

Abstract
Autoimmunity arises when immune tolerance to specific self-antigens is broken. The mechanisms leading to such a failure remain poorly understood. One hypothesis proposes that infectious agents or antigens can break B or T lymphocyte self-tolerance by expressing epitopes that mimic self. Using a transgenic immunoglobulin model, we show that challenge with self-mimicking foreign antigen rescues B cells from peripheral tolerance independent of T cell help, resulting in the accumulation of self-reactive cells in the lymph nodes and secretion of immunoglobulins that bind to a liver-expressed self-antigen. Therefore, our studies reveal a potentially important mechanism by which B lymphocytes can escape self-tolerance.
The mechanisms responsible for the onset of autoimmunity are difficult to trace, as the first signs of disease may occur long after the triggering events. Many parameters are involved in autoimmunity, including genetic predisposition (1), environmental factors such as induction of local cytokine production (2), signaling defects, or tissue damage. But because of the antigenic specificity of autoimmune diseases, it is widely believed that inappropriate activation of self-reactive B or T cells of particular specificities is the fundamental event leading to disease. Loss of tolerance to a limited spectrum of antigens may occur after pathogenic infection as a result of molecular mimicry, epitope spreading, or bystander activation (3). Tolerance breakdown can occur by activation of ignorant T or B cells upon viral infection (4), protein immunization (5), or stimulation with polyclonal activators (6). A breakdown of B cell tolerance was observed in anergic cells after removal of self-antigen (7) or when T cell help was provided at the time of initial self-antigen encounter (8). However, no tolerance breakdown was observed if T cell help was provided after self-antigen exposure (9). These and other studies have supported the two-signal model of Bretscher and Cohn, who postulated that B cells are tolerized when challenged with antigen in the absence of T cell help (10). But T-independent type 2 (TI-2) antigens provoke an antigen-specific B cell response in the absence of T cell help (11), and, as we show here, may abrogate immune tolerance.
The 3-83 immunoglobulin (Ig) transgenic (Tg) mouse expresses an antibody that binds major histocompatibility complex (MHC) class I H-2Kb and Kk molecules, but not H-2d haplotype molecules (12). On an H-2d background, 3-83 Tg mice display a monoclonal population of B cells expressing the 3-83 specificity (“nondeletor” or ND mice) (13-15). However, when 3-83 Tg mice are genetically crossed to mice engineered to express the Kb molecule specifically on the plasma membranes of liver cells (16, 17), 3-83+ B cells develop normally in the bone marrow but are deleted when they migrate into the periphery (13, 18). These mice are called “peripherally deleting” (PD) mice. In PD mice carrying a liver-expressed, albumin promoter–driven Kb gene, B cell numbers were markedly diminished in the spleens and lymph nodes (“3-83 PD,” Fig. 1A). PD mice also lacked serum Ig of 3-83 specificity (3-83 idiotype). Immunization of 3-83 ND mice with the bacteriophage P31, which expresses a 15–amino acid mimotope recognized by 3-83 Ig (19, 20), induced a strong immune response (Fig. 1B, left panel), as shown at day 4 by a 2000% increase of 3-83 circulating Ig. No such effect was seen when 3-83 ND mice were immunized with Pwt, a bacteriophage lacking the mimotope (Fig. 1B).
Fig. 1.
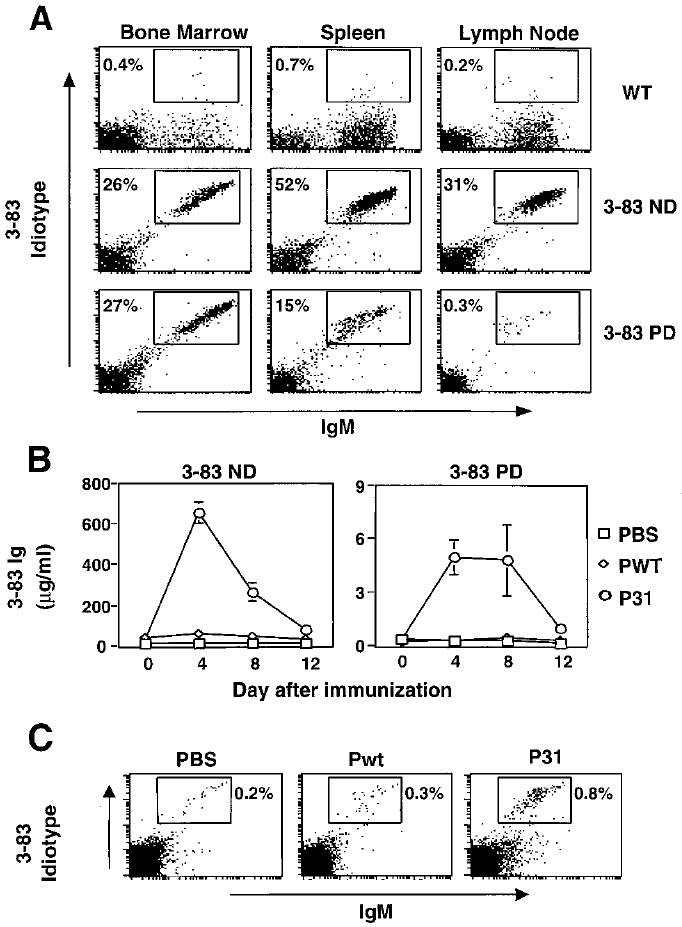
Bacteriophage P31 immunization induces a strong immune response in 3-83 nondeletor mice (ND) and breaks tolerance in 3-83 peripheral deletor (PD) mice. (A) In vivo deletion of B cells reactive to liver-expressed Kb antigen. Bone marrow, spleen, and lymph node lymphocytes of B10.D2 (WT), 3-83 nondeletor (3-83 ND), and (3-83 × pAlbumin-Kb) F1 mice (3-83 PD) were analyzed by two-color flow cytometry. Staining was done with anti-IgM and rat 3-83 idiotype antibody (54.1). (B) ND and PD mice were immunized on day 0 with 300 μg of wild-type bacteriophage (Pwt) or a phage that binds to the 3-83 BCR (P31) in PBS, or with PBS alone. After the mice were bled on the indicated days, the sera were analyzed for 3-83 Ig levels by solid-phase enzyme-linked immunosorbent assay (ELISA). Data represent mean ± SD of six independent experiments. (C) 3-83 PD mice were challenged with PBS or the indicated phage immunogens; after 24 hours, mesenteric lymph node cells were stained and analyzed as in (A).
Immunization of tolerant 3-83 PD mice with P31 stimulated the production of a small but significant level of 3-83 circulating Ig (Fig. 1B, right) that was not seen upon immunization with Pwt; this result suggested that self-tolerance was broken. In contrast, challenge of 3-83 PD mice with the known B cell polyclonal activators, lipopolysaccharide or CpG oligonucleotides, failed to increase serum levels of 3-83 Ig (21). These results suggested that a self-mimicking microbe could reverse B cell tolerance in 3-83 PD mice, and that this was dependent on specific B cell receptor (BCR) engagement.
B lymphocyte rescue from tolerance by antigenic mimicry in P31-immunized 3-83 PD mice was also apparent at the cellular level. The percentage of IgM+B220+Idiotype+ cells in the mesenteric lymph nodes was increased (by a factor of 3 to 4) 1 day after immunization with P31 (Fig. 1C), but not after phosphate-buffered saline (PBS) or Pwt injection (22).
We also evaluated the pathological consequences of B cell tolerance breakdown in the site of self-antigen expression, the liver. Four days after immunization with P31, histochemical staining of liver sections revealed the presence of massive IgM accumulation (Fig. 2A), whereas no staining was detected in the livers of Pwt-injected PD mice or P31-injected ND mice (23). Similar results were obtained with 3-83 idiotypic antibody staining. Clusters of IgMbright B cells were observed in the livers of P31-injected mice (Fig. 2B), whereas in untreated or Pwt-immunized mice only sparsely distributed IgMdull B cells were detected. Despite extensive IgM deposition in the livers of P31-immunized PD mice, which peaked at day 4, no major tissue damage was observed at the time points tested, days 4 to 12 (24).
Fig. 2.
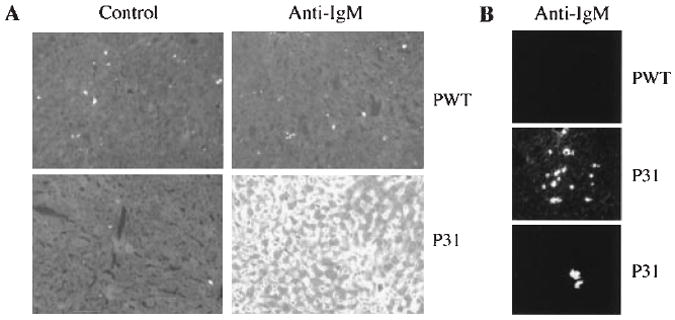
Tolerance breakdown results in massive IgM accumulation in the liver. (A) Frozen sections of livers taken 4 days after immunization were stained with FITC-conjugated anti-IgM or control antibody (rat IgG2a, κ). Photos were taken at 200× magnification with 5-s exposure. (B) Similar staining as in (A), showing clusters of B cells; photos were taken at 1-s exposure. This exposure preferentially reveals antibody-forming cells. The two lower panels are images from different mice.
To determine whether 3-83+ B cells in PD mice were activated by P31 immunization before or after encounter with Kb self-antigen, we suppressed new B cell production with inter-leukin-7 antibody (anti–IL-7) (25) before antigen challenge. These mice were treated with anti–IL-7 every third day for 9 days, then challenged with P31 and analyzed 4 days after immunization (Fig. 3A). As expected, anti–IL-7 treatment suppressed the production of immature IgM+B220low cells in the bone marrow (Fig. 3B, right panel, gate 2). Because the recirculating IgM+B220+ mature cells are deleted in the periphery, 3-83 PD mice lack these cells (Fig. 3B, center panel, gate 1). However, despite the absence of freshly exported B lymphocytes from the bone marrow, P31 bacteriophage still induced the production of circulating 3-83 Ig (Fig. 3E). After 9 days of anti–IL-7 treatment, residual B220+ B cell numbers in PD mice fell to 0.5 ± 0.4 × 106 per spleen, versus 2.7 ± 1.2 × 106 per spleen in PBS-treated controls. The residual B lymphocytes activated by P31 most likely had already encountered the self-antigen Kb in the liver, as measured by a profound down-modulation of both their idiotype and IgM levels (Fig. 3, C and D). These data also showed that very few cells carried a second receptor specificity (in addition to the idiotype) that might protect cells from tolerance. We therefore conclude that a foreign, cross-reactive antigen can block or reverse clonal deletion and can induce the activation of self-reactive B cells.
Fig. 3.

B lymphopoiesis inhibition does not affect P31-induced tolerance breakdown. (A) Experimental design. Mice were injected every third day with 1.2 mg of anti–IL-7 ip as indicated (downward arrows). Phage injection was on day 9, and analyses of antibody secretion and cell surface markers were done on day 13. (B) Anti-IgM and anti-B220 staining and flow cytometry analysis of the bone marrow of nontransgenic B10.D2 (WT), untreated 3-83 PD, and anti–IL-7–treated 3-83 PD mice taken at day 13. Gate 1, IgM+B220+ recirculating B cells; gate 2, IgM+B220low immature B cells; gate 3, IgM−B220low pro- and pre-B cells. (C and D) 3-83 idiotype expression levels on splenic B220+IgM+ cells of the indicated PBS-injected mice at day 13. Relative to ND B cells, PD cells have reduced idiotype levels and anti–IL-7–treated PD cells have very low idiotype levels. Three-color analysis (D) shows IgM and idiotype densities on B220+ gated spleen cells. (E) Sera of anti–IL-7–treated and/or immunized 3-83 PD mice were analyzed by ELISA for 3-83 Ig levels. Each symbol represents a value obtained with an individual mouse.
Because T cell help has been shown to be essential for the rescue of lysozyme-specific B cells (8), and because P31 behaves as a TI-2 antigen in vitro (19), we tested whether CD4+ T cells were also required for 3-83 B cell tolerance breakdown induced by P31 bacteriophage. We bred 3-83 PD mice onto a RAG-1 knockout background in which 3-83 B cells, but no CD4+, CD8+, or γδ+ T cells, could be generated (26). After immunization, 3-83 serum levels were increased in P31-immunized RAG-1–deficient PD mice, but not in Pwt-injected mice (Fig. 4A). Accumulation of IgM was also observed in the livers of the P31-immunized mice (24). Because immunodeficient mice might compensate for their deficiencies in unexpected ways, we independently tested the T cell independence of the P31 response in RAG-sufficient 3-83 PD mice treated before immunization with depleting anti-CD4 (GK1.5), which is effective at inhibiting T-dependent B cell responses (27). GK1.5 induced a nearly complete depletion of CD4 T helper cells (Fig. 4B). Despite this profound depletion, P31 immunization still induced production of 3-83 circulating Ig that was comparable to that seen in untreated mice (Fig. 4C). We conclude that 3-83 B cell tolerance breakdown resulted from a T cell–independent activation.
Fig. 4.

The absence of T cells does not alter B cell tolerance breakdown induced upon bacteriophage immunization. (A) 3-83 PD RAG-1−/− mice were immunized three times on days −6, −3, and 0, then analyzed on day 4 for serum level of 3-83 IgM. Each symbol represents an individual mouse. Similar results were obtained with mice injected only on day 0 (not shown). (B) 3-83 PD mice were injected with 0.5 mg of anti-CD4 ip on days −2 and −1 and immunized with phage at day 0. Spleen cells were stained with anti-CD4 and anti-CD8 on days 0 or 4 (1 or 5 days after anti-CD4 injection) to reveal the extent of T cell depletion. (C) At days 0 and 4, sera of anti-CD4–treated and immunized 3-83 PD mice were analyzed for 3-83 Ig level by ELISA.
Taken together, these data reveal a mechanism by which self-reactive B lymphocytes can escape tolerance. Tolerance breakdown resulted from specific BCR engagement, in a T-independent manner, on B cells that presumably had already been tolerized. Furthermore, because tolerance could be broken even in RAG-deficient PD mice, putative imperfect allelic exclusion is unlikely to play a role in the escape from tolerance or activation of these cells. These results underscore the need to better understand how B cells distinguish T-independent antigen from self-antigen, and they also suggest that defects in this recognition may play a role in humoral autoimmunity.
We do not observe overt autoimmune damage induced in the short time frame of our experiments. This may reflect the requirement for additional antigenic challenge, genetic factors that are lacking from the B10.D2 genetic background used here, or a need for an immunoglobulin class switch. The nature of our transgenic model, in which B cells are constrained to express only IgM or IgD and are unable to switch to other antibody classes, places inherent limits on the interpretation of the data with respect to pathogenicity of autoantibody.
Because the repetitive structures of coat and capsid proteins of many pathogenic microbes efficiently induce T-independent responses (28), it is possible that infection may trigger autoimmunity through antigenic mimicry (4). Early stages of infection, which are dominated by T-independent responses, could lead to B cell tolerance breakdown, thus setting the stage for autoimmune diseases. Several pathogenic antigens have been implicated in autoimmune diseases as a result of self-antigen mimicry (29). Bacteriophage P31 encodes a peptide that has no sequence identity to the target autoantigen, Kb (19). It remains to be seen in normal, nontransgenic systems whether antigenic mimicry can reverse tolerance to self-antigens of totally unrelated primary sequence in the course of an immune response. But if this type of molecular mimicry occurs, establishing a link between a specific infection and autoimmune disease could be extremely difficult, as has been suggested for mimicry in T cells (3).
Acknowledgments
Supported by NIH grants RO1AI33608 and PO1AI22295. We thank P. Marrack and J. Cambier for their support; N. Klinman, M. Oldstone, and D. Kono for critically reading the manuscript; and B. Arnold and G. Hämmerling for providing pAlbumin-Kb mice.
References and Notes
- 1.Vyse TJ, Todd JA. Cell. 1996;85:311. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 2.Sarvetnick N, et al. Nature. 1990;346:844. doi: 10.1038/346844a0. [DOI] [PubMed] [Google Scholar]; Gu D et al. J Exp Med. 1995;181:547. doi: 10.1084/jem.181.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]; Heath WR, et al. Nature. 1992;359:547. doi: 10.1038/359547a0. [DOI] [PubMed] [Google Scholar]
- 3.Oldstone MBA. Cell. 1987;50:819. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]; Wucherpfennig KW, Strominger JL. Cell. 1995;80:695. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; Miller SD, et al. Nature Med. 1997;3:1133. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]; Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Nature. 1992;358:155. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]; McRae BL, Vanderlugt CL, Dal CM, Miller SD. J Exp Med. 1995;182:75. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tough DF, Sprent J. Immunol Rev. 1996;150:129. doi: 10.1111/j.1600-065x.1996.tb00699.x. [DOI] [PubMed] [Google Scholar]; Yu M, Johnson JM, Tuohy VK. J Exp Med. 1996;183:1777. doi: 10.1084/jem.183.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohashi PS, et al. Cell. 1991;65:305. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]; Caton AJ, Swartzentruber JR, Kuhl AL, Carding SR, Stark SE. J Exp Med. 1996;183:13. doi: 10.1084/jem.183.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamula MJ. Immunol Rev. 1998;164:231. doi: 10.1111/j.1600-065x.1998.tb01223.x. [DOI] [PubMed] [Google Scholar]; Fritz RB, Chou CH, McFarlin DE. J Immunol. 1983;130:1024. [PubMed] [Google Scholar]; Akkaraju S, Canaan K, Goodnow CC. J Exp Med. 1997;186:2005. doi: 10.1084/jem.186.12.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hang L, et al. J Exp Med. 1983;157:874. doi: 10.1084/jem.157.3.874. [DOI] [PMC free article] [PubMed] [Google Scholar]; McHeyzer-Williams MG, Nossal GJV. J Immunol. 1988;141:4118. [PubMed] [Google Scholar]; Rocken M, Urban JF, Shevach EM. Nature. 1992;359:79. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- 7.Goodnow CC, Brink R, Adams E. Nature. 1991;352:532. doi: 10.1038/352532a0. [DOI] [PubMed] [Google Scholar]
- 8.Fulcher DA, et al. J Exp Med. 1996;183:2313. doi: 10.1084/jem.183.5.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cooke MP, et al. J Exp Med. 1994;179:425. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyster JG, Goodnow CC. Immunity. 1995;3:691. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 10.Bretscher P, Cohn M. Science. 1970;169:1042. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 11.Mond JJ, Lees A, Snapper CM. Annu Rev Immunol. 1995;13:655. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 12.Ozato K, Mayer N, Sachs DH. J Immunol. 1980;124:533. [PubMed] [Google Scholar]
- 13.Russell DM, et al. Nature. 1991;354:308. doi: 10.1038/354308a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemazee DA, Burki K. Nature. 1989;337:562. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 15.To generate nondeleting mice (3-83 ND mice), we backcrossed 3-83 transgenic mice (13), which express IgM and IgD forms of the 3-83 antibody, a minimum of 10 times onto a B10.D2nSn/J background.
- 16.Morahan G, et al. Proc Natl Acad Sci U S A. 1989;86:3782. doi: 10.1073/pnas.86.10.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schonrich G, et al. Int Immunol. 1992;4:581. doi: 10.1093/intimm/4.5.581. [DOI] [PubMed] [Google Scholar]
- 18.To generate peripheral deleting (PD) mice, we back-crossed 3-83 mice to Alb-Kb– or CRP-Kb–expressing mice (17); both types of mice were used in all the experiments and gave similar results. Both 3-83/Alb-Kb and 3-83/CRP-Kb double Tg mice presented a peripheral deletion phenotype similar to those described using 3-83 mice crossed to mice expressing Kb in the liver under the control of the sheep metallothionein promoter (13).
- 19.Kouskoff V, et al. J Exp Med. 1998;188:1453. doi: 10.1084/jem.188.8.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kouskoff V, et al. unpublished data. [Google Scholar]
- 20.Phage was purified as described (19), and mice were immunized with 300 μg of phage intraperitoneally (ip) at day 0. Anti–IL-7 (IM25) was prepared and used as described (30). Anti-CD4 (GK1.5) was obtained from ascites and injected at a dose of 0.5 mg per mouse per day. Analysis of 3-83 in the sera was performed using plates coated with rat 3-83 idiotypic antibody (54.1) at 5 μg/ml, as described (19).
- 21.Krieg AM, et al. Nature. 1995;374:546. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]; Kouskoff V, Krieg A, Nemazee D. unpublished data. [Google Scholar]
- 22.Cells were stained on ice for 20 min with the indicated antibodies in PBS supplemented with 1% bovine serum albumin and 0.02% NaN3. The following antibody reagents were used: mouse anti-IgM conjugated with fluorescein isothiocyanate (FITC) (Zymed), mouse anti-B220 conjugated with phycoerythrin (PE) (Caltag), mouse anti-CD8 conjugated with FITC, and mouse anti-CD4 conjugated with PE (Pharmingen). Flow cytometric analysis was done with a FacScan (Becton Dickinson) and the data were analyzed by Cell Quest software.
- 23.Spleens were flash-frozen in O.C.T. embedding medium (Miles), and 6-μm sections were cut and fixed in acetone. Serial sections were blocked for 10 min with PBS and 10% fetal calf serum, incubated with 2.4G2 culture supernatant for 30 min, then stained for 30 min with PE-labeled anti-B220 and FITC-labeled IgM, idiotype, or isotype control monoclonal antibodies.
- 24.Plasma transaminase levels were analyzed at days 0, 4, and 8 after immunization and were not altered in a comparison of three PBS-injected, five Pwt-injected, and four P31-immunized PD mice (V. Kouskoff, unpublished data).
- 25.Grabstein KH, et al. J Exp Med. 1993;178:257. doi: 10.1084/jem.178.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spanopoulou E, et al. Genes Dev. 1994;8:1030. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- 27.Leclerc C, Charbit A, Martineau P, Deriaud E, Hofnung M. J Immunol. 1991;147:3545. [PubMed] [Google Scholar]; Qin SX, et al. Eur J Immunol. 1990;20:2737. doi: 10.1002/eji.1830201231. [DOI] [PubMed] [Google Scholar]; Jin FS, et al. J Immunol. 1991;146:1806. [PubMed] [Google Scholar]
- 28.Szomolanyi-Tsuda E, Welsh RM. Curr Opin Immunol. 1998;10:431. doi: 10.1016/s0952-7915(98)80117-9. [DOI] [PubMed] [Google Scholar]; Bachmann MF, Zinkernagel RM. Annu Rev Immunol. 1997;15:235. doi: 10.1146/annurev.immunol.15.1.235. [DOI] [PubMed] [Google Scholar]
- 29.Bachmaier K, et al. Science. 1999;283:1335. doi: 10.1126/science.283.5406.1335. [DOI] [PubMed] [Google Scholar]; Zhao Z-S, Granucci F, Yeh L, Schaffer PA, Cantor H. Science. 1998;279:1344. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 30.Melamed D, Kench JA, Grabstein K, Rolink A, Nemazee D. J Immunol. 1997;159:1233. [PubMed] [Google Scholar]