

Abstract
The direct, asymmetric α-amination of aldehydes has been accomplished via a combination of photoredox and organocatalysis. Photon-generated, nitrogen-centered radicals undergo enantioselective α-addition to catalytically formed chiral enamines to directly produce stable α-amino aldehyde adducts bearing synthetically useful amine substitution patterns. Incorporation of a photolabile group on the amine precursor obviates the need to employ a photoredox catalyst in this transformation. Importantly, this photoinduced transformation allows direct and enantioselective access to α-amino aldehyde products that do not require post-reaction manipulation.
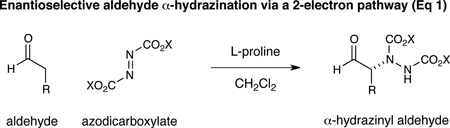
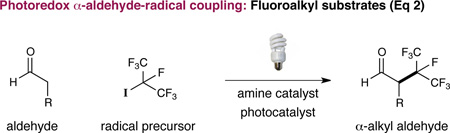
A central goal in organic synthesis is the development of methods to enantioselectively build C–N bonds within complex molecular structures. In particular, aldehydes, acids, and alcohols bearing α-amine substitution are widely distributed among pharmaceutically active compounds, and their broad representation has prompted the invention of a number of catalysis strategies for stereogenic nitrogen installation.1,2 In this context, α-amino aldehydes represent a valuable class of structural motif, mainly due to their capacity to serve as versatile synthetic handles en route to a diverse range of complex nitrogen-containing synthons.3 However, the development of robust methods for the asymmetric α-amination of aldehydes has been complicated by the requirement for electrophilic sources of nitrogen, along with the need to circumvent post-reaction racemization with relatively acid- or base-sensitive products. As a consequence, traditional α-carbonyl amination reactions often involve π-electrophile addition pathways that culminate in the installation of hydrazinyl or oxy-amino substituents,4 a class of N-stereogenicity that must be chemically modified (e.g. N–N or N–O reduction) prior to synthetic elaboration (Eq 1). In 2008, our lab introduced a versatile platform for catalytic activation, termed photoredox organocatalysis. In a common embodiment, electron-rich chiral enamines, derived from the condensation of aldehydes and secondary amine catalysts, undergo rapid and enantioselective coupling with electrophilic radical systems (e.g. CF3 •, ArCH2 •, etc; Eq 2).5 In this manuscript, we demonstrate that this “borrowed electron” catalysis strategy can be readily translated to the enantioselective α-amination of aldehydes using nitrogen-centered radicals. As a critical design element, this open shell coupling mechanism allows for the direct generation of α-amino aldehyde products of broad diversity that do not require post-reaction manipulation yet are stable to racemization.
 |
 |
 |
Design Plan
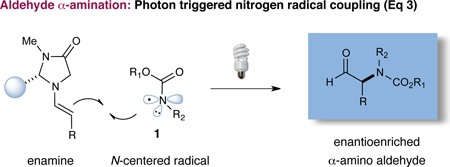
A detailed catalytic cycle for our proposed asymmetric aldehyde amination is presented in Scheme 1. We postulated that an electrophilic nitrogen-based radical 1 might be generated under mild conditions from an amine substrate 3 that incorporates a photolabile leaving group. While recent literature suggests that nitrogen-centered radicals might be formed using photoredox-active metal complexes,6 we envisioned that direct access to such open shell reaction partners might be best accomplished using a traceless activation handle such as the dinitrophenylsulfonyloxy group (a subunit that can be chemoselectively triggered using a simple household lightbulb). From the outset, it seemed plausible that an electrophilic nitrogen radical (such as 1) would rapidly undergo coupling with a transiently generated π-rich enamine 2 (derived from the condensation of an imidazolidinone catalyst with the aldehyde coupling partner). Oxidation of the resulting 3π-electron α-amino radical species 4 would then occur via single-electron transfer (SET) to a second equivalent of the photoexcited amine reagent 3*, a critical propagation step that would simultaneously deliver the iminium ion 5, while releasing the next round of the nitrogen radical coupling partner.7 Hydrolysis of iminium 5 would then reconstitute the imidazolidinone catalyst and, at the same time, deliver the enantioenriched α-amino aldehyde product. Notably, despite a growing interest in nitrogen-centered radicals as a source of electrophilic nitrogen,8 few intermolecular amine coupling processes have been reported,9 and indeed, no enantioselective applications have been described to date.
Scheme 1.

Proposed Mechanism for Aldehyde α-Amination.
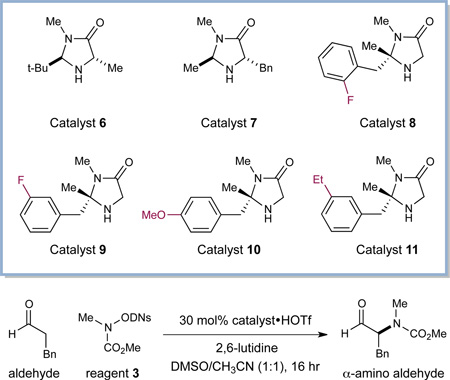
Our evaluation of the proposed aldehyde–amine coupling began with exposure of 3-phenylpropionaldehyde to a series of chiral amine catalysts and a large collection of nitrogen-based coupling partners. As revealed in Table 1, we were delighted to find that carbamate 3, incorporating a photolabile dinitrophenylsulfonyloxy (ODNs) residue, is competent at producing the requisite heteroatom-centered radical upon exposure to household light and in the presence of catalyst 6 (entry 1, 30% yield, 91% ee).10 Presumably, upon photonic excitation, amine 3*readily undergoes single-electron reduction and mesolysis of the weak N–O bond to yield the desired amine-centered radical and the ODNs anion. Initial experiments using catalyst 6 confirmed that the reaction does indeed require the use of light (cf. entries 1 and 2), and that a continuous source of photons is required for reaction propagation.11 Moreover, use of a monochromatic light source tuned to 300 nm (λmax absorption band of sulfonyloxyamine 3 = 292 nm)10 resulted in increased conversion and efficiency (entry 4, 38% yield, 90% ee, 6 h).12 These experiments provide additional evidence for the participation of the amine reagent 3* excited state in the photoredox process as described in Scheme 1.
Table 1.
Initial Studies towards α-Amination of Aldehydes.
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | temp (°C) | light | yielda | % eeb |
| 1 | 6 | rt | 26W CFL | 30% | 91% |
| 2 | 6 | rt | none | 0% | -- |
| 3c | 6 | rt | 26W CFL | 0% | -- |
| 4d | 6 | rt | LZC (300 nm) | 38% | 90% |
| 5 | 7 | rt | 26W CFL | 40% | 75% |
| 6 | 6 | −15 | 26W CFL | 47% | 92% |
| 7 | 7 | −15 | 26W CFL | 65% | 78% |
| 8 | 8 | −15 | 26W CFL | 82% | 88% |
| 9 | 9 | −15 | 26W CFL | 77% | 86% |
| 10 | 10 | −15 | 26W CFL | 72% | 86% |
| 11 | 11 | −15 | 26W CFL | 76% | 91% |
Obtained by 1H NMR analysis using methyl benzoate as internal standard.
Determined by chiral HPLC analysis of the corresponding alcohol.
Performed without 2,6-lutidine.
Reaction carried out in a photobox equipped with 10 × Luzchem LZC-UVB. DNs = 2,4-dinitrobenzenesulfonyl. CFL = compact fluorescent light.
We next evaluated amine catalysts of varying steric demand, with the supposition that higher enamine content13 and increased exposure of the reactive π-system would facilitate the critical radical addition step. Indeed, experiments performed in the presence of imidazolidinone catalyst 7, a system that generally provides higher enamine content, exhibited improved overall efficiency (entry 5, 40% yield) albeit with lower levels of stereocontrol (75% ee). Next, the effect of temperature on this α-amination protocol was evaluated. A significant improvement in reaction yield was observed at subambient temperatures, presumably due to the capacity to circumvent deleterious reduction of the carbamyl radical (cf. entries 1 and 6, 30 vs. 47% yield), a pathway that would consume amine reagent without productive C–N bond formation.14 Finally, during the course of our optimization studies, we determined that the aminal C(2)- position on the imidazolidinone framework was susceptible to hydrogen atom abstraction by the N-centered radical, leading to diminished levels of reaction efficiency. This catalyst decomposition pathway was suppressed via the design of a novel organocatalyst framework, wherein the C(2)-position incorporates a fully substituted carbon stereocenter (catalysts 8– 11, entries 8–11).15 In particular, the use of imidazolidinone 11 provided the desired α-amino aldehyde adduct with optimal levels of efficiency and enantiocontrol (entry 11, 76% yield, 91% ee).
It is notable that amine catalyst 11 was identified as the optimal organocatalyst for this transformation, given that it has not previously been utilized in enamine- or iminium-based transformations. The high levels of enantiocontrol observed in this study can be rationalized on the basis of enamine olefin geometry and π-facial selectivity. More specifically, DFT studies16 of the corresponding enamine intermediate (DFT-12, Figure 1) reveal that the (E)-configuration of the 4π-olefin system is preferred as it positions the electron-rich reaction site away from the fully substituted carbon center on the imidazolidinone framework. This preferred enamine geometry along with the meta-ethyl arene orientation (as shown) has further been confirmed by 2D NMR (NOESY) studies.17
Figure 1.

DFT structure of catalyst-derived enamine (DFT-12).
Reaction Scope
With our optimal conditions in hand, we examined the scope of this new enantioselective C–N bond-forming protocol. As shown in Table 2, this radical-based coupling is compatible with a variety of amine reaction partners adorned with an array of alkyl motifs and carbamate protecting groups (entries 1–8, 71–79% yield, 86–94% ee). It is important to note that many of these novel amine reagents are readily accessed in two steps from N-methyl hydroxylamine and are uniformly bench-stable, crystalline solids.18 Moreover, the bis- protected N-Moc, N-MOM amine reagent can be effectively used for α-amination of aldehydes with excellent enantiocontrol (entry 6, 75% yield, 94% ee). This specific example represents an important expansion of the scope of this method, offering a means to enantioselectively access orthogonally N ,N-protected α-amino aldehydes.
Table 2.
Enantioselective α-Amination: Scope of the Amine.
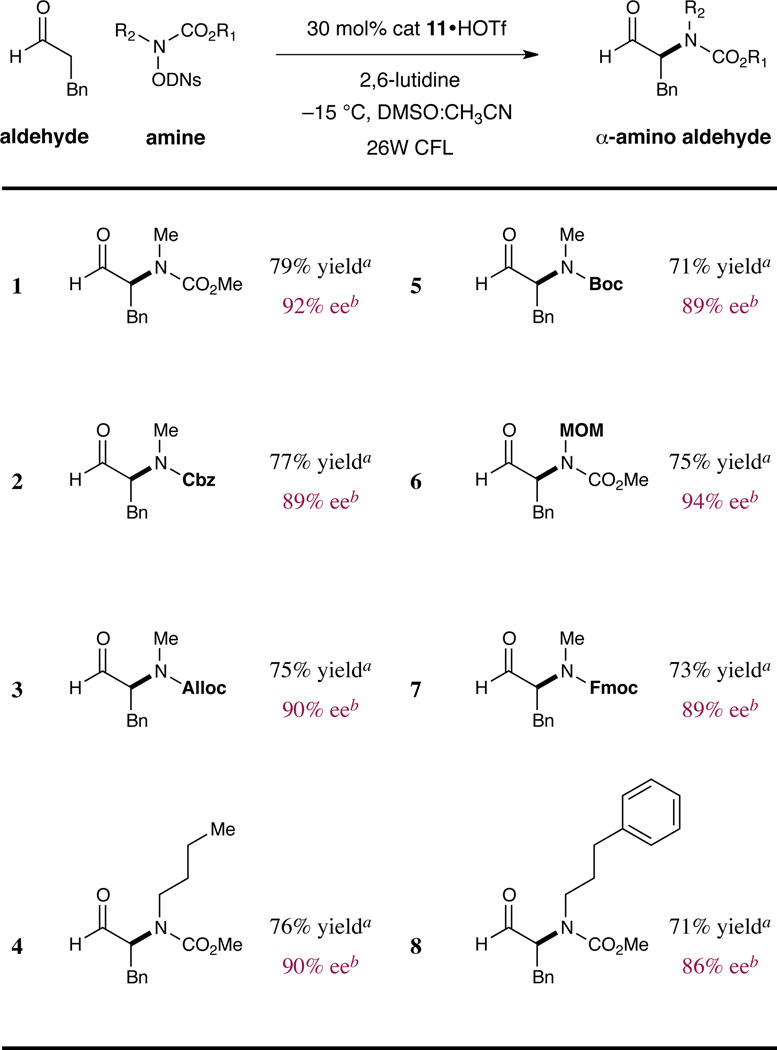 |
Stereochemistry assigned by chemical correlation or by analogy.
Enantiomeric excess determined by chiral HPLC analysis of the corresponding alcohol. DNs = 2,4-dinitrobenzenesulfonyl.
We next sought to establish the scope of the aldehyde coupling partner in this transformation. As shown in Table 3, we were pleased to find that these mild redox conditions accommodate a wide range of substituents on the aldehyde component, including ethers, amines, alkenes, and aromatic rings (entries 2–6, 71–79% yield, 88–91% ee). Moreover, excellent levels of enantiocontrol are achieved with sterically demanding formyl substrates (entries 7–8, 67–72% yield, 91–94% ee). It should be noted that α-dialkyl aldehyde systems are not useful substrates in this transformation as the imidizolidinone family of catalysts do not readily condense with α-branched aldehydes. This is an important catalyst design feature as it prevents post-reaction racemization with the products generated in Tables 2 and 3. Moreover, this protocol provides direct asymmetric access to synthetically valuable, configurationally stable α-amino aldehyde adducts which may be readily isolated and purified via column chromatography without further derivatization.
Table 3.
Enantioselective α-Amination: Scope of the Aldehyde.
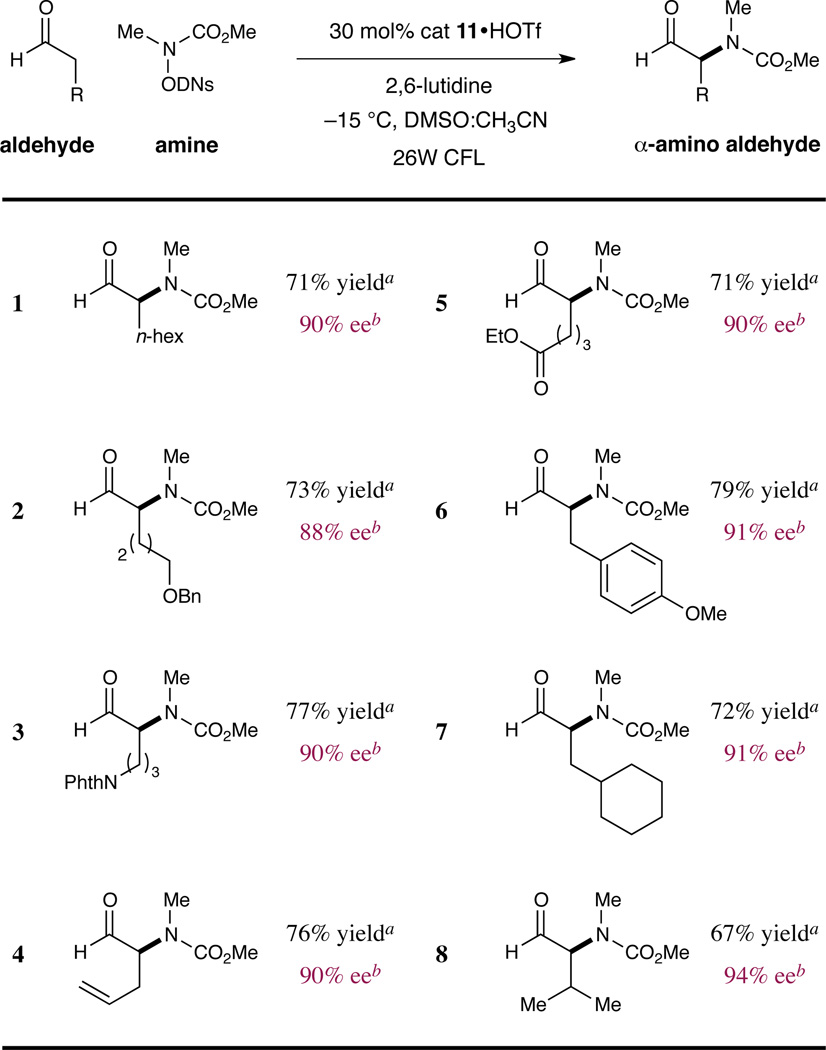 |
Stereochemistry assigned by chemical correlation or by analogy.
Enantiomeric excess determined by chiral HPLC analysis of the corresponding alcohol or 2-naphthoyl ester. DNs = 2,4-dinitrobenzenesulfonyl.
As a further demonstration of the synthetic utility of this method, we illustrate representative procedures for the conversion of these enantioenriched α-amino aldehyde adducts to either β-amino alcohol or α-amino acid motifs. As shown in Scheme 2, the crude product of the α-amination reaction (see Table 2, entry 2) may be directly converted to the corresponding β-amino alcohol in good yield and with complete stereofidelity. Alternatively, direct oxidation with buffered KMnO4 affords the Cbz-protected N-methyl phenylalanine with useful reaction efficiency and with retention of optical purity. We anticipate that this α-amination/aldehyde derivatization strategy will find broad application in the synthetic community, as a facile means by which to gain rapid access to high value non-proteogenic α-amino acids and N-alkyl α-amino acids.19–22
Scheme 2.

Telescoped synthesis of β-amino alcohols and α-amino acids from hydrocinnamaldehyde.
In summary, we have developed an organocatalytic photoredox-based approach to the asymmetric α-amination of aldehydes via the direct coupling of functionalized nitrogen and formyl precursors. This operationally facile process provides ready access to complex, N-substituted α-amino aldehyde architecture and at the same time offers a useful alternative to standard π-electron addition approaches to carbonyl α- amination. Moreover, this disclosure marks, to the best of our knowledge, the first demonstration of the use of nitrogen-based radicals as viable reagents in a catalytic enantioselective transformation. We anticipate that this α-amination method will prove widely useful in the synthesis of complex target structures bearing chiral amine fragments.
Supplementary Material
Acknowledgement
Financial support was provided by the NIHGMS (R01 GM-093213-03) and kind gifts from Merck, Amgen, and AbbVie. G. C. is grateful for a Merck overseas postdoctoral fellowship. C. M. K. thanks the German Academic Exchange Service (DAAD) for a postdoctoral fellowship. The authors thank E. Mosconi for assistance in performing Gaussian DFT calculations. Additionally, we thank C. Kraml and N. Byrne of Lotus Separations, LLC for their development of preparatory supercritical fluid chromatography (SFC) methods and gram-scale separation of the enantiomers of catalyst 11.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hili R, Yudin AK. Nat. Chem. Biol. 2006;2:284. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ricci A. Modern Amination Methods. Weinheim: Wiley–VCH; 2000. [Google Scholar]; (b) Ricci A. Amino Group Chemistry, From Synthesis to the Life Sciences. Weinheim: Wiley-VCH; 2007. [Google Scholar]; (c) Williams RM. Synthesis of Optically Active α-Amino Acids. Oxford: Pergamon; 1989. [Google Scholar]
- 3. For examples of N-protected α-amino aldehydes as chiral building blocks, see: Reetz MT. Angew. Chem. Int. Ed. 1991;30:1531. Reetz MT. Chem. Rev. 1999;99:1121. doi: 10.1021/cr980417b. Gryko D, Chalko J, Jurczak J. Chirality. 2003;15:514.
- 4.(a) Bogevig A, Juhl K, Kumaragurubaran N, Zhuang W, Jørgensen KA. Angew. Chem. Int. Ed. 2002;41:1790. doi: 10.1002/1521-3773(20020517)41:10<1790::aid-anie1790>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]; (b) List B. J. Am. Chem. Soc. 2002;124:5656. doi: 10.1021/ja0261325. [DOI] [PubMed] [Google Scholar]; (c) Konishi H, Lam TY, Malerich JP, Rawal VH. Org. Lett. 2010;12:2028. doi: 10.1021/ol1005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shih H-W, Vander Wal MN, Grange RL, MacMillan DWC. J. Am. Chem. Soc. 2010;132:13600. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lorance ED, Kramer WH, Gould IR. J. Am. Chem. Soc. 2002;124:15225. doi: 10.1021/ja020768e. [DOI] [PubMed] [Google Scholar]
- 7.Wayner DDM, Dannenberg JJ, Griller D. Chem. Phys. Lett. 1986;131:189. [Google Scholar]
- 8.(a) Zard SZ. Chem. Soc. Rev. 2008;37:1603. doi: 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]; (b) Esker JL, Newcomb M. J. Org. Chem. 1993;58:4933. [Google Scholar]; (c) Horner JH, Musa OM, Bouvier A, Newcomb M. J. Am. Chem. Soc. 1998;120:7738. [Google Scholar]; (d) Gagosz F, Moutrille C, Zard SZ. Org. Lett. 2002;4:2707. doi: 10.1021/ol026221m. [DOI] [PubMed] [Google Scholar]; (e) Baumgartner MT, Foray SG. J. Mol. Struct. 2003;633:7. [Google Scholar]; (f) Liu F, Liu K, Yuan X, Li C. J. Org. Chem. 2007;72:10231. doi: 10.1021/jo7015967. [DOI] [PubMed] [Google Scholar]; (g) Yu Y-Y, Fu Y, Xie M, Liu L, Guo Q-X. J. Org. Chem. 2007;72:8025. doi: 10.1021/jo070146h. [DOI] [PubMed] [Google Scholar]; (h) Yuan X, Liu K, Li C. J. Org. Chem. 2008;73:6166. doi: 10.1021/jo800845b. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lüring U, Kirsch A. Chem. Ber. 1993;126:1171. [Google Scholar]; (b) Tsuritani T, Shinokubo H, Oshima K. Org. Lett. 2001;3:2709. doi: 10.1021/ol016310j. [DOI] [PubMed] [Google Scholar]; (c) Kemper J, Studer A. Angew. Chem. Int. Ed. 2005;44:4914. doi: 10.1002/anie.200463032. [DOI] [PubMed] [Google Scholar]; (d) Guin J, Muck-Lichtenfeld C, Grimme S, Studer A. J. Am. Chem. Soc. 2007;129:4498. doi: 10.1021/ja0692581. [DOI] [PubMed] [Google Scholar]; (e) Guin J, Frohlich R, Studer A. Angew. Chem. Int. Ed. 2008;47:779. doi: 10.1002/anie.200703902. [DOI] [PubMed] [Google Scholar]
- 10. Syntheses of amine reagents are detailed in Supporting Information (Section XI) and recorded UV-Vis spectra of amine reagents are provided in Supporting Information (Section XII).
- 11. The transformation did not undergo propagation upon removal of the light source, therefore implicating a photon-induced electron transfer event. It was further determined that generation of the nitrogen-centered radical requires the presence of electron-rich enamine (Table 1, entry 3). More specifically, UV-Vis analysis of the radical precursor and the catalytically activated enamine (generated separately and concurrently), clearly demonstrates that disproportionation of a charge transfer complex is not the mechanism for carbamyl radical production (Scheme 1). Lastly, we have determined that N–O bond homolysis of reagent 3 does not occur in the absence of a SET event.
- 12. We chose to optimize this transformation using a household light source in lieu of the slightly more efficient LZC system, to allow operational convenience for practitioners of this chemistry.
- 13. The enamine concentration during the reaction was evaluated by 1H NMR analysis of crude reaction mixture performed in deuterated solvent (CD3CN/DMSO-d6).
- 14. 1H NMR analysis of the crude reaction mixture showed decreased levels of MeNHCO2Me (10–15%) for couplings performed at −15 °C. We recognize that MeNHCO2Me can be formed from the corresponding radical by either hydrogen abstraction from the solvent or SET reduction to the amine anion followed by protonation from the medium.
- 15. A detailed synthesis of imidazolidinone catalyst 11 is reported in Supporting Information (Section IV and V).
- 16. DFT calculations were performed at the B3LYP/6-31G* level of theory as implemented in Gaussian 03 package.
- 17. Structural studies on the enamine by 1H NMR and 2D NOESY are reported in Supporting Information (Section IX).
- 18. These amine reagents can be stored in the presence of moisture or light at ambient temperatures without decomposition.
- 19.(a) Sieber SA, Marahiel MA. Chem. Rev. 2005;105:715. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]; (b) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Disc. Today. 2010;15:40. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 20.(a) Yu J, Butelman ER, Woods JH, Chait BT, Kreek MJ. J. Pharm. Exp. Ther. 1997;280:1147. [PubMed] [Google Scholar]; (b) Harris KS, Casey JL, Coley AM, Karas JA, Sabo JK, Tan YY, Dolezal O, Norton RS, Hughes AB, Scanlon D, Foley M. J. Biol. Chem. 2009;284:9361. doi: 10.1074/jbc.M808762200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tulla-Puche J, Marcucci E, Prats-Alfonso E, Bayol-Puxan NR, Albericio F. J. Med. Chem. 2009;52:834. doi: 10.1021/jm800784k. [DOI] [PubMed] [Google Scholar]
- 21.(a) Teixido M, Zurita E, Malakoutikhah M, Tarrago T, Giralt E. J. Am. Chem. Soc. 2007;129:11802. doi: 10.1021/ja073522o. [DOI] [PubMed] [Google Scholar]; (b) Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew. Chem. Int. Ed. 2008;47:2595. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 22.a) Ron D, Gilon C, Hanani M, Vromen A, Selinger Z, Chorev M. J. Med. Chem. 1992;35:2806. doi: 10.1021/jm00093a013. [DOI] [PubMed] [Google Scholar]; (b) Dechantsreiter MA, Planker E, Matha B, Taylor JE, Coy DH. J. Med. Chem. 2001;44:1305. [Google Scholar]; (d) Chatterjee J, Ovadia O, Zahn G, Marinelli L, Hoffman A, Gilon C, Kessler H. J. Med. Chem. 2007;50:5878. doi: 10.1021/jm701044r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.