

Abstract
Despite rapid progress in anticancer drug development and improvement in clinical outcomes, the survival rate for many types of cancer is still unacceptably low. Therefore, it is crucial to discover novel anticancer drugs to both prevent and treat the disease. In recent years, the advent of combinatorial chemistry allows the design and parallel synthesis of millions of small compounds that have drug-like properties. In vitro high throughput screening of such compound libraries has allowed the identification of many new drug candidates that may be further evaluated for their efficacy and mechanism of action. The overall objective of this study was to identify small molecule compounds as candidates for anti-cancer drug development. We first used cell proliferation and cytotoxicity assays to identify compounds exhibiting anti-cancer activity in vitro in a leukemia cell line (K562). Six top compounds selected from the initial screening of a library of 2,560 compounds were further evaluated in multiple cancer cell lines to rank the drug candidates. The top candidate was further investigated to elucidate the molecular mechanism underlying its anticancer activity. Our studies suggest that this piperazine derivative effectively (GI50 = 0.06-0.16 μM) inhibits cancer cell proliferation and induces caspase-dependent apoptosis via inhibiting multiple cancer signaling pathways including the PI3K/AKT, the Src family kinases and the BCR-ABL pathways.
Keywords: Drug discovery, high throughput screening, apoptosis, piperazine, cancer, leukemia
Introduction
Cancer is a group of diseases that is characterized by uncontrollable cell growth. Cancer accounts for 1 in every 4 deaths in the United States [1,2]. Thus it is not surprising that cancer is the second leading cause of death in the US., with an estimated 577,000 Americans having died in 2012 due to some form of cancer [1]. In the year 2012 alone, there were an estimated 1,638,000 new cases of cancer in the United States [1]. Although survival rates for cancer are improving, the 5-year survival rate for all types of cancer diagnosed between 2001 and 2007 is only 67% [1]. Assuming the same percentage, 540,000 Americans who were diagnosed with cancer in 2012 will not live to see 2018. Therefore, it is crucial to discover novel anticancer compounds to both prevent and treat cancer.
Previous generations of drugs for cancer treatment were discovered largely from a limited number of chemical entities and these drugs usually target the general cellular machinery related to cell proliferation [3-6]. Such therapies are toxic to not only cancer cells but also normal fast-growing cells. In recent years, two major paradigm shifts have occurred in drug discovery. First, the advent of combinatorial chemistry allows the design and parallel synthesis of millions of small chemical compounds that have drug-like properties [7-12]. In vitro high throughput screening (HTS) of such compound libraries has allowed scientists to identify a large number of candidate compounds that may be further evaluated for their efficacy and toxicity, greatly speeding up the development of new drugs. Second, the advent of high throughput genomic and proteomic technologies has generated large numbers of new molecules that may be targeted for drug development. The new drug targets may be specifically altered only in cancer cells, allowing development of drugs specially targeted towards cancer cells.
The overall objective of this study was to identify small molecule compounds synthesized through combinatorial chemistry as candidates for anti-cancer drug development. We first used a cell proliferation assay to identify compounds exhibiting anti-cancer activity in vitro in a leukemia cell line (K562). Top compounds selected from the initial screening of a library of 2,560 compounds were further evaluated in multiple cancer cell lines to rank the drug candidates. A top candidate was further investigated to elucidate the molecular mechanism of action underlying its anticancer activity. Our studies suggest that this piperazine derivative effectively inhibits cancer cell proliferation and induces caspase-dependent apoptosis via inhibiting multiple signaling pathways implicated in cancer.
Materials and methods
Cell culture
Cancer cell lines K562 (human chronic myelogenous leukemia (CML) cells), HeLa (cervical cancer cells), and AGS (gastric adenocarcinoma cells) were obtained from the American Type Culture Collection (ATCC). K562, HeLa, and AGS cells were cultured in RPMI 1640 medium (Invitrogen) with 10% Fetal Bovine Serum and 1% antibiotics (PSA). Cells were incubated in 5% CO2 at 37°C and were given medium and plate changes as needed.
Drug library and screening
A library of 2,560 small compounds was purchased from ChemBridge Corporation (California). One-hundred nanoliters of drug was transferred into 100 μl of culture medium in 96-well plates (Thermo Scientific) using a Quadra 3 robotic system (Tomtec, Inc.) and the final concentration was approximately 10 μM for each drug. K562 cells were then added into the wells at a density of 3,000 cells/well. Plates were incubated for three days at 37°C and 5% CO2. Viable cell number was assessed using a Dojindo CCK-8 kit (Dojindo Molecular Technologies, Inc.) according to manufacturer’s instructions. Plates were read using a Synergy HT Microplate Reader (BioTek Instruments, Inc.), and the optical density (OD) values were recorded.
OD values were converted to cell numbers by comparing the experimental OD values to OD values for known cell numbers. For this purpose, a standard curve was constructed for each cell type. Briefly, cells were counted and diluted to 5 × 105 cells/mL. A total of 100 μL (5 × 104 cells) were seeded into the first well of a 96-well plate. Each successive well was seeded with half as many cells as the previous well and supplemented with RPMI medium to a total volume of 100 μL. Plates were read using the same procedure as the experimental assays. A standard curve was constructed using an excel spreadsheet. OD value for each experimental well was converted to cell number based on the standard curve. Growth inhibition was calculated by first subtracting the original number of cells seeded (3,000), and then using the following formula: GI = (control cell number – experimental cell number)/control cell number.
Evaluation of 50% growth inhibition (GI50) by proliferation assay
Cancer cells (K562, HeLa, and AGS) were prepared and diluted to a concentration of 3.5 × 104 cells/mL. The center 6 × 10 wells of the 96-well plates were seeded with 100 μL of the cell suspension, totaling 3.5 × 103 cells/well. The border wells of the 96-well plates were seeded with 150 μL of autoclaved water to minimize evaporation of the center wells. The plates were incubated in 5% CO2 at 37°C overnight, giving ample time for adherent cell lines to attach to the plates. The following day, 1 μL of the prepared compound dilutions (2-fold dilutions) was added to each well, so that the highest final concentration was 10 μM with 5 successive 2x dilutions. Plates were then incubated for another three days. A Dojindo CCK-8 kit was used to estimate the number of viable cells in each well according to the manufacturer’s instructions. The data was then analyzed to construct a GI50 curve and calculate the GI50 value using Sigmaplot 10.0 software.
Analysis of apoptosis and DNA contents
K562 cells were prepared and seeded in 24-well multidishes (Thermo Scientific) at a concentration of 5 × 104 cells/well in 2 mL RPMI medium. Compounds were prepared and added to the wells at a concentration of 5 μM. Cells were incubated in 5% CO2 at 37°C for 24 hours. Then, cells were collected and washed twice with PBS, centrifuged, and the supernatant was removed. Cells were resuspended in 1x Annexin V Binding Buffer (BD Biosciences). One hundred microliters of the suspension was transferred to a 5 mL polystyrene round-bottom tube (BD Biosciences). Five microliters of PE Annexin V (BD Pharmingen) and 5 μL of 7-AAD was added to the suspension. The suspension was gently mixed and incubated for 15 minutes in the dark at room temperature. An additional 400 μL of binding buffer was added to each tube. Samples were analyzed by flow cytometry using FACSDiva Version 6.1 software (BD Biosciences).
K562 cells were also analyzed for DNA contents by propidium iodide (PI) staining. Briefly, cells were seeded in 6-well multidishes (Thermo Scientific) at a concentration of 1 × 106 cells/well in 3 mL of RPMI medium. Cells were incubated in 5% CO2 at 37°C. Samples were collected at 24 hour time intervals for 3 days. Cell suspensions were transferred to a 5 mL polystyrene round-bottom tube (BD Biosciences), centrifuged, and had the supernatant removed. Cells were washed with phosphate-buffered saline (PBS). Afterwards, cells were fixed with 70% ethanol and stored in 4°C until assay. Samples were then centrifuged and ethanol was discarded. A 10x Permeabilization Buffer (eBioscience) was diluted to 1x in deionized water and added to each sample. Samples were gently vortexed before being centrifuged. The supernatant was removed and cells were treated with a PI/RNase Staining Buffer (BD Pharmingen) and incubated for 15 minutes at room temperature in darkness. Cells were analyzed for DNA contents using flow cytometry and data was analyzed using FACSDiva Version 6.1 software (BD Biosciences).
Western blotting
Cells were harvested, washed with ice-cold PBS, and lysed in M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) supplemented with 1% Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) for 15 minutes. Samples were then centrifuged at 13,000 rpm for 10 minutes at 4°C and the supernatant was collected. Protein concentration was measured using a Nanodrop 1000 Spectrophotometer (Thermo Scientific) and adjusted with cell lysis buffer. Proteins were separated by SDS-PAGE and transferred to PVDF membranes through electroblotting. Membranes were then blocked in Tris-Buffered Saline (TBS) supplemented with 2% non-fat dry milk and 0.1% Tween 20. Membranes were incubated overnight at 4°C with primary antibodies (1:1000 dilution or as recommended) in TBS supplemented with 5% w/v Bovine Serum Albumin (BSA) and 0.1% Tween 20, washed with a TBST buffer (TBS supplemented with 0.1% Tween 20) three times over 25 minutes, and incubated for one hour with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (Santa Cruz Biotechnology) at a dilution of 1:10,000 in TBST buffer. Finally, membranes were washed again three times over 25 minutes. Blots were developed with an enhanced chemiluminescent substrate (Pierce ECL Substrate; Thermo Scientific). Immunopositive bands were photographed (Versadoc; Bio-Rad) on X-ray films obtained from Phenix Research Products. Antibodies recognizing phospho-Lyn (04-375) were obtained from Millipore. Antibodies to Crk-L (sc-319), STAT5 (sc-835), c-Abl (sc-887), and GAPDH (sc-25778) were obtained from Santa Cruz Biotechnology. Antibodies to Akt (9272), phospho-Akt (9271), Bcl-2 (4223), Bcl-xl (2764), Caspase-3 (9662), cleaved Caspase-3 (9664), PARP (9542), PI3 Kinase p85 (4257), Caspase-8 (4790), cleaved Caspase-8 (9496), Src (2123), GSK-3β (9315), phospho-c-Abl (2861), phospho-STAT5 (9351), Lyn (2796), phospho-CrkL (3181), and β-Actin (4970) were obtained from Cell Signaling Technology.
Results
High throughput screening of compound library
In order to identify compounds with potential anti-cancer activity, we used a high-throughput cell proliferation assay to test 2,560 compounds with drug-like properties. This assay allows for rapid in vitro screening of large numbers of compounds in 96-well plates. K562 cells were cultured with compounds at a final concentration of 10 μM for 72 hours and cell numbers were then estimated using a Dojindo CCK-8 kit. To evaluate the activity of each compound, the number of cells in the presence of compound was compared to the cell numbers without compounds to calculate the percentage of growth inhibition (GI) as described in the method section. In summary, 92.9% of the tested compounds had little to no activity (GI = 0-20%) against K562 cells, while 32 compounds (1.25% of 2560) had potent inhibitory activity (GI > 80%) against K562 cells (Figure 1). These results were then used to select compounds for further evaluations.
Figure 1.
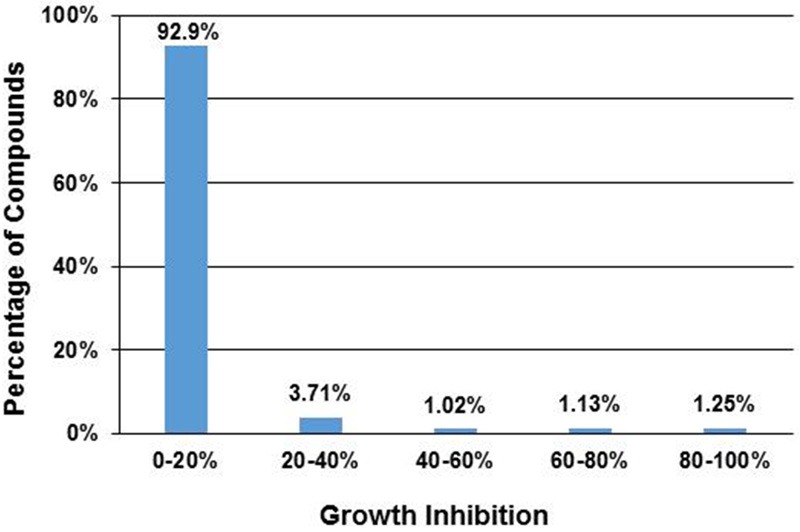
High throughput screening of drug-like compounds for their anti-cancer activity. A total of 2,560 compounds were assayed using a cell proliferation assay with K562 cells. Shown are the percentages of compounds with different ranges of growth inhibition.
Secondary evaluation of nine selected compounds using 3 cancer cell lines
Six compounds with potent inhibitory activity (GI > 80%) and three compounds with less inhibitory activity (GI < 80%) in the initial screening assay were selected for further evaluation. These nine compounds were tested against 3 different cancer cell lines (HeLa, K562, and AGS) to confirm their efficacy at different drug concentrations. Serial dilutions (10 μM, 5 μM, 2.5 μM, 1.25 μM, 0.63 μM, and 0.31 μM) were used to determine the growth inhibition. As expected, the three compounds exhibiting little inhibitory activity in the original screening assay (C501, C503 and C506) exhibited little inhibitory activity in the confirmation tests against all 3 cell lines, although C506 exhibited good inhibition at the highest concentration (10 μM) (Figure 2). The other six compounds that exhibited potent inhibitory activity in the original screens also exhibited potent inhibitory activity at the 10 μM concentration on all 3 cell lines in the confirmation studies (Figure 2). However, five of these compounds exhibited significantly lower inhibitory activity at lower drug concentrations. For HeLa and K562 cells, only C505 and C508 exhibited greater than 50% inhibition at the lowest concentration tested in this study (0.31 μM). C505 also had much higher potency (GI > 90% for all three cell lines) than C508 (GI = 58% for HeLa and 67% for K562) (Figure 2). Therefore, C505 is the most potent compound for all three cell lines.
Figure 2.
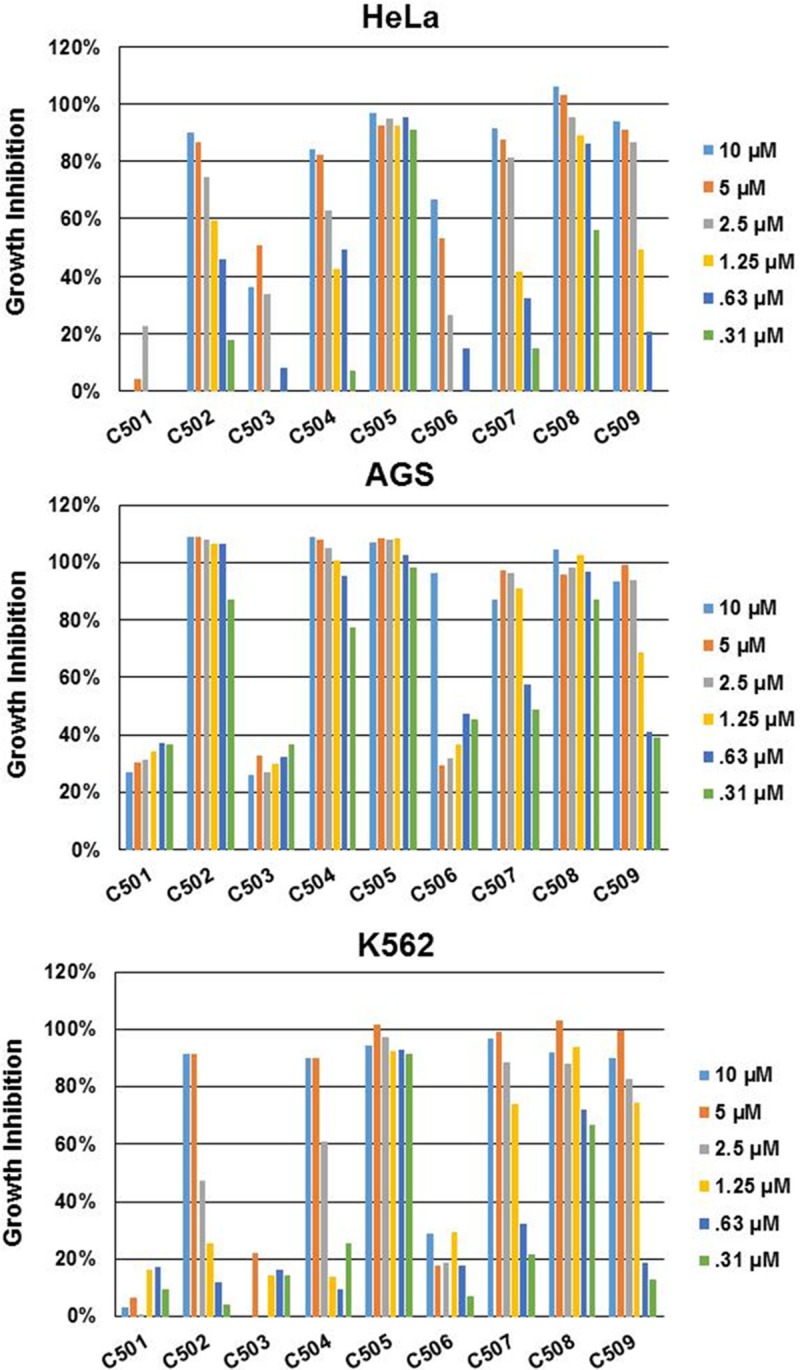
Growth inhibition of nine compounds against three different cancer cell lines. Six different concentrations were tested for each compound. Growth inhibition varies according to compounds, compound concentration, and cell lines. Generally, growth inhibition decreases as compound concentrations decreases.
Determination of GI50 for compound C505
A wider range of drug dilutions was used to construct a growth inhibition (GI) curve and determine the GI50 value, which is the concentration required to achieve 50% growth inhibition. GI curves generally follow a logistic distribution, characterized by the S-shaped curve they resemble. As shown in Figure 3, a concentration below 0.01 μM results in practically no inhibition. On the higher end of drug concentration, the curve plateaus around 3 μM, achieving maximum inhibition (100%). Inhibition steadily increases with drug concentration between the extremes. The point where inhibition reaches 50% is denoted the GI50 value and is often used as a measure of potency. These studies further confirmed the efficacy of C505 through the identification of the GI50 value for each cell line: HeLa = 0.155 μM; K562 = 0.058 μM; AGS = 0.055 μM (Figure 3). These results suggest that C505 has very potent anti-cancer activity and is an excellent anti-cancer drug candidate.
Figure 3.
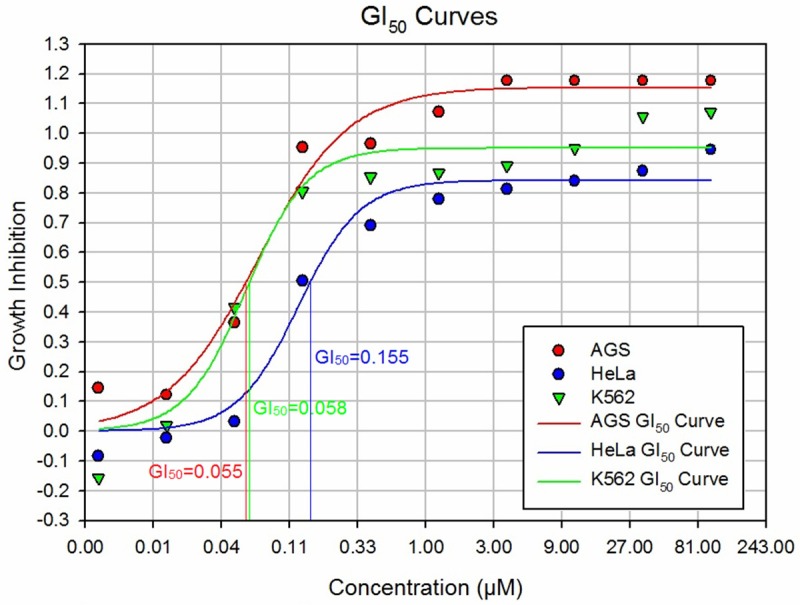
Growth inhibition curves and GI50 values for compound C505. Nine different concentrations of C505 were used to treat three different cancer cell lines for 72 hours to establish growth inhibition curves (plots of growth inhibition by compound concentration). The GI curves allow the estimation of GI50 value, which is the compound concentration required to achieve 50% inhibition.
C505 kills cancer cells via induction of apoptosis
Having confirmed C505’s efficacy towards multiple cancer cell lines, further studies were carried out to elucidate the mechanism underlying the anticancer activity of C505. Apoptosis and necrosis are two major types of cell death. Necrosis is the premature death of cells caused by factors external to the cell or tissue such as infection, toxins, or trauma. Necrosis results in membrane disruption, cell swelling and rupture. Apoptosis is the process of programmed cell death which involves a series of biochemical events leading to cell destruction and death. It can prevent tumor formation by achieving homeostasis between cell death rate and mitosis rate. Apoptosis leads to cell membrane blebbing, shrinkage of cell, nuclear collapse (nuclear fragmentation, chromatin condensation, chromosomal DNA fragmentation), and apoptotic body formation. Annexin V is a 35-36 kDa Ca2+ dependent phospholipid-binding protein that has a high affinity for the plasma membrane phospholipid phosphatidylserine (PS), and binds to cells with exposed PS, one of the earliest features of apoptosis. Annexin V staining precedes the loss of membrane integrity which accompanies the latest stages of cell death resulting from either apoptotic or necrotic processes. Therefore, staining with Annexin V is typically used in conjunction with a DNA-binding dye such as 7-amino-actinomycin (7-AAD) [13], which is excluded by viable cells with intact membranes, while the membranes of dead or damaged cells are permeable to 7-AAD. Therefore, viable cells are 7-AAD and Annexin V negative; early apoptotic cells are Annexin V positive and 7-AAD negative, while cells in late apoptosis and dead cells are both Annexin V and 7-AAD positive. This technique was used to investigate cell death as a result of C505 treatment. For these experiments, K562 cells were treated with C505 and imatinib at a concentration of 5 μM for 24, 48 and 72 hours. As shown in Figure 4, the vast majority of the untreated control cells stained negative for both 7-AAD and Annexin V at all three time points, indicating that the cells are viable. However, after treatment with C505 or a positive control drug (imatinib) for 48 or 72 hours, a significantly higher proportion of cells are undergoing early apoptosis (positive for Annexin V and negative for 7-AAD) and an increasingly higher proportion of treated cells become double positive for Annexin V and 7-AAD (late apoptotic or dead cells). The results suggest that C505 and imatinib both kill K562 cells via induction of apoptosis.
Figure 4.
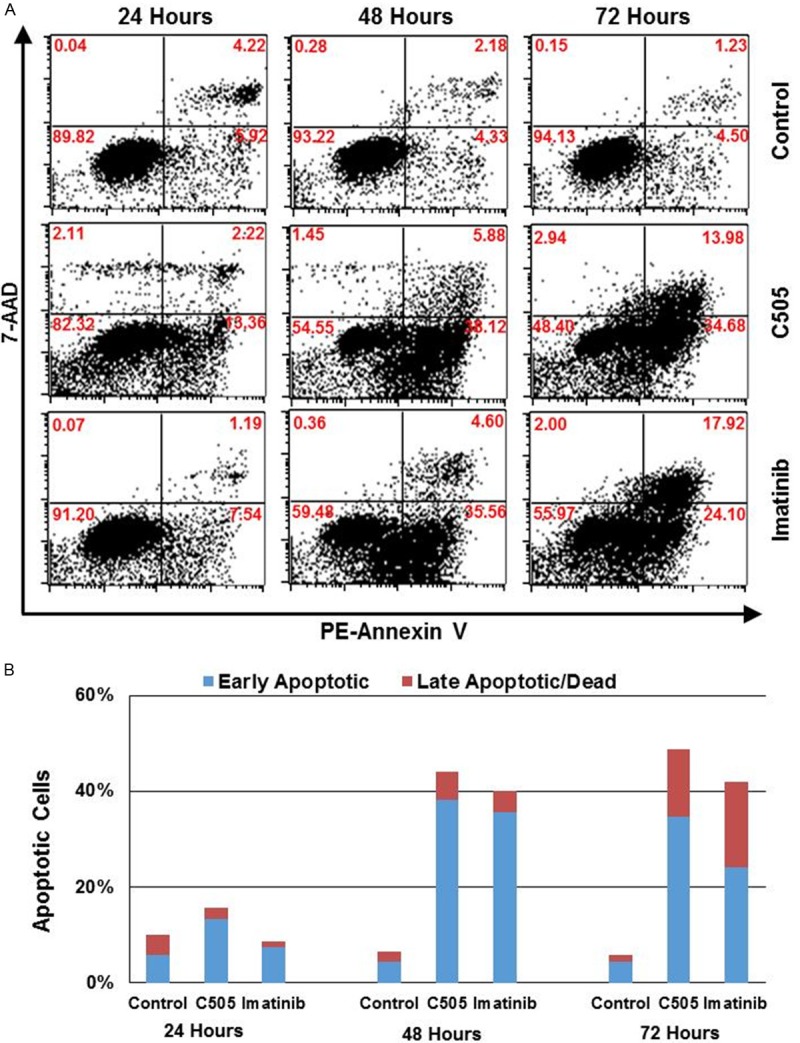
Annexin V and 7-AAD double staining. K562 cells were treated with 5 μM of C505 for 24, 48 and 72 hours before staining. A. Flow cytometry profiles showing staining of cells for Annexin V (x-axis) and 7-AAD (y-axis). The number in each quadrant shows the percentage of cells that are located in that quadrant. Lower left quadrant (double negative) cells are viable cells; lower right quadrant (Annexin V positive and 7-AAD negative) cells are early apoptotic cells; and upper right quadrant (double positive) cells are late apoptotic or dead cells. B. Bar chart presentation of the data shown in Figure 4A. Shown are the percentages of early apoptotic cells (blue) and late apoptotic or dead cells (red).
Propidium iodide (PI) is an intercalating agent and a fluorescent molecule that can be used to stain cells. PI can be used as a DNA stain for both flow cytometry, to evaluate DNA content in cell cycle analysis and cell viability, and microscopy to visualize DNA containing organelles such as the nucleus. PI can be used to differentiate viable, apoptotic and necrotic cells [14]. PI is the most commonly used dye to quantitatively assess DNA content. In this study, K562 cells were treated with 5 μM of compound C505 for 24, 48 and 72 hours, in addition to an untreated control (0 hour). PI staining reveals three peaks that mark sub-G0/G1 phase cells (apoptotic cells), G0/G1 phase cells, and S/G2/M phase cells (Figure 5). Comparing to the untreated cells, the treated cells clearly have different PI staining profiles that change over the course of treatment. At 24 hours, most cells (74.2%) are arrested at the S/G2/M (74.2%) phase, higher than the untreated cells (52%). As duration of treatment lengthened to 48 and 72 hours, a new peak arose in the region of the G0/G1 gate that was not seen in untreated cells. These results indicate that C505 can induce cell cycle arrest. The untreated cells exhibited minimal apoptotic activity (2%), while cells treated with C505 exhibited steadily increasing apoptotic activity: 22.0% at 24 hours, 31.2% at 48 hours, and 37.9% at 72 hours (Figure 5), further confirming our finding that C505 kills cancer cells via apoptosis.
Figure 5.
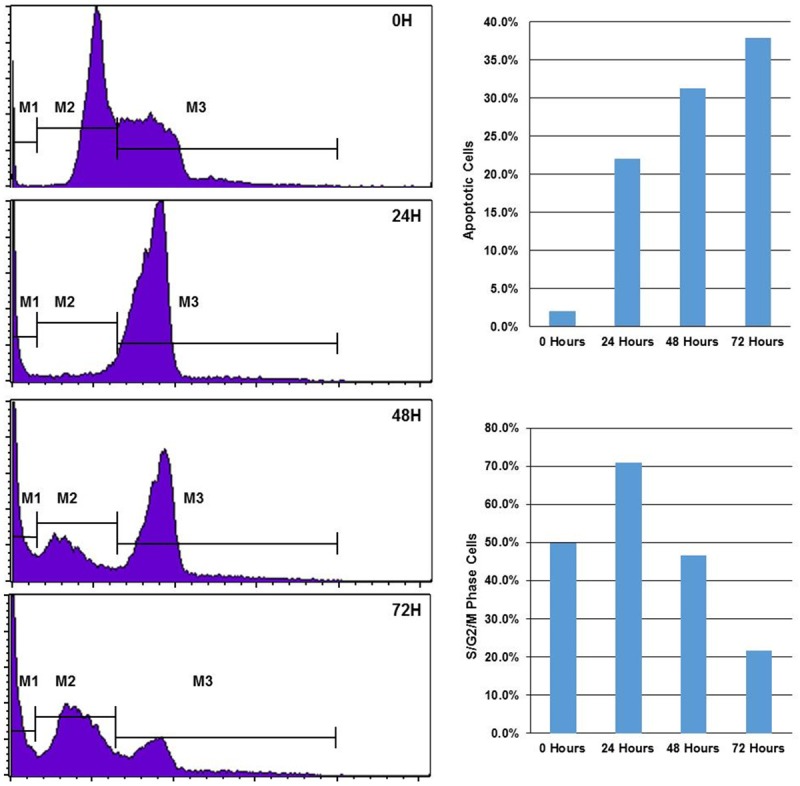
Propidum iodide (PI) staining for apoptosis and cell cycle analysis. K562 cells were treated with 5 μM of C505 for 0, 24, 48 and 72 hours before staining with PI. After treatment, cells were analyzed using flow cytometry. Shown on the left panel are frequencies of cells with increasing PI intensity (or DNA content) from left to right. The cells can generally be classified into three groups corresponding to different cell cycles and viability. Cells in the M1 gate are apoptotic cells (sub G0/G1); cells in the M2 gate are cells at early cell cycle before DNA replication (G0/G1 phase); and cells at the M3 gate are cells at the late stage of cell cycle with active DNA synthesis (S/G2/M). Bar charts on the right panel summarize the percentage of apoptotic cells (M1 gate) and late cell cycle cells (S/G2/M) at different treatment time points.
C505 induces caspase-dependent apoptosis
Next, we attempted to elucidate the apoptotic pathways induced by C505 treatment. Caspases are proteolytic enzymes that play critical roles in apoptosis. In this study we used Western blotting techniques to determine the expression levels of several important caspases and the cleaved (or activated) caspases. As shown in Figure 6A, caspase-8 and caspase-3 are clearly activated through proteolytic cleavage, suggesting that C505 kills cancer cells through induction of caspase-dependent apoptosis. Poly (ADP-ribose) polymerase (PARP) is activated by caspase cleavage. Activation of PARP expedites cellular disassembly by rapid catalysis of NAD+ and subsequent ATP depletion. It is also a robust indicator of cells undergoing apoptosis [15,16]. PARP is significantly altered after 48 h treatment time point, which is consistent with the apoptotic phenotype observed in this study. However, C505 treatment did not significantly alter the protein expression of two anti-apoptotic members of the Bcl family (namely Bcl-2 and Bcl-xl) (Figure 6A).
Figure 6.
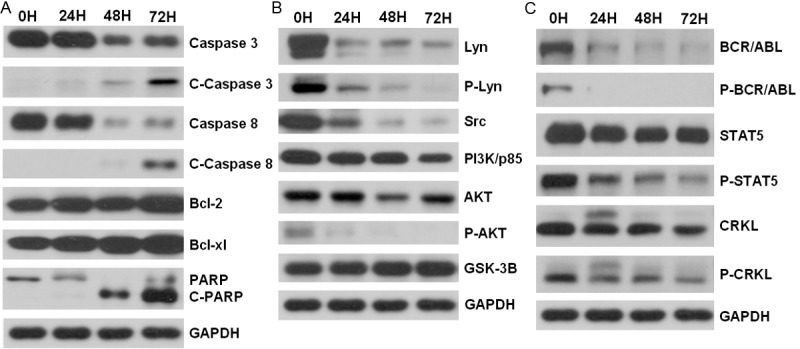
Western blotting analyses of proteins after C505 treatment. K562 cells were treated with 5 μM of C505 for 0, 24, 48 and 72 hours and cell extracts were prepared. GAPDH was used to indicate equal amount of protein for each preparation. A. Analyses of proteins involved in apoptosis. C-caspase 3 = cleaved caspase-3, C-caspase 8 = cleaved caspase-8, C-PARP = cleaved PARP. B. Proteins involved in cell proliferation and apoptosis pathways. P-Lyn and P-AKT indicate phosphorylated Lyn and AKT, respectively. C. Western blotting for the BCR-ABL signaling pathway.
Signaling pathways inhibited by C505
Cell proliferation and apoptosis are two opposing but highly related biological processes that share common molecular signaling pathways. In order to understand how C505 inhibits cancer cell proliferation and induces apoptosis, we further investigated the signaling proteins that were potentially involved in these processes using Western blotting. As shown in Figure 6B, C505 treatment significantly reduced the levels of Src and phosphorylated AKT (p-AKT), two critical signaling pathways involved in cell proliferation and apoptosis. C505 treatment slightly reduced the levels of PI3K/p85 but did not significantly alter the expression of GSK-3B.
We also examined the proteins in the signaling of BCR-ABL, which is a fusion kinase that only occurs in CML patients. As shown in Figure 6C, C505 is highly effective in reducing the expression of the BCR-ABL protein and completely eliminated phosphorylated BCR-ABL. Consistent with these findings, the signaling cascade down-stream of BCR-ABL signaling is also blocked as indicated by the reduction of phosphorylated STAT5 and phosphorylated CRKL.
Discussion
In this study, we identified one compound with potent anti-cancer activity from a library of 2,560 potential drug compounds. The selected compound C505 exhibited toxicity at very low concentrations (GI50 < 0.16 μM) against all three cancer cell lines (K562, HeLa, and AGS) tested. C505 is a piperazine-containing compound that was shown in this study for the first time to possess potent anticancer activity. Piperazine consists of a six-membered ring containing two nitrogen atoms at opposite positions in the ring [17,18]. The piperazines are a large class of chemical compounds, many of which have important pharmacological properties. Many currently notable drugs contain a piperazine ring as part of their molecular structure. Examples include antianginals, antidepressants,antihistamines, antipsychotics, urologicals,and recreational drugs. Indeed, imatinib, which has potent anti-leukemia activity and is used as a positive control drug in this study, is a piperazine derivative [19,20]. Given the high potency of C505 and the potentially excellent pharmacological properties, C505 is an excellent candidate that may be further developed into an anticancer drug.
A series of experiments was also conducted to determine the cellular and molecular mechanism underlying the killing activity of C505. Annexin V/7-AAD staining as well as PI staining indicates that C505 can kill cancer cells via the induction of cell cycle arrest and apoptosis. Furthermore, Western blotting analyses suggest that C505-induced apoptosis is caspase-dependent as several caspases including caspase-8 and caspase-3 are activated by C505 treatment (Figure 6A).
In order to understand how C505 induces caspase-dependent apoptosis, we investigated several cell proliferation and apoptosis signaling pathways commonly implicated in cancer. Our studies indicate that multiple signaling pathways are altered after C505 treatment. The PI3K-AKT pathway is an intracellular signaling pathway critical in cell proliferation and apoptosis and hence in cancer [21-24]. The PI3K/AKT pathway is activated by receptor tyrosine kinases (RTK). The PI3K p85beta protein is down-regulated by C505 treatment, while AKT phosphorylation is severely reduced by C505 treatment (Figure 6B). These results suggest that inhibition of the PI3K/AKT pathway may be a major mechanism through which C505 inhibits cancer cell proliferation and induces apoptosis. In many cancers, the PI3K/AKT pathway is overactive, allowing proliferation of cancer cells [21-23]. A number of experimental anti-cancer drugs aim to inhibit this signaling pathway. Our results suggest that C505 is an effective inhibitor of the PI3K/AKT pathway.
Src family kinases are a family of non-receptor tyrosine kinases with nine members: Src, Yes, Fyn, Fgr, Lck, Hck, Blk, Lyn and Frk [25,26]. Src family kinases interact with many cellular cytosolic, nuclear and membrane proteins, modifying these proteins by phosphorylation of tyrosine residues [27-29]. These kinases participate in signaling pathways that control a diverse spectrum of biological activities including cell cycle progression, apoptosis, cell adhesion, migration, and transformation. There are functional redundancies between members of the Src kinase family. In this study, we analyzed two Src family kinases (Src and Lyn) commonly implicated in various cancers and our results indicate that C505 can dramatically reduce the expression levels of both kinases as well as phosphorylated Lyn (Figure 6B). Src is one of the primary kinases activated following engagement of receptors and plays a role in the activation of other protein tyrosine kinase families. Src plays a role in PDGF-mediated tyrosine phosphorylation of both STAT1 and STAT3, leading to increased DNA binding activity of these transcription factors. It is involved in the RAS pathway that is a critical cell proliferation pathway.
Lyn, depending on the cellular context, activates or inhibits several signaling cascades. It regulates PI3K and AKT activation [30,31]. It also regulates activation of the MAP kinase signaling cascade [31-34]. Lyn can also mediate activation of STAT5A and STAT5B [35]. Consistent with the reduced Lyn expression after C505 treatment, STAT5 phosphorylation is also significantly reduced after C505 treatment.
BCR-ABL is a kinase resulted from the fusion of two different proteins, BCR and ABL1, a phenomenon that only occurs in CML patients. It has been shown that the formation of this fusion kinase is responsible for CML and inhibition of the BCR-ABL kinase activity by specific inhibitors such as imatinib is highly effective in treating CML [35,36]. Since C505 has potent activity against the CML cell line K562, we examined whether C505 could also reduce the expression or inhibit the activity of BCR-ABL. Our results indicate that the total BCR-ABL protein and phosphorylated BCR-ABL are both dramatically reduced by C505 treatment. Furthermore, the substrates of BCR-ABL such as phosphorylated STAT5 and phosphorylated CRKL are both reduced after C505 treatment. These results suggest that inhibition of the BCR-ABL pathway is another mechanism that can lead to inhibition of cancer cell proliferation and induction of apoptosis after C505 treatment.
Conclusion
In conclusion, we discovered a novel piperazine compound with potent anticancer activity through high throughput screening of a drug-like compound library. This compound effectively inhibits cancer cell proliferation and induces caspase-dependent apoptosis via inhibition of multiple cancer signaling pathways including PI3K-AKT, Src family kinases and the BCR-ABL signaling pathways.
Acknowledgements
The authors wish to thank Bing Yi, Haitao Liu, Hongfang Yu, Bo Chen and Wei Xiao for their technical support and advice.
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Hermeking H. The MYC oncogene as a cancer drug target. Curr Cancer Drug Targets. 2003;3:163–175. doi: 10.2174/1568009033481949. [DOI] [PubMed] [Google Scholar]
- 4.Holtick U, Vockerodt M, Pinkert D, Schoof N, Sturzenhofecker B, Kussebi N, Lauber K, Wesselborg S, Loffler D, Horn F, Trumper L, Kube D. STAT3 is essential for Hodgkin lymphoma cell proliferation and is a target of tyrphostin AG17 which confers sensitization for apoptosis. Leukemia. 2005;19:936–944. doi: 10.1038/sj.leu.2403750. [DOI] [PubMed] [Google Scholar]
- 5.Theodoropoulou M, Stalla GK. Somatostatin receptors: From signaling to clinical practice. Front Neuroendocrinol. 2013;34:228–252. doi: 10.1016/j.yfrne.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi-Yanaga F. Activator or inhibitor? GSK-3 as a new drug target. Biochem Pharmacol. 2013;86:191–199. doi: 10.1016/j.bcp.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 7.He W, She PW, Fang Z, Guo K. [The research progress of dynamic combinatorial chemistry] . Yao Xue Xue Bao. 2013;48:814–823. [PubMed] [Google Scholar]
- 8.Chaguturu R. Combinatorial Chemistry & High Throughput Screening. Editorial. Comb Chem High Throughput Screen. 2013;16:1. doi: 10.2174/1386207311316010001. [DOI] [PubMed] [Google Scholar]
- 9.Cougnon FB, Sanders JK. Evolution of dynamic combinatorial chemistry. Acc Chem Res. 2012;45:2211–2221. doi: 10.1021/ar200240m. [DOI] [PubMed] [Google Scholar]
- 10.Kodadek T. The rise, fall and reinvention of combinatorial chemistry. Chem Commun (Camb) 2011;47:9757–9763. doi: 10.1039/c1cc12102b. [DOI] [PubMed] [Google Scholar]
- 11.Xiang J, Yang H, Che C, Zou H, Wei Y, Quan J, Zhang H, Yang Z, Lin S. Identifying tumor cell growth inhibitors by combinatorial chemistry and zebrafish assays. PLoS One. 2009;4:e4361. doi: 10.1371/journal.pone.0004361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wildonger RA, Deegan TL, Lee JW. Is combinatorial chemistry on the right track for drug discovery? J Autom Methods Manag Chem. 2003;25:57–61. doi: 10.1155/S1463924603000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Chen H, Patel DJ. Solution structure of actinomycin-DNA complexes: drug intercalation at isolated G-C sites. J Biomol NMR. 1991;1:323–347. doi: 10.1007/BF02192858. [DOI] [PubMed] [Google Scholar]
- 14.Lecoeur H. Nuclear apoptosis detection by flow cytometry: influence of endogenous endonucleases. Exp Cell Res. 2002;277:1–14. doi: 10.1006/excr.2002.5537. [DOI] [PubMed] [Google Scholar]
- 15.Uchida M, Hanai S, Uematsu N, Sawamoto K, Okano H, Miwa M, Uchida K. Genetic and functional analysis of PARP, a DNA strand break-binding enzyme. Mutat Res. 2001;477:89–96. doi: 10.1016/s0027-5107(01)00110-5. [DOI] [PubMed] [Google Scholar]
- 16.Bursztajn S, Feng JJ, Berman SA, Nanda A. Poly (ADP-ribose) polymerase induction is an early signal of apoptosis in human neuroblastoma. Brain Res Mol Brain Res. 2000;76:363–376. doi: 10.1016/s0169-328x(00)00026-7. [DOI] [PubMed] [Google Scholar]
- 17.Yevich JP, New JS, Smith DW, Lobeck WG, Catt JD, Minielli JL, Eison MS, Taylor DP, Riblet LA, Temple DL Jr. Synthesis and biological evaluation of 1-(1,2-benzisothiazol-3-yl)- and (1,2-benzisoxazol-3-yl)piperazine derivatives as potential antipsychotic agents. J Med Chem. 1986;29:359–369. doi: 10.1021/jm00153a010. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Joseph DB, Bowen WD, Flippen-Anderson JL, Dersch CM, Rothman RB, Jacobson AE, Rice KC. Synthesis and biological evaluation of tropane-like 1-[2-[bis(4-fluorophenyl)methoxy] ethyl] -4-(3-phenylpropyl)piperazine (GBR 12909) analogues. J Med Chem. 2001;44:3937–3945. doi: 10.1021/jm0101592. [DOI] [PubMed] [Google Scholar]
- 19.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 20.Majsterek I, Sliwinski T, Poplawski T, Pytel D, Kowalski M, Slupianek A, Skorski T, Blasiak J. Imatinib mesylate (STI571) abrogates the resistance to doxorubicin in human K562 chronic myeloid leukemia cells by inhibition of BCR/ABL kinase-mediated DNA repair. Mutat Res. 2006;603:74–82. doi: 10.1016/j.mrgentox.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 22.Cortot A, Armand JP, Soria JC. [PI3K-AKT-mTOR pathway inhibitors] . Bull Cancer. 2006;93:19–26. [PubMed] [Google Scholar]
- 23.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Paul R, Herbert JM, Maffrand JP, Lansen J, Modat G, Pereillo JM, Gordon JL. Inhibition of vascular smooth muscle cell proliferation in culture by pentosan polysulphate and related compounds. Thromb Res. 1987;46:793–801. doi: 10.1016/0049-3848(87)90071-5. [DOI] [PubMed] [Google Scholar]
- 25.Bergman M, Mustelin T, Oetken C, Partanen J, Flint NA, Amrein KE, Autero M, Burn P, Alitalo K. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 1992;11:2919–2924. doi: 10.1002/j.1460-2075.1992.tb05361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun G, Budde RJ. Expression, purification, and initial characterization of human Yes protein tyrosine kinase from a bacterial expression system. Arch Biochem Biophys. 1997;345:135–142. doi: 10.1006/abbi.1997.0236. [DOI] [PubMed] [Google Scholar]
- 27.Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–678. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 29.Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, Mirosevich J, Lee FY, Jove R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9189. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 30.Iqbal MS, Tsuyama N, Obata M, Ishikawa H. A novel signaling pathway associated with Lyn, PI 3-kinase and Akt supports the proliferation of myeloma cells. Biochem Biophys Res Commun. 2010;392:415–420. doi: 10.1016/j.bbrc.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 31.Liu WH, Chang LS. Suppression of ADAM17-mediated Lyn/Akt pathways induces apoptosis of human leukemia U937 cells: Bungarus multicinctus protease inhibitor-like protein-1 uncovers the cytotoxic mechanism. J Biol Chem. 2010;285:30506–30515. doi: 10.1074/jbc.M110.156257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Congleton J, MacDonald R, Yen A. Src inhibitors,PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 2012;26:1180–1188. doi: 10.1038/leu.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su N, Peng L, Xia B, Zhao Y, Xu A, Wang J, Wang X, Jiang B. Lyn is involved in CD24-induced ERK1/2 activation in colorectal cancer. Mol Cancer. 2012;11:43. doi: 10.1186/1476-4598-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sainz-Perez A, Gary-Gouy H, Portier A, Davi F, Merle-Beral H, Galanaud P, Dalloul A. High Mda-7 expression promotes malignant cell survival and p38 MAP kinase activation in chronic lymphocytic leukemia. Leukemia. 2006;20:498–504. doi: 10.1038/sj.leu.2404073. [DOI] [PubMed] [Google Scholar]
- 35.Kim BH, Won C, Lee YH, Choi JS, Noh K, Han S, Lee H, Lee CS, Lee DS, Ye SK, Kim MH. Sophoraflavanone G induces apoptosis of human cancer cells by targeting upstream signals of STATs. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.08.009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 36.Qin YZ, Jiang Q, Jiang H, Li JL, Li LD, Zhu HH, Lai YY, Lu XJ, Liu YR, Jiang B, Huang XJ. Which method better evaluates the molecular response in newly diagnosed chronic phase chronic myeloid leukemia patients with imatinib treatment, BCR-ABL(IS) or log reduction from the baseline level? Leuk Res. 2013;37:1035–1040. doi: 10.1016/j.leukres.2013.06.003. [DOI] [PubMed] [Google Scholar]