells were starved in 0.2% serum overnight and then induced with 20% serum for four hours. Firefly luciferase levels were normalized to the Renilla pRLSV40P levels and are the means of three experiments +/− the standard deviation. *** indicates a p-value ≤ 0.05. C) Conservation of sequence elements in the conserved 100 bp Id1 promoter region −1150 to −1050. m, mouse; h, human; c, chicken. M8 and M16/M17 indicated mutated regulatory elements from reference [43].
Figure 4. Mapping the serum responsive element of the mId1 promoter.

A) Sequence of the conserved regulatory region of mId1 with the indicated putative regulatory elements. B) Left (L), mutations in the BRE region and Right (R), mutations in the BRE region. C) Alignment of the BMP responsive elements (BRE) in the mouse Id1, Id2 and Id3 promoters. D, E) The indicated mutants were transfected into NIH3T3 cells and assayed for luciferase activities as in figure 3.
The conserved region of −1150 to −1050 was sufficient to mediate serum induction when placed upstream of an SV40 promoter (Fig. 5B). However, a single copy of the BRE was not sufficient, as a −1177 to −1050 construct (that lacks the YY1-Egr1-GC region) did not mediate serum induction. When four copies of the BRE were placed in front of the SV40 promoter, there was basal expression but it was not inducible by serum (Fig. 5). While the SBE region was also not sufficient, the combination of the BRE and SBE was sufficient for serum induction. This is not surprising as the BRE-SBE combination is conserved in the Id1, 2 and 3 genes (Fig. 4C) and the SBE sequence matches the sequence which is thought to be important for the binding of the co-Smad, Smad4 [69]. It is likely that while the R-Smads bind the BRE, the SBE is bound by co-Smad4. Our efforts to show specific Smad binding in this region of DNA were unsuccessful. We and others have seen weak specific binding of a factor in the BRE – SBE region by electrophoretic mobility shift assays (EMSA) (unpublished results and [43]), but we were unable to successfully identify the factor(s) which, bind this region. Nevertheless, given previous analysis of Smad binding to BRE sequences, it is likely that Smad4 binds with Smad1, 5 or 8 [67, 69].
Figure 5. The BRE-SBE elements are sufficient for serum induction.

A) Four copies of the indicated elements of the mId1 promoter were cloned upstream of the SV40 promoter in the pGL3 luciferase reporter. B) These constructs were co-transfected with pRLS40P into NIH 3T3 cells, serum-induced and assayed as in figure 3.
It has been reported that bovine serum contains a BMP-like factor [70]. This and the requirement of the BRE sequence suggest that the BMP-Smad pathway is mediating serum induction of Id1 expression. To further test this model, we utilized purified BMP2 and known inhibitors of the BMP pathway. BMP2 induced Id1 expression in NIH3T3 cells about twice as strongly as serum (Fig. 6A), confirming that the BMP pathway is sufficient for induction of Id1 and that there are receptors for BMP2 on NIH3T3 cells. BMP2 was also able to activate the Id1 promoter similar to serum in a reporter assay (Fig. 6B). We also found that BMP2 induced Id1 independently of new protein synthesis, consistent with its identification as an immediate early gene (supplemental Fig. S3). In fact induction in the presence of the protein synthesis inhibitor cycloheximide was higher, similar to the result with c-fos induction where cycloheximide treatment had the effect of stabilizing c-fos mRNA [71, 72].
Figure 6. The BMP pathway is necessary and sufficient for serum induction of Id1.

A) NIH3T3 cells were serum-starved overnight and induced with 20% serum or 20 ng/ml BMP2 as indicated. Where indicated, 100 ng/ml noggin was added during serum-starvation. Gene expression was measured by qPCR as in figure 1. B) NIH3T3 cells were transfected with the mId1 −1150 and pRLSV40P luciferase reporter constructs, serum starved with or without noggin, and induced with serum or BMP2 as in A. Luciferase levels were measured as in figure 3. C) NIH3T3 cells were starved in 0.2% serum overnight then induced with 20% serum or 20 ng/ml BMP2 for 1 hour. The lysates were immunoblotted with phospho-Smad1/5/8 specific antibodies or anti-actin as a loading control. D) NIH3T3 cells were serum-starved, treated with Dorsomorphin at the indicated concentrations for 1 hour, and then induced with 20% NCS for 1 hour. Id1 mRNA levels were measured by qPCR as in figure 1. E) Cells were transfected with control or Smad4 siRNAs. The next day the media was changed to 0.2% serum overnight and the cells serum-induced for 1 hour. Smad4 and Id1 mRNA levels were measured by qPCR as in figure 1.
Not surprisingly, serum and BMP2 both induced the phosphorylation of Smad1/5/8 in starved NIH3T3 cells (Fig 6C). We also used the BMP antagonist noggin to study its effect on Id1 serum induction. Noggin-treatment of NIH3T3 cells abolished serum induction of Id1 (Fig. 6A). Noggin also abolished serum activation of the Id1 promoter in a reporter assay (Fig. 6B). This was confirmed by the use of the small molecule inhibitor, dorsomorphin, which is a known inhibitor of BMP receptors [73]. Dorsomorphin strongly inhibited induction of Id1 mRNA in response to serum treatment (Fig. 6D). Finally, we depleted NIH3T3 cells of the co-Smad regulator, Smad4, with siRNAs. Smad4 mRNA was reduced at least 80% by the siRNAs. This strongly impaired serum induction of Id1 mRNA (Fig. 6E). Therefore, these experiments show that the BMP pathway is indispensable for serum induction of Id1.
The src inhibitor AZD0530 inhibits Id1 expression
We were prompted to examine the possibility of the tyrosine kinase Src's involvement in Id1 regulation as the Src inhibitor AZD0530 (sarcatinib) was found to inhibit Id1 expression in the A549 lung carcinoma cell line, while the Src inhibitor PP2 inhibited Id1 expression in the MDA-MB-231 breast carcinoma line [38, 74]. In A549 cells, AZD0530 also prevented activation of Smad1/5 [38]. We investigated whether AZD0530 inhibits the serum induction of Id1. NIH3T3 cells pretreated with AZD0530 showed a complete loss of Id1 serum induction both in mRNA expression and promoter activation (Fig. 7A and B). Induction of both the longer −1150 Id1 reporter and the shorter 4X BRE-SBE reporter were blocked by AZD0530, while as a control, there was little effect on induction of the c-fos promoter (Fig. 7B). We tested for AZD0530 sensitivity on other IEG expression. Id3 was strongly reduced (Fig. 7A), consistent with it containing a BRE-like sequence [68]. There was no effect on c-fos induction (Fig. 7A). The IEG, plasminogen activator inhibitor-1 (PAI-1), on the other hand, showed a decrease in serum activation, suggesting that it is partially regulated by an AZD0530 sensitive pathway, similar to Id1.
Figure 7. src inhibitor AZD0530 inhibits serum induction of Id1.

A) NIH3T3 cells were starved in 0.2% serum overnight and AZD0530 (100 nM) was added to the cells for 1 hour. Cells were then induced with 20% serum for the indicated times. Gene expression of indicated genes was analyzed by qPCR as in figure 1. B) The indicated luciferase reporters (described in figures 3 and 5) as well as a c-fos promoter reporter were transfected into NIH3T3 cells as in figure 3. The cells were treated with AZD0530 (100 nM) for 1 hour and then with 20% NCS for 4 hours. Luciferase assays were as in figure 3. *** indicates a p-value ≤ 0.05.
Surprisingly, the serum-induced autophosphorylation of Src at Tyrosine 416 was unaffected at the concentration of AZD0530 that inhibited Id1 expression (Fig. 8A). We sought to further examine whether src family members are involved in Id1 serum regulation. Neither another Src inhibitor, PP2, nor the BCR-abl inhibitor, imatinib, had any effect on the response of the Id1 gene to serum (data not shown). In addition, we tested SYF cells, which are fibroblasts deficient in the expression of src family members src, yes, and fyn [75]. Serum induction of Id1 was normal in the SYF null cells and there was actually higher serum induction of Id3 in these cells (Figure 8B). src, yes and fyn are the three predominant Src family members expressed in fibroblasts, though it is possible that low expression of other family members could be involved in Id1 regulation. However, since Src phosphorylation was not affected by AZD0530, since loss of the three Src family members had no effect, and since other src inhibitors did not block Id1 expression, we considered the possibility that AZD0530 was inhibiting Id1 expression through another mechanism.
Figure 8. AZD0530 inhibits Smad phosphorylation in vivo.

A) NIH3T3 cells were serum-starved overnight and then treated with DMSO, 100 nM AZD0530 or 1μM Dorsomorphin for 1 hour followed by serum for hour. Serum-induced phosphorylation was measured by immunoblotting using phospho-specific antibodies for phospho-Smad1/5/8, Smad1/5/8, phospho ERK1/2, and phospho srcY416. Anti-actin antibodies were used as a loading control. B) SYF+/+ (wt fibroblasts) and SYF−/− cells (null for src, yes and fyn) were starved in 0.2% serum overnight and induced with 20% serum for 1 hour. Expression of Id1 and Id3 was measured by qPCR as in figure 1.
Since earlier studies have shown that Smad1/5/8 are involved in Id1 expression and that these Smads are necessary for BMP signaling [36, 38], we studied whether treating NIH3T3 cells with AZD0530 affects the phosphorylation status of these Smads. AZD0530 blocked the phosphorylation of phospho-Smad1/5/8 suggesting that it blocks a component upstream of Smad1/5/8 phosphorylation (Fig. 8A). The total level of Smad1/5/8 was not affected by treatment with AZD0530. As an additional control, there was no effect on phospho-ERK1/2 induction.
One possibility is that AZD0530 directly inhibits the activity of BMP receptors involved in BMP signaling. We tested this hypothesis by testing the inhibition of BMP receptor kinase activity by AZD0530 in vitro. The BMP family of receptors consists of two subunits. The type 2 receptor is common, BMPR-2, while there are seven type 1 receptors, termed ALK1 to 7 [44, 76, 77]. There was no inhibition of BMPR-2 kinase activity in vitro, however there was strong inhibition of ALK1, 2, and 3 with IC50 values in the 3–30 nM range (Fig. 9). Inhibition of ALK4, 5 and 6 was less sensitive, with IC50s in the 300–800 nM range. For comparison, inhibition by a non-specific protein kinase inhibitor, staurosporine, varied, but was closer to the micromolar range (Fig. 9B). ALK1, 2 and 3 are receptor subunits for the BMP family members BMP2, 4 and 6, which can each activate Id1 [78, 79].
Figure 9. AZD0530 inhibits BMP responsive ALKs in vitro.

A) In vitro kinase assays were performed with the indicated purified ALK protein kinases at 10 different AZD0530 concentrations in triplicate. The relative activities are shown. B) The IC50s of AZD0530 and a non-specific protein kinase inhibitor, staurosporine, are shown for each BMP receptor subunit. ND, not done. Alternative names for ALKs are shown.
We sought to confirm this inhibition of BMP receptors by AZD0530 in vivo using a reporter assay. We transfected constitutively active forms of each of the BMP receptors [13, 63] with the Id1 reporter gene. We found that the BMP responsive ALKs, ALK1, 2 and 3 activated the Id1 promoter in the absence of serum, while there was no activation by ALK4, 5 and 7 (Fig. 10). The activation by ALK1, 2 and 3 was significantly decreased in the presence of AZD0530. The limited inhibition compared to that with rapid serum induction may have to do with the long-term transfection of the constitutively activated receptors. It is also possible that AZD0530 is less effective at inhibiting the constitutively activated forms compared to the normal forms of these proteins.
Figure 10. AZD0530 inhibits the constitutive activation of the Id1 promoter by type 1 BMP receptors.
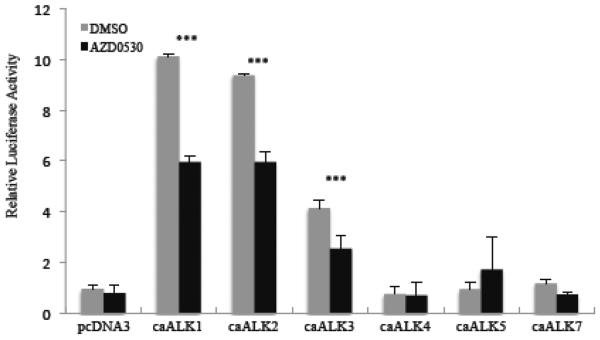
Constitutively active type 1 BMP receptors, ALK1 to 5 and ALK7 were transfected with the Id1 −1150 luciferase reporter gene along with pRLSV40P into NIH3T3 cells. The next day the media was changed to 0.2% serum containing AZD0530 (100 nM) or DMSO as a control and the cells were incubated overnight. Luciferase levels were measured as in figure 3. *** indicates a p-value ≤ 0.05.
Finally we sought to check the efficacy of AZD0530 in blocking Id1 expression in a colon cancer cell line, HCT116, which exhibits high Id1 expression. We found that Id1 expression and Smad1/5/8 phosphorylation were constitutively elevated in these cells (Fig. 11A and B). Upon starvation of HCT116 cells and serum stimulation, there was some further increase in phospho-Smad1/5/8 levels, but there was no further increase in Id1 expression, demonstrating that Id1 expression is misregulated in these cells. Treatment with AZD0530 or dorsomorphin resulted in the inhibition of phospho-Smad1/5/8 levels as well as inhibition of Id1 expression. These results show that AZD0530 can be used to reduce Id1 levels in cancer cells and suggest that altered Id1 expression is dependent upon the BMP pathway.
Figure 11. AZD0530 inhibits constitutive Id1 expression in HCT116 colon cancer cells.

HCT116 cells were serum starved in 0.2% serum overnight with DMSO, AZD0530 (100 nM) or Dorsomorphin (1 μm). These chemicals were added again the next day one hour before addition of 20% serum for one hour. A) Immunoblots for phospho-Smad 1/5/8 and actin control. B) Id1 mRNA levels were measured by qPCR as in figure 1. *** indicates a p-value ≤ 0.05.
Discussion
There are many IEGs that do not have clear SREs in their promoters. Whether there is a common pathway/DNA element by which some of these genes are regulated by serum is yet unknown. We sought to identify genes which do not contain SREs and whose serum regulation is independent of SRF. Many SRE containing genes were found to be SRF dependant. Unexpectedly c-fos, the canonical SRF regulated gene, was not affected by SRF depletion. This may indicate that the residual SRF is sufficient to allow c-fos induction. SRF binding to the c-fos SRE in the SRF depleted cells was reduced by 50–70% in chromatin immunoprecipitation assays such that this residual binding may be sufficient for induction (unpublished results). We also found that a c-fos reporter gene could still be induced by serum in the SRF depleted cells (unpublished results). As mutation of the SRE abolishes serum induction of reporter genes [80], this result also suggests that residual SRF in the cell is sufficient for induction of the c-fos reporter gene. It is possible that certain IEGs, such as c-fos, require less SRF due to the different affinities of SRF for specific SREs. Alternatively, there may be another pathway for c-fos induction in NIH3T3 cells. Loss of the SRF alleles in ES cells resulted in a strong reduction of serum-induced c-fos expression [81]. However, there was low residual serum induction in these cells, supporting the existence of an alternative pathway. Understanding whether there is a lower SRF threshold or an alternative pathway for c-fos regulation will be important.
We found that the serum regulation of Id1 and Id3, members of the HLH group of transcription factors, was independent of SRF. The serum induction of Id1 and Id3 both increased when SRF was depleted by shRNAs and was independent of the ERK1/2, RhoA and PI3K pathways. This suggests that there is a novel pathway through which these genes are regulated by serum. SRF and many IEGs are regulated by one or more of these pathways [5, 22]. In addition, there are no SRE sequences found in the Id1 promoter and no SRF binding was observed in genomic chromatin immunoprecipitation experiments [82, 83].
We subsequently identified a BRE in the promoter of Id1 as being responsible for its serum activation. Although this element in Id1 was identified previously [13], its link to serum activation of Id1 was unknown. A BRE is a GC rich sequence, i.e. GCCGNC or GRCGNC to which BMP responsive Smads bind [41, 84, 85]. It also pairs with a Smad4 binding element (SBE)(GTCT) with a spacer of five bases between the sites (Fig. 4C; [86]). These BMP responsive elements are found in BMP target genes such as Id1 [13], Id2 [67] and Id3 [68]. BREs have been identified across the phylogenetic spectrum, found for example in Drosophila, Xenopus and mammals [42, 87]. The demonstration of this element as a BRE clears up some confusion in the literature where elements at different positions in Id1 were referred to as BMP-responsive [40–42]. In many cases the Egr1 binding region upstream (see Figure 4) was reported to be BMP-responsive. Our evidence suggests that the Egr1 site functions with the BRE, but is not sufficient for the response. In addition, the element mutated by Tournay and Benezra (m16 and m17) [43] that affected Id1 reporter expression, corresponds to the BRE site discussed here.
We were able to show that the BRE-SBE element is necessary and sufficient for the serum activation of Id1. This BRE located at −1073 to −1056 in the mId1 promoter, is located within a highly conserved 100 bp region, −1150 to −1050. Although many other transcription factors bind to the Id1 promoter within this 100 bp region, mutation of the BRE had the greatest effect on serum activation of the Id1 promoter. Mutations in other sites suggest that the BRE works with other sequences to obtain optimal serum induction. There were partial effects on Id1 expression when most of these sequences were mutated. Considerable evidence supports the serum activation of Id1 through the BMP pathway and the BRE. The mId1 promoter was activated by BMP2, inhibited by the BMP antagonist noggin, and blocked by the BMP receptor inhibitor dorsomorphin. We show that the BMP receptor inhibitor dorsomorphin inhibits the phosphorylation of Smad1/5/8 in NIH3T3 cells while the total level of Smad1/5/8 was unchanged. Depleting the co-Smad, Smad4, also resulted in decreased activation of Id1. These results confirmed the involvement of Smads and BMP in serum induced Id1 expression. Recently, it was shown that BMP signaling is also required for serum induction of the Id2 gene [67]. It will be interesting to determine whether the BMP pathway also activates other IEGs. We found strong dependence with the Id3 gene and partial dependence with the PAI-1 gene, but no requirement with the other SRF-independent genes tested.
AZD0530 is a well-documented inhibitor of the Src family. As reported previously [38], AZD0530 caused a dramatic decrease in Id1 expression in several cancer cell lines. However, we found that AZD0530 under our conditions (a lower concentration and briefer incubation) did not block Src phosphorylation at a site thought to be due to autophosphorylation. In addition, serum induction of Id1 was unaffected in SYF cells, which lack the three most abundant Src family members, src, yes and fyn. On the other hand, we found that AZD0530 specifically blocked serum induced Id1 expression, Id1 reporter gene expression and Smad1/5/8 phosphorylation. In vitro kinase assays showed that AZD0530 directly inhibits the BMP type I receptors, ALK1, 2 and 3. We confirmed inhibition of these ALKs in vivo by AZD0530 using constitutively activated forms of the receptors with the Id1 reporter genes. Together these experiments suggest that AZD0530 blocks activation of the Id1 promoter by direct inhibition of the BMP receptor subunit rather than by inhibition of the Src family.
Increased Id family expression is a hallmark of many cancers [88–90] and Id1 overexpression and BMP constitutive activation [61] is associated with tumor angiogenesis in human pancreatic [91] and other cancers [48, 92, 93]. As we have shown that the small molecule AZD0530 inhibits the serum induction of Id1 at low concentrations through inhibition of the BMP receptors, AZD0530 may have therapeutic potential in targeting this pathway in addition to its effects on Src.
To check this hypothesis we looked in HCT116 colon cancer cells, which are known to have elevated levels of Id1 [94]. We found constitutive, serum-independent, expression of Id1 in these cells and high basal phospho-Smad1/5/8 levels. Treatment of these cells with AZD0530 or dorsomorphin blocked phosphorylation of Smad1/5/8 and expression of Id1. These results suggest that AZD0530 may be a promising drug for treatment of cancers in which Id1 is constitutively active due to deregulation of the BMP pathway. Our preliminary results indicate that AZD0530 did not reduce the proliferation of HCT116 cells. It is possible that these cells have other pathways activated such that they do not require Id1 or the BMP pathway. Furthermore, Id1 activation may be required for other cancer related properties such as cell migration or angiogenesis where Id1 has previously shown a role [54, 56, 57]. The role of the BMP pathway in cancer appears complicated as BMP4 can suppress cell growth in many systems, yet it can also induce migration, invasion and epithelial to mesenchymal transitions [95]. Therefore, it will be important to identify types of cancer where the BMP pathway or Id1 are activated and to demonstrate a beneficial effect of inhibiting these factors.
Finally, we propose a model of IEG activation in which members of the HLH family of transcription factors are activated by serum through the presence of BREs in their promoters. When quiescent cells are treated with serum, BMPs in serum bind to and activate the BMP receptors. This results in the phosphorylation of the BMP responsive R-Smads, Smad1/5/8, by the type 1 BMP receptors. The R-Smads then recruit co-Smad 4; they migrate to the nucleus, bind BREs in the Id promoter regions and activate transcription of these HLH family members.
Conclusion
The Id family of transcription factors is regulated by a novel pathway of serum induction involving BMP receptors and their downstream effectors, in particular Smads. This identifies a novel way by which SRF-independent IEGs are regulated. In addition, we found that AZD0530 is an inhibitor of BMP type 1 receptors. This drug, currently used as a Src inhibitor, may also be useful as a BMP pathway inhibitor, suggesting additional therapeutic uses.
Supplementary Material
Highlights
Many immediate early genes (IEG) are SRF-independent.
Id1 is an SRF-independent IEG.
Serum induction of Id1 is mediated by a BMP responsive element.
Induction of Id1 is dependent upon the BMP/Smad pathway.
The src inhibitor AZD0530 inhibits Id1 induction by direct inhibition of BMP receptor kinase activity.
Acknowledgements
This work was partially supported by a National Institute of Health Grant CA50329 to R.P. We thank Dr. Peter Ten Dijke and Dr. Kristina Bostrom for their gracious gift of plasmids. We also thank Dr. Mike Sheetz for providing SYF cells and Dr. Carol Prives for providing HCT116 cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Smith MR, Ryu SH, Suh PG, Rhee SG, Kung HF. S-phase induction and transformation of quiescent NIH 3T3 cells by microinjection of phospholipase C. Proc Natl Acad Sci U S A. 1989;86:3659–3663. doi: 10.1073/pnas.86.10.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Adolph S, Brusselbach S, Muller R. Inhibition of transcription blocks cell cycle progression of NIH3T3 fibroblasts specifically in G1. J Cell Sci. 1993;105(Pt 1):113–122. doi: 10.1242/jcs.105.1.113. [DOI] [PubMed] [Google Scholar]
- [3].Koskinen PJ, Sistonen L, Bravo R, Alitalo K. Immediate early gene responses of NIH 3T3 fibroblasts and NMuMG epithelial cells to TGF beta-1. Growth Factors. 1991;5:283–293. doi: 10.3109/08977199109000292. [DOI] [PubMed] [Google Scholar]
- [4].Cochran BH, Reffel AC, Stiles CD. Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell. 1983;33:939–947. doi: 10.1016/0092-8674(83)90037-5. [DOI] [PubMed] [Google Scholar]
- [5].O'Donnell A, Odrowaz Z, Sharrocks AD. Immediate-early gene activation by the MAPK pathways: what do and don't we know? Biochem Soc Trans. 2012;40:58–66. doi: 10.1042/BST20110636. [DOI] [PubMed] [Google Scholar]
- [6].Lau LF, Nathans D. Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci U S A. 1987;84:1182–1186. doi: 10.1073/pnas.84.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dragunow M. A role for immediate-early transcription factors in learning and memory. Behav Genet. 1996;26:293–299. doi: 10.1007/BF02359385. [DOI] [PubMed] [Google Scholar]
- [8].Shore P, Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- [9].Beedles KE, Sharpe PT, Wagner EF, Grigoriadis AE. A putative role for c-Fos in the pathophysiology of Paget's disease. J Bone Miner Res. 1999;14(Suppl 2):21–28. doi: 10.1002/jbmr.5650140206. [DOI] [PubMed] [Google Scholar]
- [10].Dunn KL, Espino PS, Drobic B, He S, Davie JR. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol. 2005;83:1–14. doi: 10.1139/o04-121. [DOI] [PubMed] [Google Scholar]
- [11].Niu Z, Li A, Zhang SX, Schwartz RJ. Serum response factor micromanaging cardiogenesis. Curr Opin Cell Biol. 2007;19:618–627. doi: 10.1016/j.ceb.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Jr., Miano JM. Defining the mammalian CArGome. Genome Res. 2006;16:197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- [14].Selvaraj A, Prywes R. Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol Biol. 2004;5:13. doi: 10.1186/1471-2199-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olson EN, Perry M, Schulz RA. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev Biol. 1995;172:2–14. doi: 10.1006/dbio.1995.0002. [DOI] [PubMed] [Google Scholar]
- [16].Treisman R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J. 1987;6:2711–2717. doi: 10.1002/j.1460-2075.1987.tb02564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prywes R, Roeder RG. Purification of the c-fos enhancer-binding protein. Mol Cell Biol. 1987;7:3482–3489. doi: 10.1128/mcb.7.10.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herrera RE, Shaw PE, Nordheim A. Occupation of the c-fos serum response element in vivo by a multi-protein complex is unaltered by growth factor induction. Nature. 1989;340:68–70. doi: 10.1038/340068a0. [DOI] [PubMed] [Google Scholar]
- [19].Fowler T, Sen R, Roy AL. Regulation of primary response genes. Mol Cell. 2011;44:348–360. doi: 10.1016/j.molcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Janknecht R, Ernst WH, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993;12:5097–5104. doi: 10.1002/j.1460-2075.1993.tb06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kalita K, Kuzniewska B, Kaczmarek L. MKLs: co-factors of serum response factor (SRF) in neuronal responses. Int J Biochem Cell Biol. 2012;44:1444–1447. doi: 10.1016/j.biocel.2012.05.008. [DOI] [PubMed] [Google Scholar]
- [22].Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004;93:74–82. doi: 10.1002/jcb.20199. [DOI] [PubMed] [Google Scholar]
- [23].Lee SM, Vasishtha M, Prywes R. Activation and repression of cellular immediate early genes by serum response factor cofactors. J Biol Chem. 2010;285:22036–22049. doi: 10.1074/jbc.M110.108878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;23:6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kuwahara K, Barrientos T, Pipes GC, Li S, Olson EN. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol. 2005;25:3173–3181. doi: 10.1128/MCB.25.8.3173-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guettler S, Vartiainen MK, Miralles F, Larijani B, Treisman R. RPEL motifs link the serum response factor cofactor MAL but not myocardin to Rho signaling via actin binding. Mol Cell Biol. 2008;28:732–742. doi: 10.1128/MCB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- [28].Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- [29].Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316:1749–1752. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- [30].Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- [31].Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–418. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- [32].Pesce S, Benezra R. The loop region of the helix-loop-helix protein Id1 is critical for its dominant negative activity. Mol Cell Biol. 1993;13:7874–7880. doi: 10.1128/mcb.13.12.7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jen Y, Manova K, Benezra R. Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Dev Dyn. 1996;207:235–252. doi: 10.1002/(SICI)1097-0177(199611)207:3<235::AID-AJA1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [34].Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- [35].Liang YY, Brunicardi FC, Lin X. Smad3 mediates immediate early induction of Id1 by TGF-beta. Cell Res. 2009;19:140–148. doi: 10.1038/cr.2008.321. [DOI] [PubMed] [Google Scholar]
- [36].Ueki Y, Reh TA. Activation of BMP-Smad1/5/8 signaling promotes survival of retinal ganglion cells after damage in vivo. PLoS One. 2012;7:e38690. doi: 10.1371/journal.pone.0038690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang J, Li X, Al-Lamki RS, Southwood M, Zhao J, Lever AM, Grimminger F, Schermuly RT, Morrell NW. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res. 2010;107:252–262. doi: 10.1161/CIRCRESAHA.109.209940. [DOI] [PubMed] [Google Scholar]
- [38].Gautschi O, Tepper CG, Purnell PR, Izumiya Y, Evans CP, Green TP, Desprez PY, Lara PN, Gandara DR, Mack PC, Kung HJ. Regulation of Id1 expression by SRC: implications for targeting of the bone morphogenetic protein pathway in cancer. Cancer Res. 2008;68:2250–2258. doi: 10.1158/0008-5472.CAN-07-6403. [DOI] [PubMed] [Google Scholar]
- [39].Vinals F, Ventura F. Myogenin protein stability is decreased by BMP-2 through a mechanism implicating Id1. J Biol Chem. 2004;279:45766–45772. doi: 10.1074/jbc.M408059200. [DOI] [PubMed] [Google Scholar]
- [40].Katagiri T, Imada M, Yanai T, Suda T, Takahashi N, Kamijo R. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells. 2002;7:949–960. doi: 10.1046/j.1365-2443.2002.00573.x. [DOI] [PubMed] [Google Scholar]
- [41].Lopez-Rovira T, Chalaux E, Massague J, Rosa JL, Ventura F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. The Journal of biological chemistry. 2002;277:3176–3185. doi: 10.1074/jbc.M106826200. [DOI] [PubMed] [Google Scholar]
- [42].Karaulanov E, Knochel W, Niehrs C. Transcriptional regulation of BMP4 synexpression in transgenic Xenopus. The EMBO journal. 2004;23:844–856. doi: 10.1038/sj.emboj.7600101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tournay O, Benezra R. Transcription of the dominant-negative helix-loop-helix protein Id1 is regulated by a protein complex containing the immediate-early response gene Egr-1. Mol Cell Biol. 1996;16:2418–2430. doi: 10.1128/mcb.16.5.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- [45].Ebara S, Nakayama K. Mechanism for the action of bone morphogenetic proteins and regulation of their activity. Spine (Phila Pa 1976) 2002;27:S10–15. doi: 10.1097/00007632-200208151-00004. [DOI] [PubMed] [Google Scholar]
- [46].Hara E, Yamaguchi T, Nojima H, Ide T, Campisi J, Okayama H, Oda K. Id-related genes encoding helix-loop-helix proteins are required for G1 progression and are repressed in senescent human fibroblasts. J Biol Chem. 1994;269:2139–2145. [PubMed] [Google Scholar]
- [47].Maw MK, Fujimoto J, Tamaya T. Overexpression of inhibitor of DNA-binding (ID)-1 protein related to angiogenesis in tumor advancement of ovarian cancers. BMC Cancer. 2009;9:430. doi: 10.1186/1471-2407-9-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schindl M, Schoppmann SF, Strobel T, Heinzl H, Leisser C, Horvat R, Birner P. Level of Id-1 protein expression correlates with poor differentiation, enhanced malignant potential, and more aggressive clinical behavior of epithelial ovarian tumors. Clin Cancer Res. 2003;9:779–785. [PubMed] [Google Scholar]
- [49].O'Brien CA, Kreso A, Ryan P, Hermans KG, Gibson L, Wang Y, Tsatsanis A, Gallinger S, Dick JE. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell. 2012;21:777–792. doi: 10.1016/j.ccr.2012.04.036. [DOI] [PubMed] [Google Scholar]
- [50].Singh J, Murata K, Itahana Y, Desprez PY. Constitutive expression of the Id-1 promoter in human metastatic breast cancer cells is linked with the loss of NF-1/Rb/HDAC-1 transcription repressor complex. Oncogene. 2002;21:1812–1822. doi: 10.1038/sj.onc.1205252. [DOI] [PubMed] [Google Scholar]
- [51].Clement JH, Marr N, Meissner A, Schwalbe M, Sebald W, Kliche KO, Hoffken K, Wolfl S. Bone morphogenetic protein 2 (BMP-2) induces sequential changes of Id gene expression in the breast cancer cell line MCF-7. J Cancer Res Clin Oncol. 2000;126:271–279. doi: 10.1007/s004320050342. [DOI] [PubMed] [Google Scholar]
- [52].Kebebew E, Treseler PA, Duh QY, Clark OH. The helix-loop-helix protein, Id-1, is overexpressed and regulates growth in papillary thyroid cancer. Surgery. 2003;134:235–241. doi: 10.1067/msy.2003.227. [DOI] [PubMed] [Google Scholar]
- [53].Kebebew E, Treseler PA, Duh QY, Clark OH. The helix-loop-helix transcription factor, Id-1, is overexpressed in medullary thyroid cancer. Surgery. 2000;128:952–957. doi: 10.1067/msy.2000.111082. [DOI] [PubMed] [Google Scholar]
- [54].Wang H, Yu Y, Guo RW, Shi YK, Song MB, Chen JF, Yu SY, Yin YG, Gao P, Huang L. Inhibitor of DNA binding-1 promotes the migration and proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2010;335:19–27. doi: 10.1007/s11010-009-0236-9. [DOI] [PubMed] [Google Scholar]
- [55].Hui CM, Cheung PY, Ling MT, Tsao SW, Wang X, Wong YC, Cheung AL. Id-1 promotes proliferation of p53-deficient esophageal cancer cells. Int J Cancer. 2006;119:508–514. doi: 10.1002/ijc.21874. [DOI] [PubMed] [Google Scholar]
- [56].Nishiyama K, Takaji K, Kataoka K, Kurihara Y, Yoshimura M, Kato A, Ogawa H, Kurihara H. Id1 gene transfer confers angiogenic property on fully differentiated endothelial cells and contributes to therapeutic angiogenesis. Circulation. 2005;112:2840–2850. doi: 10.1161/CIRCULATIONAHA.104.516898. [DOI] [PubMed] [Google Scholar]
- [57].Benezra R. Role of Id proteins in embryonic and tumor angiogenesis. Trends Cardiovasc Med. 2001;11:237–241. doi: 10.1016/s1050-1738(01)00117-7. [DOI] [PubMed] [Google Scholar]
- [58].Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, Benezra R. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- [59].Langenfeld EM, Kong Y, Langenfeld J. Bone morphogenetic protein 2 stimulation of tumor growth involves the activation of Smad-1/5. Oncogene. 2006;25:685–692. doi: 10.1038/sj.onc.1209110. [DOI] [PubMed] [Google Scholar]
- [60].Fong S, Itahana Y, Sumida T, Singh J, Coppe JP, Liu Y, Richards PC, Bennington JL, Lee NM, Debs RJ, Desprez PY. Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc Natl Acad Sci U S A. 2003;100:13543–13548. doi: 10.1073/pnas.2230238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shepherd TG, Mujoomdar ML, Nachtigal MW. Constitutive activation of BMP signalling abrogates experimental metastasis of OVCA429 cells via reduced cell adhesion. J Ovarian Res. 2010;3:5. doi: 10.1186/1757-2215-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wang Y, Falasca M, Schlessinger J, Malstrom S, Tsichlis P, Settleman J, Hu W, Lim B, Prywes R. Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ. 1998;9:513–522. [PubMed] [Google Scholar]
- [63].Yao Y, Zebboudj AF, Torres A, Shao E, Bostrom K. Activin-like kinase receptor 1 (ALK1) in atherosclerotic lesions and vascular mesenchymal cells. Cardiovasc Res. 2007;74:279–289. doi: 10.1016/j.cardiores.2006.09.014. [DOI] [PubMed] [Google Scholar]
- [64].Manak JR, Prywes R. Phosphorylation of serum response factor by casein kinase II: evidence against a role in growth factor regulation of fos expression. Oncogene. 1993;8:703–711. [PubMed] [Google Scholar]
- [65].Latinkic BV, O'Brien TP, Lau LF. Promoter function and structure of the growth factor-inducible immediate early gene cyr61. Nucleic Acids Res. 1991;19:3261–3267. doi: 10.1093/nar/19.12.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].O'Connell CB, Wheatley SP, Ahmed S, Wang YL. The small GTP-binding protein rho regulates cortical activities in cultured cells during division. J Cell Biol. 1999;144:305–313. doi: 10.1083/jcb.144.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kurooka H, Nakahiro T, Mori K, Sano K, Yokota Y. BMP signaling is responsible for serum-induced Id2 expression. Biochem Biophys Res Commun. 2012;420:281–287. doi: 10.1016/j.bbrc.2012.02.150. [DOI] [PubMed] [Google Scholar]
- [68].Shepherd TG, Theriault BL, Nachtigal MW. Autocrine BMP4 signalling regulates ID3 proto-oncogene expression in human ovarian cancer cells. Gene. 2008;414:95–105. doi: 10.1016/j.gene.2008.02.015. [DOI] [PubMed] [Google Scholar]
- [69].Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- [70].Kodaira K, Imada M, Goto M, Tomoyasu A, Fukuda T, Kamijo R, Suda T, Higashio K, Katagiri T. Purification and identification of a BMP-like factor from bovine serum. Biochemical and biophysical research communications. 2006;345:1224–1231. doi: 10.1016/j.bbrc.2006.05.045. [DOI] [PubMed] [Google Scholar]
- [71].Cochran BH, Zullo J, Verma IM, Stiles CD. Expression of the c-fos gene and of an fos-related gene is stimulated by platelet-derived growth factor. Science. 1984;226:1080–1082. doi: 10.1126/science.6093261. [DOI] [PubMed] [Google Scholar]
- [72].Muller R, Bravo R, Burckhardt J, Curran T. Induction of c-fos gene and protein by growth factors precedes activation of c-myc. Nature. 1984;312:716–720. doi: 10.1038/312716a0. [DOI] [PubMed] [Google Scholar]
- [73].Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sun X, Li C, Zhuang C, Gilmore WC, Cobos E, Tao Y, Dai Z. Abl interactor 1 regulates Src-Id1-matrix metalloproteinase 9 axis and is required for invadopodia formation, extracellular matrix degradation and tumor growth of human breast cancer cells. Carcinogenesis. 2009;30:2109–2116. doi: 10.1093/carcin/bgp251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 1999;18:2459–2471. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23:609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- [77].Sieber C, Kopf J, Hiepen C, Knaus P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009;20:343–355. doi: 10.1016/j.cytogfr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- [78].ten Dijke P, Korchynskyi O, Valdimarsdottir G, Goumans MJ. Controlling cell fate by bone morphogenetic protein receptors. Mol Cell Endocrinol. 2003;211:105–113. doi: 10.1016/j.mce.2003.09.016. [DOI] [PubMed] [Google Scholar]
- [79].Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- [80].Wang Y, Falasca M, Schlessinger J, Malstrom S, Tsichlis P, Settleman J, Hu W, Lim B, Prywes R. Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ. 1998;9:513–522. [PubMed] [Google Scholar]
- [81].Schratt G, Weinhold B, Lundberg AS, Schuck S, Berger J, Schwarz H, Weinberg RA, Ruther U, Nordheim A. Serum response factor is required for immediate-early gene activation yet is dispensable for proliferation of embryonic stem cells. Mol Cell Biol. 2001;21:2933–2943. doi: 10.1128/MCB.21.8.2933-2943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cooper SJ, Trinklein ND, Nguyen L, Myers RM. Serum response factor binding sites differ in three human cell types. Genome Res. 2007;17:136–144. doi: 10.1101/gr.5875007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Brugger SM, Merrill AE, Torres-Vazquez J, Wu N, Ting MC, Cho JY, Dobias SL, Yi SE, Lyons K, Bell JR, Arora K, Warrior R, Maxson R. A phylogenetically conserved cis-regulatory module in the Msx2 promoter is sufficient for BMP-dependent transcription in murine and Drosophila embryos. Development. 2004;131:5153–5165. doi: 10.1242/dev.01390. [DOI] [PubMed] [Google Scholar]
- [85].Ishida W, Hamamoto T, Kusanagi K, Yagi K, Kawabata M, Takehara K, Sampath TK, Kato M, Miyazono K. Smad6 is a Smad1/5-induced smad inhibitor. Characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. The Journal of biological chemistry. 2000;275:6075–6079. doi: 10.1074/jbc.275.9.6075. [DOI] [PubMed] [Google Scholar]
- [86].Johnson K, Kirkpatrick H, Comer A, Hoffmann FM, Laughon A. Interaction of Smad complexes with tripartite DNA-binding sites. J Biol Chem. 1999;274:20709–20716. doi: 10.1074/jbc.274.29.20709. [DOI] [PubMed] [Google Scholar]
- [87].Yao LC, Blitz IL, Peiffer DA, Phin S, Wang Y, Ogata S, Cho KW, Arora K, Warrior R. Schnurri transcription factors from Drosophila and vertebrates can mediate Bmp signaling through a phylogenetically conserved mechanism. Development. 2006;133:4025–4034. doi: 10.1242/dev.02561. [DOI] [PubMed] [Google Scholar]
- [88].Wong YC, Wang X, Ling MT. Id-1 expression and cell survival. Apoptosis. 2004;9:279–289. doi: 10.1023/b:appt.0000025804.25396.79. [DOI] [PubMed] [Google Scholar]
- [89].Sumida T, Murase R, Onishi-Ishikawa A, McAllister SD, Hamakawa H, Desprez PY. Targeting Id1 reduces proliferation and invasion in aggressive human salivary gland cancer cells. BMC Cancer. 2013;13:141. doi: 10.1186/1471-2407-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].de Candia P, Benera R, Solit DB. A role for Id proteins in mammary gland physiology and tumorigenesis. Adv Cancer Res. 2004;92:81–94. doi: 10.1016/S0065-230X(04)92004-0. [DOI] [PubMed] [Google Scholar]
- [91].Lee KT, Lee YW, Lee JK, Choi SH, Rhee JC, Paik SS, Kong G. Overexpression of Id-1 is significantly associated with tumour angiogenesis in human pancreas cancers. Br J Cancer. 2004;90:1198–1203. doi: 10.1038/sj.bjc.6601684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lin CQ, Singh J, Murata K, Itahana Y, Parrinello S, Liang SH, Gillett CE, Campisi J, Desprez PY. A role for Id-1 in the aggressive phenotype and steroid hormone response of human breast cancer cells. Cancer Res. 2000;60:1332–1340. [PubMed] [Google Scholar]
- [93].Schindl M, Oberhuber G, Obermair A, Schoppmann SF, Karner B, Birner P. Overexpression of Id-1 protein is a marker for unfavorable prognosis in early-stage cervical cancer. Cancer Res. 2001;61:5703–5706. [PubMed] [Google Scholar]
- [94].Qian Y, Chen X. ID1, inhibitor of differentiation/DNA binding, is an effector of the p53-dependent DNA damage response pathway. J Biol Chem. 2008;283:22410–22416. doi: 10.1074/jbc.M800643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kallioniemi A. Bone morphogenetic protein 4-a fascinating regulator of cancer cell behavior. Cancer Genet. 2012;205:267–277. doi: 10.1016/j.cancergen.2012.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.