

Abstract
Pemphigus vulgaris (PV) is an autoimmune blistering disease characterized by autoantibodies to the keratinocyte adhesion protein desmoglein (Dsg) 3. Previous studies suggest that PV pathogenesis involves p38 mitogen activated protein kinase-dependent and -independent pathways. However, p38 is a difficult protein to study and therapeutically target because it has four isoforms and multiple downstream effectors. In the current study, we identify MAPKAP kinase 2 (MK2) as a downstream effector of p38 signaling in PV and describe MK2-dependent and -independent mechanisms of blister formation using passive transfer of human anti-Dsg IgG4 mAbs to neonatal mice. In human keratinocytes, PV mAbs activate MK2 in a dose-dependent manner. MK2 is also activated in human pemphigus skin blisters, causing translocation of MK2 from the nucleus to the cytosol. Small molecule inhibition of MK2 and silencing of MK2 expression block PV mAb-induced Dsg3 endocytosis in human keratinocytes. Additionally, small molecule inhibition and genetic deletion of p38α and MK2 inhibit spontaneous, but not induced, suprabasal blisters by PV mAbs in mouse passive transfer models. Collectively, these data suggest that MK2 is a key downstream effector of p38 that can modulate PV autoantibody pathogenicity. MK2 inhibition may be a valuable adjunctive therapy for control of pemphigus blistering.
Keywords: autoimmunity, desmosome, cell adhesion, cadherin, cell signaling
Introduction
Pemphigus vulgaris (PV) is a potentially life-threatening blistering disease caused by autoantibodies against desmoglein (Dsg) adhesion proteins. Pathogenic anti-Dsg antibodies disrupt the assembly of Dsg into nascent desmosomes, resulting in desmosome disassembly and loss of intercellular adhesion (Kitajima, 2002;Calkins et al., 2006;Cirillo et al., 2006;Mao et al., 2009), evidenced histologically as suprabasal acantholysis. Pathogenic serum IgG antibodies bind calcium-sensitive conformational epitopes in the Dsg extracellular amino-terminal domains, while nonpathogenic serum IgG bind non-conformational epitopes throughout the extracellular domains (Amagai et al., 1995;Amagai et al., 1992). Anti-Dsg monoclonal antibodies (mAbs) that bind amino-terminal calcium-sensitive epitopes and cause suprabasal acantholysis have been identified from an active immune mouse model of PV (Tsunoda et al., 2003), as well as human PV patients (Payne et al., 2005;Yeh et al., 2006).
Skin adhesion is weakened even in the non-blistered skin of pemphigus patients, as blisters can be induced in normal-appearing skin by mechanical shear stress (known as the Nikolsky sign). However, blisters also spontaneously occur in pemphigus patients, even in the absence of mechanical shear stress. Past studies have implicated diverse signaling pathways in PV IgG-induced acantholysis, including p38 mitogen-activated protein kinase (Berkowitz et al., 2005), rho family GTPases (Waschke et al., 2006), protein kinase C (Sanchez-Carpintero et al., 2004), and c-myc (Williamson et al., 2006), among others (Sharma et al., 2007). Of these, a role for p38 has been the best established, with data from multiple laboratories supporting necessary and sufficient roles for p38 in various aspects of pemphigus pathology. p38 is activated in in vitro studies using PV serum IgG and mAbs (Berkowitz et al., 2005;Kawasaki et al., 2006;Mao et al., 2011) and in vivo in pemphigus patient skin (Berkowitz et al., 2007). Exogenous p38 activation by oxidative stress or ultraviolet light causes Dsg3 endocytosis in keratinocytes (Mao et al., 2011), and p38 inhibition prevents pathologic effects of PV serum IgG and mAbs, including Dsg3 endocytosis and blistering in a mouse passive transfer model (Berkowitz et al., 2006;Jolly et al., 2010;Mao et al., 2011). Recent studies by our laboratory and others have proposed that steric hindrance of Dsg3 adhesion (pathophysiologically equivalent to the Nikolsky sign in patients) is signaling-independent (Mao et al., 2011;Saito et al., 2012;Heupel et al., 2008), whereas spontaneous blistering is amplified by p38 signaling, which may explain the therapeutic benefit of p38 inhibition in some but not all PV models.
Currently, rapid control of pemphigus blistering is achieved with corticosteroids, although the immunosuppressive and hyperglycemic side effects of corticosteroids can cause complications, making p38 a promising adjunctive target for therapy in PV. However, p38 inhibitors have systemic toxicities in humans (Goldstein et al., 2010), due to p38’s involvement in a broad range of cellular processes and p38 inhibitor promiscuity, leading to off-target effects. Additionally, p38 has four isoforms (α, β, γ, and δ), which complicate its study in biological systems. Therefore, we sought to identify downstream effectors that regulate Dsg3 endocytosis and keratinocyte adhesion, which could be more specific targets for therapeutic intervention. MAPKAP kinase 2 (MK2) regulates several cellular events downstream of p38 signaling, including tumor necrosis factor alpha production and cytoskeletal remodeling, both of which may contribute to PV pathogenesis (Kotlyarov et al., 1999;Kayyali et al., 2002). In the present study, we identify MK2 as the major downstream effector of p38 signaling in PV and describe MK2-dependent and -independent mechanisms of blister formation.
Results
MK2 is activated in lesional keratinocytes of pemphigus patients
To determine whether MK2 is activated in vivo in pemphigus, we performed immunohistochemistry on skin biopsies from 4 PV patients and 2 patients with the related blistering disease, pemphigus foliaceus (PF), to detect activated (phospho-Thr222) MK2. Normal rabbit serum was a primary antibody control (Figure 1, left panels). Activated MK2 was observed in keratinocytes in the blister roof and base of PV and PF patients (arrows). MK2 phosphorylation was not observed in PV non-lesional epidermis, as compared to normal human epidermis, although slight elevation of activated MK2 was observed in perilesional keratinocytes (PV non-lesional, arrows).
Figure 1. MK2 is activated in lesional skin keratinocytes from pemphigus vulgaris (PV) and foliaceus (PF) patients.
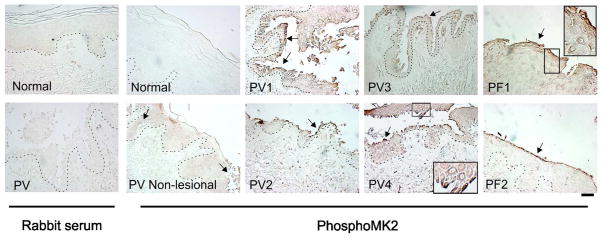
Activated MK2, demonstrated by immunohistochemical staining of pemphigus skin biopsy samples with an antibody specific for phospho-MK2, was markedly increased in PV (PV1-4) and PF (PF1-2) lesional skin keratinocytes. No significant activation was observed in normal human skin or PV non-lesional keratinocytes (arrows indicate focal activation in perilesional keratinocytes). Normal rabbit serum was used as antibody negative control. Activated MK2 is primarily observed in the cytoplasm of keratinocytes (PV4 and PF1, inset). Scale bar=100μm.
Pathogenic anti-Dsg PV mAb activates MK2 in a dose-dependent manner in primary human epidermal keratinocytes (PHEK), causing translocation of MK2 from the nucleus to the cytoplasm
Previously, we cloned human anti-Dsg single chain variable fragment (scFv) mAbs from PV patients (Payne et al., 2005). Similar to PV serum IgG, pathogenic scFv mAbs cause Dsg3 endocytosis, dissociation of PHEK, and suprabasal blisters in human skin explants and neonatal mice after passive transfer (Payne et al., 2005;Mao et al., 2009). Pathogenic scFv, IgG1, and IgG4 mAbs expressing the same variable region activate p38 MAPK in PHEK with equivalent dose-dependency (Mao et al., 2011). In contrast, nonpathogenic mAbs bind Dsg3 but do not activate p38, cause Dsg endocytosis, or induce skin blisters. We first determined whether a pathogenic IgG4 mAb (recognizing both Dsg3 and Dsg1) that activates p38 and causes skin blisters also activates MK2, by immunoblotting PHEK lysates with an antibody specific for activated MK2, using total MK2 protein as a loading control. Oxidative stress (200 μM H2O2) was a positive control and nonpathogenic anti-Dsg3/1 IgG4 mAb a negative control for stimulating p38/MK2 signaling. Both the pathogenic mAb and H2O2 activated MK2 in a dose-dependent manner (Figure 2A, upper panels), with peak activation at 2 hours (Figure 2B). Activation of p38 showed a similar pattern (Figure 2A, lower panels).
Figure 2. Pathogenic but not nonpathogenic PV mAbs activate MK2 in primary human keratinocytes.

A) Pathogenic (P) mAb activates MK2 in a dose-dependent manner (treatment 2 hours), while nonpathogenic (NP) mAb does not, similar to positive controls for p38 activation by P mAb and oxidative stress (H2O2). B) Peak activation of MK2 by 50 μg/mL P mAb occurs at 2 hours. C) MK2 translocates from the nucleus to the cytosol after treatment of keratinocytes with P mAb. MK2 protein levels in the cytosolic and nuclear fractions (co-fractionation with beta-tubulin and histone, respectively) were detected by immunoblot. D) P mAb and H2O2, but not NP mAb, cause MK2 translocation from the nucleus to the cytosol at 4 hours, demonstrated by immunofluorescence. Scale bar=20μm.
Activation of MK2 causes its translocation from the nucleus to the cytoplasm (Engel et al., 1998). We therefore performed subcellular fractionation of PHEK after pathogenic mAb stimulation. Before PV mAb treatment, MK2 is predominantly located in the nucleus (Figure 2B, 0 hrs). 4–16 hours after pathogenic PV mAb stimulation, MK2 protein shifts from the nuclear to cytosolic fractions. Similarly, MK2, detected by immunofluorescence staining, remains in the nucleus in PHEK treated with nonpathogenic mAb (Figure 2C). Upon activation by pathogenic mAb or H2O2, MK2 translocates to the cytoplasm. Similar findings are observed in vivo, as activated MK2 is predominantly localized in the cytoplasm of lesional keratinocytes in pemphigus patient skin (Figure 1, PV4 and PF1 insets).
Inhibition or knockdown of MK2 prevents PV mAb-mediated loss of cell surface Dsg3 in human keratinocytes
If MK2 is the primary downstream effector of p38 signaling in PV, then MK2 inhibition should prevent the pathologic effects of PV mAbs that are regulated by p38, including Dsg3 endocytosis and loss of Dsg3 from the desmosomal fraction of PHEK (Jolly et al., 2010;Mao et al., 2011). We first examined the efficacy and specificity of a pyrrolopyridinyl MK2 inhibitor. The MK2 inhibitor (MK2I) decreases phosphorylation of HSP27, a direct downstream target of MK2, in a dose-dependent pattern (Figure 3A; total HSP27 as a loading control), but does not affect the phosphorylation of the upstream kinase p38. Treatment of PHEK with the p38 inhibitor SB202190 or MK2I decreases the loss of Dsg3 and desmoplakin from the Triton-insoluble fraction of PHEK (Figure 3B, upper panels). Consistent with these findings, MK2 inhibition prevents the pathogenic PV mAb-mediated loss of cell-surface Dsg3, similar to the effects of p38 inhibition (Figure 3C).
Figure 3. MK2 inhibition blocks loss of cell surface Dsg3 by pathogenic (P) PV mAb.

A) Pretreatment of PHEK with an MK2-specific inhibitor (MK2I) for 2 hours, then PV mAb for 2 hours, blocks phosphorylation of HSP27 but not p38 in a dose-dependent pattern. B) MK2I rescues the loss of desmosomal Dsg3 caused by P mAb. PHEK were treated with DMSO, 2μM SB202190 (p38 inhibitor) or 2.5 μg/mL MK2I for 2 hours, followed by 50 μg/ml PV mAbs (P or NP) for 6 hours. Triton X-100-insoluble fractions were immunoblotted with antibodies as indicated. Data are representative of three independent experiments. C) P but not NP mAb (as in B) causes loss of cell surface Dsg3 (green), which is inhibited by SB202190 and MK2I. Scale bar=20μm.
To confirm the inhibitor studies, we used shRNA to silence MK2 expression in HaCat human keratinocytes. MK2 protein levels were determined by immunoblotting, using histone H3 as a control. Compared to the control (Ctl) shRNA-transfected cell line, MK2 expression was almost completely silenced in the MK2 shRNA stable cell line (Figure 4A, upper panel). p38 expression is also significantly decreased in MK2 shRNA cells, which was unanticipated but consistent with prior reports indicating that MK2 stabilizes p38 protein (Kotlyarov et al., 2002). MK2 knockdown was further confirmed by immunofluorescence staining (Figure 4B). MK2 shRNA and control cells were treated with PBS, H202, or PV pathogenic mAb, and the subcellular location of Dsg3 was determined by immunostaining. Both H202 and PV mAb-induced loss of cell surface Dsg3 were decreased in MK2 shRNA cells compared to control cells (Figure 4C), consistent with the results of p38 and MK2 small molecule inhibition.
Figure 4. Silencing of MK2 expression prevents PV mAb-induced loss of cell surface Dsg3.
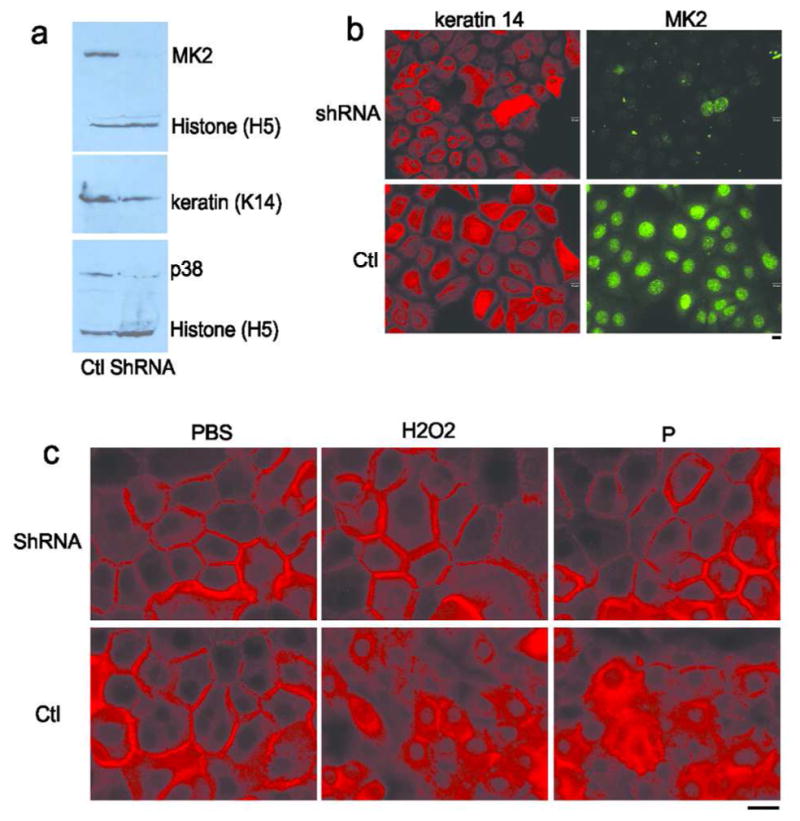
A) HaCat cells that stably express MK2 shRNA (shRNA) show markedly reduced MK2 and p38 protein levels compared to cells expressing control (Ctl) shRNA. Levels of histone H3 are shown as a loading control. B) Immunofluorescence staining of HaCat cells expressing MK2 or control (Ctl) shRNA confirms knockdown of MK2 expression 72 hours after transduction. C) shRNA silencing of MK2 expression decreases loss of cell surface Dsg3 (shown in red) 16 hours after treatment with pathogenic PV mAb (P) and oxidative stress (H2O2). Scale bar=20μm.
Genetic deletion or small molecule inhibition of MK2 does not inhibit induced blisters in a mouse passive transfer model
Mice can develop both Nikolsky and spontaneous suprabasal blisters after passive transfer of PV mAbs. 4–6 hours after passive transfer, Nikolsky skin fragility occurs (induction of blisters by mechanical shear stress). 16–18 hours after passive transfer, spontaneous blisters appear (in the absence of applied mechanical shear stress). Previously, we demonstrated that Nikolsky skin fragility is p38α-independent (Mao et al., 2011). Similarly, mice with a genetic deletion of MK2 (Kotlyarov et al., 1999) are susceptible to induced suprabasal blisters after passive transfer of pathogenic PV mAb (Figure 5A). 6/6 wild-type and 4/4 MK2-knockout mice demonstrated gross blisters, with suprabasal acantholysis and similar histology scores (Figure 5B–C).
Figure 5. Nikolsky (induced) blisters are not inhibited by genetic deletion or small molecule inhibition of MK2.
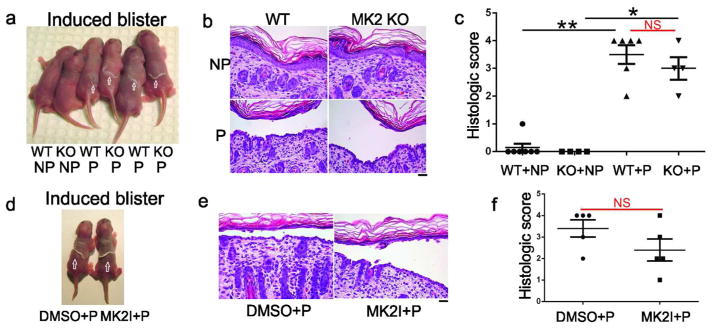
25 μg of pathogenic (P), but not nonpathogenic (NP), mAb caused Nikolsky blisters (arrows) 6 hours after mAb injection then mechanical shear stress in MK2-knockout (KO) and wild-type (WT) littermate control mice (A), and wild-type mice pretreated with DMSO or MK2I (D). (B, E) Histologic analysis demonstrates suprabasal acantholysis in mice injected with P but not NP mAb. The extent of histologic blistering was significantly different between P and NP mAb, but similar in WT and MK2 KO mice injected with P mAb (C), as well as wild-type mice injected with P mAb after pretreatment with DMSO or MK2I (F). Scale bar=100 μm. **p<0.01; *p<0.05; NS, non-significant
Because MK2-deficient mice demonstrate decreased p38 levels, we also tested whether inhibition of MK2 signaling, which does not affect p38 activation (Figure 3A), can block induced blisters. We injected wild-type mice with PV mAbs, two hours after injection with 5μg of p38 inhibitor (SB202190) or 6.25μg of MK2I (doses determined by in vitro assays confirming sufficiency of kinase inhibition (Figure 3A) relative to effective in vivo doses of p38 inhibitors (Berkowitz et al., 2006)). Similar to MK2-knockout mice, wild-type mice pretreated with DMSO or MK2I then pathogenic (P) mAb develop blisters after mechanical shear stress (Figure 5D). 5/5 DMSO+P and 5/5 MK2I+P mice demonstrated gross blisters, again with suprabasal acantholysis and similar histology scores (Figure 5E–F). Direct immunofluorescence analysis confirmed mAb binding (data not shown).
Genetic deletion or inhibition of MK2 inhibits spontaneous blister formation in a mouse passive transfer model
We next determined whether MK2 is required for spontaneous blistering in the mouse passive transfer model. In contrast to induced blisters, spontaneous blistering was significantly reduced in MK2 knockout mice compared to wild-type littermate controls (Figure 6A). 25/30 wild-type mice showed gross blisters after passive transfer of pathogenic mAb, compared to 4/15 MK2-knockout mice (p=0.0005). Direct immunofluorescence analysis of skin confirmed mAb binding (data not shown). All 30 wild-type mice demonstrated suprabasal acantholysis, with significantly increased histologic blistering (Figure 6B–C). MK2-knockout mice without gross blisters did not demonstrate microscopic blisters. Unexpectedly, MK2-knockout mice with gross blisters demonstrated predominantly superficial, with focal areas of suprabasal, acantholysis (Figure 6B, bottom right). This phenotype was also observed in mice with a K14-Cre mediated deletion of p38α in the epidermis after injection with pathogenic mAb (see Supplemental Text, Figure S1, for full discussion.) The extent of suprabasal blistering in MK2-knockout mice was significantly reduced compared to wild-type mice (Figure 6C).
Figure 6. Spontaneous blistering is inhibited by genetic deletion of MK2 and p38 or MK2 small molecule inhibition.
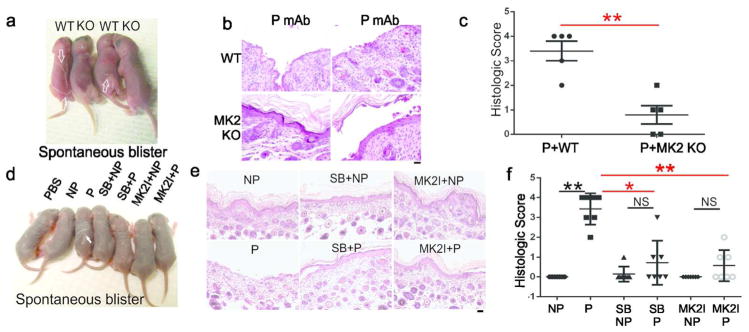
A) 16 hours after pathogenic (P) mAb injection, spontaneous blisters occur in wild-type (WT) (arrows) but not MK2-knockout (KO) mice. B) Histology shows suprabasal acantholysis in WT mice. 11/15 KO mice did not blister (left). 4/15 KO mice with gross blisters demonstrated superficial, not suprabasal, acantholysis (right). C) Suprabasal histologic blistering was significantly decreased in KO versus WT mice. D) SB202190 or MK2I prevents spontaneous blistering by P mAb (arrow). E) P but not NP mAbs cause suprabasal acantholysis, inhibited by SB202190 or MK2I, with focal blistering in some sections. F) Histologic blistering was significantly decreased by SB202190 and MK2I. Scale bar=100 μm. **p<0.01; *p<0.05; NS, non-significant.
We then tested whether spontaneous blisters are dependent on MK2 signaling. Wild-type mice injected with pathogenic but not nonpathogenic mAb developed spontaneous blisters 16–18 hours after passive transfer (Figure 6D). Pre-treatment with SB202190 (a p38 inhibitor) or MK2I inhibited blister formation. 7/8 mice injected with pathogenic mAb demonstrated gross blistering, whereas 2/8 mice injected with MK2I and 1/4 with SB202190 then pathogenic mAb blistered (p<0.05). Histologic analysis confirmed inhibition of blistering (Figure 6E–F), although focal suprabasal acantholysis was observed on the edge of some tissue specimens, consistent with Nikolsky skin fragility due to tissue processing. Direct immunofluorescence confirmed binding of mAbs to the keratinocyte cell surface (data not shown).
We attempted to evaluate whether PV serum IgG is similarly blocked by MK2 deficiency or inhibition. However, two PV serum IgG samples that caused suprabasal blisters in human skin explants did not cause suprabasal blisters in mice, due to lack of cross-reactivity or low affinity binding to mouse compared to human Dsg3 (data not shown).
Discussion
In this study, we describe signaling-dependent and –independent models of blister formation in PV and identify MK2 as the major downstream effector of p38-dependent pathology in PV. Pathogenic PV autoantibodies activate MK2 in vivo in the skin of PV patients and mice after passive transfer (Figure 1 and Figure S1) and in vitro in PHEK (Figure 2A). MK2 is activated by phosphorylation of residue Thr222 and Thr334 by p38. Phosphorylation of Thr334, which regulates the nuclear localization and nuclear export signals of MK2, results in translocation of MK2 from the nucleus to the cytoplasm (Kotlyarov et al., 2002). Accordingly, MK2 translocates from the nucleus to cytoplasm in response to pathogenic PV mAb (Figure 2) and is predominantly localized in the cytoplasm of lesional keratinocytes in pemphigus patient skin (Figure 1).
Additionally, we demonstrate that MK2 inhibition, using both small molecule and genetic approaches, prevents loss of cell surface Dsg3 (Figures 3, 4) and spontaneous blister formation in a mouse passive transfer PV model (Figure 6). The specificity of MK2 small molecule inhibition is supported by our studies using shRNA knockdown or genetic deletion of MK2, although MK2 cannot be uniquely targeted by genetic approaches since its knockdown results in simultaneous destabilization of p38 (Figure 4A) (Kotlyarov et al., 2002;Ronkina et al., 2007). Nevertheless, the MK2 silencing, knockout, and inhibitor studies all demonstrate a consistent protective phenotype in regard to maintenance of keratinocyte cell surface Dsg3 and spontaneous blistering, which collectively support a role for the p38-MK2 signaling pathway in PV pathogenesis. It is interesting that genetic loss of MK2 and p38α in mouse epidermis may be more protective for suprabasal than superficial blistering (Figure 6 and S1). This selective phenotype was not observed with MK2 and p38 inhibitors, suggesting that small molecule inhibition of p38 and MK2 may be a more effective method than genetic targeting for suppression of blistering. The expression pattern of Dsg3 in the epidermis in WT, MK2 KO, and p38 KO mice were similar, as determined by direct immunofluorescence staining of Dsg3 (data not shown). It is possible that the requirement for p38 or MK2 is different downstream of Dsg3 and Dsg1 in the suprabasal versus superficial epidermis (Spindler et al., 2011), for example due to differential compensation by other proteins such as MK3, MK5, or other p38 isoforms, or that the superficial epidermis is more subject to frictional shear stress than the suprabasal epidermis.
Consistent with our studies on p38, we observe MK2-dependent and independent mechanisms of blister formation. In our models, Nikolsky skin fragility is p38- and MK2-independent, evidenced in mice with a genetic deletion of p38α in the epidermis (Mao et al., 2011), MK2 knockout mice, and wild type mice treated with MK2 inhibitors (Figure 5). We believe this is the first step in disease pathology, reflecting steric hindrance of Dsg adhesion by binding of pathogenic anti-Dsg antibodies followed by mechanical shear stress. In contrast, signaling-dependent pathology is evaluated in our mouse models by spontaneous blistering, which occurs later than Nikolsky skin fragility and is inhibited by genetic deletion of MK2 or small molecule inhibition of p38 and MK2 (Figure 6). These late events may reflect amplification of pathology by activation of p38, which causes endocytosis of multiple desmosomal molecules including Dsg3 and desmocollin 3, leading to desmosome disassembly (Mao et al., 2009;Mao et al., 2011).
There are some discrepancies between our models and studies using PV IgG. Blister formation by PV serum IgG, but not a pathogenic mouse AK23 mAb, was blocked by p38 inhibition in human skin explants, which is primarily a test of Nikolsky fragility (Saito et al., 2012). PV serum IgG may have additional signaling-dependent pathogenic mechanisms related to Dsg3 clustering, causing more effective disassembly of desmosomes (whereas mAbs primarily deplete non-desmosomal Dsg3, with indirect effects on desmosome disassembly (Mao et al., 2009)). Induced blisters after passive transfer of PV serum IgG to mice were also blocked by p38 inhibition (Berkowitz et al., 2006). The latter study used lower doses for histologic versus gross blister evaluation, suggesting that the p38-dependence of induced blisters may be dose-dependent. Nevertheless, our models using human anti-Dsg IgG4 mAbs offer the advantage of being able to directly identify pathologic effects due to anti-Dsg antibodies in PV serum IgG and consistently reproduce key aspects of disease pathology caused by PV serum IgG, including activation of p38, Dsg endocytosis, and suprabasal blisters in human and mouse skin.
In summary, our results suggest that MK2 is the major pathologic downstream effector of p38 signaling in pemphigus. Future studies aim to establish a direct connection between MK2 activation and the cell pathologic effects of PV autoantibodies, including potential effects on other desmosomal proteins such as desmocollin, desmoplakin, and keratin. MK2 directly phosphorylates the actin-modulating protein HSP27 (Stokoe et al., 1992), which is activated in pemphigus (Berkowitz et al., 2007). Additionally, MK2 phosphorylates keratins 18 and 20 in intestinal epithelial cells (Menon et al., 2010). Since collapse of the keratin intermediate filament network is a prominent cytologic finding in PV, it is possible that spontaneous blistering may be directly linked to regulation of the cytoskeletal network by MK2.
Therapeutically, inhibition of MK2 activity represents a more targeted strategy aimed to block the pathologic response of the skin to pemphigus autoantibodies, without generally suppressing patients’ immunity. MK2 knockout mice are relatively resistant to endotoxic shock due to deficient production of tumor necrosis factor alpha, and this function is partially redundant with MAPKAP kinase 3 (Kotlyarov et al., 1999;Ronkina et al., 2007). Therefore, MK2 inhibition may preserve systemic immunity while still inhibiting the pathologic effects of MK2 in skin keratinocytes. Inhibition of MK2, either topically or systemically, may represent a fast treatment option for PV during disease flare, allowing time for immunosuppressive agents to decrease pathogenic autoantibodies. Such interventions may be safer than topical or oral corticosteroids, which are currently used for rapid disease control.
Materials and Methods
Antibodies and reagents
Antibodies included rabbit anti-p38, anti-MK2, anti-phospho-p38 (Thr180/Tyr182), anti-phospho-MK2 (Thr222, Cell Signaling Technology, Danvers, MA), mouse anti-Dsg3 (5G11, Invitrogen, Grand Island, NY), mouse anti-desmoplakin (BioDesign International, Cincinnati, OH), mouse anti-keratin 14 (K14, Abcam Inc., Cambridge, MA), rabbit anti-histone H3 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), HRP-conjugated donkey anti-mouse or -rabbit IgG (Jackson ImmunoResearch, West Grove, PA), Alexa 488-conjugated donkey anti-rabbit IgG, Alexa 488-conjugated goat anti-mouse and rabbit IgG, and Alexa 594-conjugated goat anti-mouse, rat, or rabbit IgG (Invitrogen).
PV IgG4 mAbs were produced as described, expressing the (D31)2/29 pathogenic and (D31)12b/6 nonpathogenic variable regions (also previously described as P1 and NP1 (Mao et al., 2009)) with an IgG4 heavy chain (Payne et al., 2005;Payne et al., 2007).
Other reagents included p38 inhibitor (SB202190), hydrogen peroxide, and puromycin (Sigma-Aldrich, St. Louis, MO); MK2 inhibitor III, MK2 shRNA and control shRNA lentiviral particles (Santa Cruz Biotechnology).
Cell culture and treatments
Primary human epidermal keratinocytes (PHEK) were obtained from the Penn Skin Disease Research Center and cultured as described (Mao et al., 2009;Mao et al., 2011). For p38 or MK2 inhibition assays, cells were treated with 0–2 μM SB202190 or 0–5 μg/ml MK2 inhibitor, followed by 200 μg/mL PV mAbs or 200 μM H2O2. HaCat cells were maintained in 50% DK-SFM:50% DMEM containing 10% fetal bovine serum and penicillin/streptomycin.
Immunofluorescence
Immunofluorescence was performed as described for PHEK (Mao et al., 2009) and mouse skin (Mao et al., 2011).
Subcellular fractionation/immunoblotting
After incubation with PV mAbs, PHEK were chilled on ice and washed with ice-cold PBS. Cells were harvested in ice-cold PBS by centrifugation at 2,500 × g for 1 minute at 4°C, then lysed in buffer containing 10 mM Hepes (pH 7.6), 0.4 mM CaCl2, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 0.5 mM PMSF, 10 μg/mL aprotinin and leupeptin on ice for 10 minutes. Lysates were passaged 20 times through a 26-G insulin syringe, followed by centrifugation at 7,200 × g for 1 minute at 4°C. The supernatant (cytoplasmic fraction) was removed, and the remaining nuclear pellet was washed and solubilized in Laemmli sample buffer. Triton X-100 cell fractionation and immunoblotting were performed as described (Mao et al., 2011).
RNA interference
MK2 and control lentiviral shRNA were used to infect HaCat cells according to manufacturer’s protocols. 72 hours after transduction, cells were selected with media containing 5 Jg/ml puromycin. Transduced cells were treated with 50 μg/ml PV mAbs or 100 μM H2O2 for 24 hours prior to analysis.
Neonatal mouse passive transfer
p38 fl/fl;+/+ (WT) or p38 fl/fl;K14cre/+ (KO) mice (Mao et al., 2011), or MK2 KO mice (Kotlyarov et al., 1999) and wild type littermate controls were used for passive transfer studies. Newborn mice (1–3 days, 1.2–2.5 g) were injected intradermally with 25μg purified PV monoclonal IgG4, +/− intradermal injection of p38 or MK2 inhibitor 2 hours prior, and sacrificed 16–18 hours after the last injection for evaluation. Skin sections were processed as described (Mao et al., 2011). Histology scores on serial sections were quantified as described (Spindler et al., 2013).
Histology/immunohistochemistry
Formalin-fixed, paraffin-embedded mouse skin sections were stained with hematoxylin and eosin according to standard procedures. Human pemphigus skin sections were obtained from the Penn Skin Disease Research Center Tissue Bank. Immunohistochemistry was performed to evaluate MK2 activation as described (Mao et al., 2011), incubating a rabbit anti-phospho-MK2 antibody (1:500) with sections overnight at 4°C, followed by peroxidase-conjugated donkey anti-rabbit IgG for one hour at room temperature.
Statistical Analysis
Figures 5A,5D,6A,6D: Fisher’s exact test. Figures 5F,6C: Wilcoxon rank sum test. Figures 5C,6F: Kruskal-Wallis test with Dunn’s post-hoc multiple comparisons analysis to identify significant differences. p<0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Tzvete Dentchev and Eun Jung Choi for technical support, and John Seykora and John Stanley for reagents and scientific discussions. This publication was made possible by AR053505, AR057001 (ASP) and AR057217 (Penn Skin Disease Research Center) from NIAMS/NIH, AI074957 (JMP) from NIAID/NIH, Deutsche Forschungsgemeinschaft (MG), and the Dermatology Foundation (XM). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Amagai M, Ishii K, Hashimoto T, et al. Conformational epitopes of pemphigus antigens (Dsg1 and Dsg3) are calcium dependent and glycosylation independent. J Invest Dermatol. 1995;105:243–247. doi: 10.1111/1523-1747.ep12317587. [DOI] [PubMed] [Google Scholar]
- Amagai M, Karpati S, Prussick R, et al. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992;90:919–926. doi: 10.1172/JCI115968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz P, Diaz LA, Hall RP, et al. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 2007;128:738–740. doi: 10.1038/sj.jid.5701080. [DOI] [PubMed] [Google Scholar]
- Berkowitz P, Hu P, Liu Z, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280:23778–23784. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- Berkowitz P, Hu P, Warren S, et al. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci USA. 2006;103:12855–12860. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins CC, Setzer SV, Jennings JM, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. 2006;281:7623–7634. doi: 10.1074/jbc.M512447200. [DOI] [PubMed] [Google Scholar]
- Cirillo N, Femiano F, Gombos F, et al. Serum from pemphigus vulgaris reduces desmoglein 3 half-life and perturbs its de novo assembly to desmosomal sites in cultured keratinocytes. FEBS Lett. 2006;580:3276–3281. doi: 10.1016/j.febslet.2006.04.089. [DOI] [PubMed] [Google Scholar]
- Engel K, Kotlyarov A, Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 1998;17:3363–3371. doi: 10.1093/emboj/17.12.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DM, Kuglstatter A, Lou Y, et al. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. doi: 10.1021/jm9012906. [DOI] [PubMed] [Google Scholar]
- Heupel WM, Zillikens D, Drenckhahn D, et al. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 2008;181:1825–1834. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- Jolly PS, Berkowitz P, Bektas M, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 2010;285:8936–8941. doi: 10.1074/jbc.M109.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Aoyama Y, Tsunoda K, et al. Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity. 2006;39:587–590. doi: 10.1080/08916930600971943. [DOI] [PubMed] [Google Scholar]
- Kayyali US, Pennella CM, Trujillo C, et al. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem. 2002;277:42596–42602. doi: 10.1074/jbc.M205863200. [DOI] [PubMed] [Google Scholar]
- Kitajima Y. Mechanisms of desmosome assembly and disassembly. Clin Exp Dermatol. 2002;27:684–690. doi: 10.1046/j.1365-2230.2002.01116.x. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Yannoni Y, Fritz S, et al. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22:4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. 2009;129:908–918. doi: 10.1038/jid.2008.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Sano Y, Park JM, et al. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. 2011;286:1283–1291. doi: 10.1074/jbc.M110.172874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MB, Schwermann J, Singh AK, et al. p38 MAP kinase and MAPKAP kinases MK2/3 cooperatively phosphorylate epithelial keratins. J Biol Chem. 2010;285:33242–33251. doi: 10.1074/jbc.M110.132357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne AS, Ishii K, Kacir S, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne AS, Siegel DL, Stanley JR. Targeting pemphigus autoantibodies by their heavy chain variable region genes. J Invest Dermatol. 2007;127:1681–1691. doi: 10.1038/sj.jid.5700790. [DOI] [PubMed] [Google Scholar]
- Ronkina N, Kotlyarov A, Dittrich-Breiholz O, et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Stahley SN, Caughman CY, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One. 2012;7:e50696. doi: 10.1371/journal.pone.0050696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Carpintero I, Espana A, Pelacho B, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol. 2004;151:565–570. doi: 10.1111/j.1365-2133.2004.06147.x. [DOI] [PubMed] [Google Scholar]
- Sharma P, Mao X, Payne AS. Beyond steric hindrance: The role of adhesion signaling pathways in the pathogenesis of pemphigus. J Dermatol Sci. 2007;48:1–14. doi: 10.1016/j.jdermsci.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Spindler V, Endlich A, Hartlieb E, et al. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am J Pathol. 2011;179:1905–1916. doi: 10.1016/j.ajpath.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler V, Rotzer V, Dehner C, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest. 2013 doi: 10.1172/JCI60139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Engel K, Campbell DG, et al. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313:307–313. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- Waschke J, Spindler V, Bruggeman P, et al. Inhibition of Rho A activity causes pemphigus skin blistering. Journal of Cell Biology. 2006;175:721–727. doi: 10.1083/jcb.200605125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L, Raess NA, Caldelari R, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25:3298–3309. doi: 10.1038/sj.emboj.7601224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SW, Cavacini LA, Bhol KC, et al. Pathogenic human monoclonal antibody against desmoglein 3. Clin Immunol. 2006;120:68–75. doi: 10.1016/j.clim.2006.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.