

Abstract
Molecular probe tool compounds for the Sphingosine 1-phosphate receptor 2 (S1PR2) are important for investigating the multiple biological processes in which the S1PR2 receptor has been implicated. Amongst these are NF-κB-mediated tumor cell survival and fibroblast chemotaxis to fibronectin. Here we report our efforts to identify selective chemical probes for S1PR2 and their characterization. We employed high throughput screening to identify two compounds which activate the S1PR2 receptor. SAR optimization led to compounds with high nanomolar potency. These compounds, XAX-162 and CYM-5520, are highly selective and do not activate other S1P receptors. Binding of CYM-5520 is not competitive with the antagonist JTE-013. Mutation of receptor residues responsible for binding to the zwitterionic headgroup of sphingosine 1-phosphate (S1P) abolishes S1P activation of the receptor, but not activation by CYM-5520. Competitive binding experiments with radiolabeled S1P demonstrate that CYM-5520 is an allosteric agonist and does not displace the native ligand. Computational modeling suggests that CYM-5520 binds lower in the orthosteric binding pocket, and that co-binding with S1P is energetically well tolerated. In summary, we have identified an allosteric S1PR2 selective agonist compound.
1. Introduction
Sphingolipids are an important family of bioactive molecules with cell signaling properties. Sphingosine 1-phosphate (S1P) is a pleiotropic lysophospholipid mediator present in plasma and is released in large amounts from activated platelets. S1P regulates various biological processes such as cell proliferation, migration, survival, and differentiation. The five sphingosine-1-phosphate receptors S1PR1 through S1PR5 mediate cellular functions upon S1P binding.1 S1PR1, S1PR2 and S1PR3 are widely expressed on various tissues and cell types, whereas the expression of S1PR4 and S1PR5 is prominent in cells of the immune and nervous systems, respectively.
The S1pr2 gene was cloned from rat smooth muscle cDNA library as an orphan receptor homologous to the S1pr1 gene.2 In K562 cells transiently transfected with S1pr2 cDNA, S1P increased intracelluar calcium levels from intra- and extracellular reserves.3 S1PR2 is a high affinity subnanomolar receptor for S1P and has been implicated in multiple biological functions, including Rho activation, inhibition of Rac and cell migration, and in “feed forward” autocrine signaling in NF-κB survival signaling of tumor cells.4, 5 S1PR2 promiscuously couples to the heterotrimeric G proteins Gq, Gs, Gi/o, and G12/13.1 Studies with genetic deletions can provide insights into the physiologic functions of the targeted gene product. Kono et al reported that S1PR2 expression is essential for proper functioning of the auditory and vestibular system.6 Skoura et al reported the essential role of S1PR2 in pathological angiogenesis of the mouse retina.7 About one half of S1pr2 gene null mice develop clonal B-cell lymphomas by age 1.5 to 2 years.8 In addition to these observations, it is further expected that S1PR2 exerts other unknown physiological functions.
S1P is a unique, amphiphilic GPCR ligand, consisting of a hydrophilic, polar zwitterionic phosphoamine headgroup and a hydrophobic aliphatic straight chain C18 tail (Figure 1A). The flexibility of the acyl chain may allow binding to many diverse sites. Along with genetic manipulations, chemical approaches provide novel insights into the function of S1P receptors. In the case of S1PR1, SEW-2871 is well recognized as an S1PR1-agonist and demonstrates the essential role of S1PR1 in lymphocyte trafficking Recently a S1PR1 subtype selective agonist provided insights into the pulmonary response to viral infection.9 Further, Sanna et al reported that W146, a chiral S1PR1 antagonist, enhances capillary leakage and restores lymphocyte egress in vivo.10 Therefore S1P receptor subtype selective agonists and antagonists will be of broad utility in understanding the regulatory mechanism of cell functions in vitro and physiological phenomenon in vivo.
Figure 1.
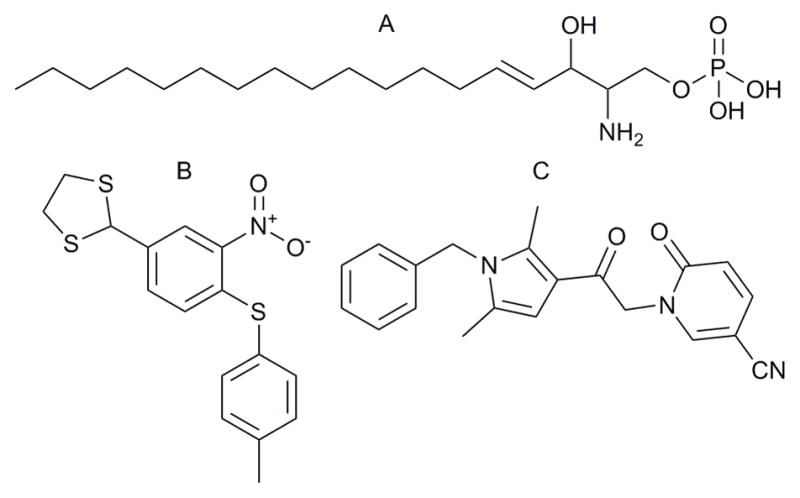
Chemical structure of ligands used in this study.
A. Sphingosine-1-phosphate, B. XAX-162, C. CYM-5520
There are few existing chemical probes for S1PR2. JTE-013 has been developed as an S1PR2 selective antagonist.11 However, to our knowledge there is no known S1PR2 selective agonist. An S1P analogue, DS-SG-44, (2S,3R)-2-amino-3-hydroxy-4-(4-octylphenyl)butyl phosphoric acid), was reported to block isoprenaline-mediated morphological changes in rat C6 glioma cells, and was hypothesized to acted as an S1PR2 receptor agonist.12 However, no receptor pharmacology for DS-SG-44 is reported, and is likely that it will be problematic for biological studies because of solubility and metabolic liabilities.
We report here HTS-driven identification of novel chemical scaffolds for S1PR2, chemical optimization and characterization of receptor activation via binding to the hydrophobic portion of the S1P biotopic, orthosteric binding site. Receptor binding and activation were delineated by radioligand binding competition, antagonist inhibition of CRE-reporter responses, cAMP biosensor detection of activation of wild type and head group mutant S1PR2 receptors, and molecular modeling studies.
2. Materials and Methods
2.1 Chemicals
S1P was purchased from Biomol (Plymouth Meeting, PA) and dissolved in methanol (1 mM) and stored at −80 °C. Forskolin was purchased from Sigma-Aldrich and stored as a 10 mM DMSO solution at −35 °C. JTE-013 was purchased from Calbiochem (San Diego, CA) and Cayman Chemical (Ann Arbor, MI) and stored as a 10 mM DMSO solution at −35 °C.
2.2 S1P Reporter and Counterscreen Assays
S1pr1 CRE-bla CHO, S1pr2 CRE-bla CHO, S1pr3-Gα16 NFAT-bla CHO, S1pr4–TANGO, S1pr4-TANGO and the counterscreen CRE-bla CHO reporter assays were performed as described.13 PubChem assays are listed in table S1.
2.3 Jump-In CHO S1PR2 wild type and head group triple mutant cell lines
Multisite Gateway cloning was utilized to generate in-frame S1pr2-eGFP expression constructs from pEnter-15- S1pr2 and pENTER-52-eGFP. The S1pr2-eGFP fusion protein expression vector was cloned into pDEST-CMV-JTI (Invitrogen). S1PR2 head group binding side chains were identified by alignment with S1PR1. These mutations were generated by overlapping oligonucleotide PCR.14 The triple mutant S1PR2 (R108A, E109A and K269A) was generated by overlapping oligonucleotide PCR mutagenesis. All constructs were confirmed by DNA sequencing. These vectors were transfected into CHO JumpIn cells(Invitrogen) and selected with 10 microgram/mL blasticidin as described.15 A homogenous pool of cells was generated by FACS sorting of GFP positive cells.
2.4 Glo-sensor cAMP transient transfection assay
The GloSensor vector (pGLoSensor-20FcAMP, Promega) was transfected using Fugene HD into S1PR2-eGFP or S1PR2-TM-eGFP Jump-In CHO cells. The following day, cells were harvested with 0.05% trypsin EDTA, resuspended to 500,000 cells/mL in CO2 independent Media (Invitrogen) containing 2% charcol dextran stripped serum (CDS, Invitrogen) and 20 μL of the cell suspension was added to 384 well tissue culture treated white plates (Corning, part number 3570). These plates were incubated overnight at 37 °C, 5% CO2. 25 uL of CO2 independent media containing 2%CDS and 4% GloSensor Reagent (Promega) were then added and the plates were incubated for 2 hours at room temperature. Antagonist (JTE-013) or vehicle were added and incubated for 20 minutes followed by agonist compounds or S1P. After 15 minutes, luminescence was read on a Perkin Elmer Envision plate reader.
2.5 33P-S1P Radioligand Competition Binding Assay
Sphingosine, D-erythro [33P] 1-phosphate was purchased from American Radiolabeled Chemicals, Inc (St Louis). S1PR2-CRE bla cells were seeded into wells of a 24 well plate at 200,000 cells in 1.0 mL growth media and the plate incubated overnight in an incubator with 100% humidity, 5% C02, 37°C. The media was replaced with 1% CDS serum media for 4 hours prior to the assay. At 4°C, the media was removed and replaced with test compounds or vehicle controls in binding buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 15 mM NaF with freshly added 1 mM Na3VO4 and protease inhibitors).
2.6 Compound Synthesis and Characterization
1 (CYM 5482)
1-(1-benzyl-2,5-dimethyl-1H-pyrrol-3-yl)-2-chloroethanone (50 mg, 0.191 mmol) in DMF were added sequentially DIPEA (66 μl, 0.38 mmol) and succinamide (38 mg, 0.38 mmol). The reaction was stirred 40 minutes in the microwave at 130°C. The mixture was diluted in water and extracted with Ethyl acetate (4×50 ml). The combined organic phase was washed with brine (2×50 mL) and concentrated under reduced pressure. The mixture was purified by column chromatography using DCM/MeOH to yield 12 mg (0.036 mmol, 19%) of product as pale yellow powder.
1H NMR (400 MHz, CDCl3): δ 7.34-7.25 (m, 3H), 6.87 (d, J = 7.68 Hz, 2H), 6.34 (s, 1H), 5.05 (s, 2H), 4.73 (s, 2H), 2.84 (s, 4H), 2.45 (s, 3H), 2.14 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 186.46, 177.91, 137.72, 138.00, 137.20, 129.83, 129.64, 128.42, 126.35, 117.68, 107.64, 47.52, 46.36, 29.20, 13.08, 12.70. IR (cm−1): 1702 s, 1663 s. MS (EI) m/z: 325 (M+H)).
9 (CYM 5477)
To a stirred solution of potassium tert-butoxide (20 mg, 0.076 mmol) in THF at 0°C was added slowly the 2-pyrrolidone (12 μl, 0.15 mmol) and the mixture was stirred at 0°C for 20 minutes. The reaction was warmed to room temperature and stirred for additional 20 minutes. The mixture was cooled to 0°C followed by addition of 1-(1-benzyl-2,5-dimethyl-1H-pyrrol-3-yl)-2-chloroethanone (17 mg, 0.15 mmol), the reaction was stirred for 20 minutes at 0°C and overnight at room temperature. The mixture was diluted with water, and the product extracted with ethyl acetate. The product was purified by column chromatography using DCM/MeOH to yield 14 mg (0.045 mmol, 60%) as pale yellow powder
1H NMR (400 MHz, CDCl3): δ 7.33-7.25 (m, 3H), 6.87 (d, J = 7.28 Hz, 2H), 6.31 (s, 1H), 5.04 (s, 2H), 4.51 (s, 2H), 3.53 (t, J = 7.08 Hz, 2H), 2.52 (t, J = 8.04 Hz, 2H), 2.47 (s, 3H), 2.13-2.06 (m, 5H); 13C NMR (125 MHz, CDCl3): δ 189.89, 176.58, 136.82, 136.82, 129.32, 129.00, 127.90, 125.87, 117.80, 107.21, 50.29, 48.72, 47.00, 30.92, 18.37, 12.58, 12.20. IR (cm−1): 1740 s, 1663s. MS (EI) m/z: 311 (M+H)).
10 (CYM 5478)
To a stirred solution of 1-(1-benzyl-2,5-dimethyl-1H-pyrrol-3-yl)-2-chloroethanone (50 mg, 0.191 mmol) in DMF (2.5 mL) were added sequentially DIPEA (66 μl, 0.38 mmol) and 2-hydroxy-5-trifluoromethyl pyridine (62 mg, 0.38 mmol). The reaction was stirred 48 hours at 70°C. The mixture was diluted in water and extracted with ethyl acetate (4×50ml). The combined organic phase was washed with brine (2×50 mL) and concentrated under reduced pressure. The mixture was purified by column chromatography using DCM/MeOH to yield 20 mg (0.052 mmol, 27%) of product as a pale yellow powder.
1H NMR (400 MHz, CDCl3): δ 7.63 (bs, 1H), 7.48 (dd, J = 9.5, 2.4 Hz, 1H), 7.34-7.26 (m, 3H), 6.89 (d, J = 7.2 Hz, 2H), 6.66 (d, J = 9.5 Hz, 1H), 6.40 (s, 1H), 5.18 (s, 2H), 5.06 (s, 2H), 2.48 (s, 3H), 2.16 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 187.07, 162.26, 138.86 (q, J3 = 5 Hz), 137.71, 136.59, 135.77 (q, J3 = 2 Hz), 129.47, 129.37, 128.00, 125.89, 123.77 (q, J1 = 268 Hz), 121.48, 117.25, 109.65 (q, J2 = 35 Hz), 107.17, 55.44, 47.11, 12.62, 12.27. IR (cm−1): 1740 s, 1675 s, 1330 s. MS (EI) m/z: 389 (M+H)).
11 (CYM 5491)
Product obtained in 32% yield.
1H NMR (400 MHz, CDCl3): δ 7.39-7.24 (m, 4H), 6.89 (d, J = 8.16 Hz, 2H), 6.88 (s, 1H), 6.40 (s, 1H), 6.33 (d, J = 8.6 Hz, 1H), 5.19 (s, 2H), 5.06 (s, 2H), 2.47 (s, 3H), 2.15 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 187.28, 161.81, 141.58 (q, J2 = 34 Hz), 140.77, 137.72, 136.59, 129.56, 129.44, 128.00, 125.89, 122.53 (q, J1 = 273 Hz), 118.57 (q, J3 = 4 Hz), 117.32, 107.19, 101.12 (q, J3 = 3 Hz), 55.26, 47.11, 12.62, 12.26. IR (cm−1): 1679 s, 1609 s, 1167 s, 1133 s. MS (EI) m/z: 321 (M+H)).
12 (CYM 5481)
Product obtained in 46% yield.
1H NMR (400 MHz, CDCl3): δ 7.39-7.22 (m, 5H), 6.89 (d, J = 7.08 Hz, 2H), 6.61 (d, J = 9.08 Hz, 1H), 6.43 (s, 1H), 6.20 (td, J = 7.9, 1.2, 1H), 5.18 (s, 2H), 5.06 (s, 2H), 2.47 (s, 3H), 2.15 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 188.80, 163.46, 140.63, 139.54, 137.86, 137.21, 129.82, 129.63, 128.42, 126.39, 121.64, 118.09, 107.80, 106.46, 55.64, 47.54, 13.10, 12.72. IR (cm−1): 1740 s, 1655 s. MS (EI) m/z: 321 (M+H)).
13 (CYM 5473)
Product obtained in 34% yield.
1H NMR (400 MHz, CDCl3): δ 7.66 (bs, 1H), 7.54-7.47 (m, 4H), 7.17 (d, J = 7.8 Hz, 2H), 6.66 (d, J = 8.6 Hz, 1H), 6.44 (s, 1H), 5.19 (s, 2H), 2.30 (s, 3H), 2.01 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 187.25, 162.20, 138.89 (q, J3 = 5 Hz), 138.40, 137.28, 135.79 (q, J3 = 5 Hz), 130.18, 129.95, 129.35, 128.30, 123.78 (q, J1 = 268 Hz), 121.50, 117.47, 109.62 (q, J2 = 35 Hz), 106.80, 55.50, 13.40, 13.10. IR (cm−1): 1675 s, 1650 s, 1334 s. MS (EI) m/z: 375 (M+H)).
14 (CYM-5520)
Product obtained in 27% yield.
1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 2.4 Hz, 1H), 7.43 (dd, J = 9.5, 2.4 Hz, 1H), 7.34-7.25 (m, 3H), 6.88 (d, J = 7.0 Hz, 2H), 6.61 (d, J = 9.5 Hz, 1H), 6.38 (s, 1H), 5.18 (s, 2H), 5.06 (s, 2H), 2.47 (s, 3H), 2.15 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 186.46, 161.28, 146.83, 139.55, 138.00, 136.49, 129.65, 129.40, 128.05, 125.89, 121.67, 117.05, 116.73, 107.13, 91.52, 55.22, 47.16, 12.64, 12.31. IR (cm−1): 2227 s, 1736 s, 1659s. MS (EI) m/z: 346 (M+H)).
2.7 Computational Studies
2.7.1 S1PR1/S1PR2 Structure wild type (WT) and mutant Models
The initial S1PR1 receptor structure was taken from the antagonist x-ray co-crystal structure (PDB code 3V2W).16 The structure was prepared using the protein preparation workflow in Maestro (Schrodinger Inc.) to assign hydrogens, optimize hydrogen bonds and to perform constraint minimization (impref). The homology model of S1PR2 was built using the Uniprot sequence S1PR2_Human (accession O95136) in Prime (Schrodinger Inc.). This initial S1PR2 model was optimized using the same protein preparation workflow above. Both the S1PR1 and the S1PR2 model with the antagonist sphingolipid mimic ML5 ligand were then optimized using a multi-step all-atom minimization and molecular dynamics (MD) simulation implemented in the software package Desmond (DE Shaw Research).17 Prior to the MD multi-step simulation, a membrane bilayer model (POPC 300K) was added to both the S1PR1 and S1PR2 models. The system was set up using the OPLS-AA force field, the TIP4P explicit solvent model in an orthorhombic simulation box 10Å distance in all directions and adding counter ions. Simulations were performed at 300K and 1.01325 bar using the NPT ensemble class. All other settings were default. The production simulation time was 12 ns. Simulations were run on an IBM E-server 1350 cluster (36 nodes of 8 Xeon 2.3 GHZ cores and 12 GB of memory). Several later simulation frames were extracted from the S1PR1 and S1PR2 simulations based on conformational diversity, low (stable) RMSD, and a stable ML5 (ligand) pose with maximum H-bonds. To avoid clashing side chains, constraint (impref) minimization (in Maestro, Ref: Schrodinger Inc.) was performed for the WT and mutant S1PR1 and S1PR2 receptor structures. These structures were then used for further modeling.
2.7.2 Ligand Receptor Binding Models
Using the optimized S1PR1 and S1PR2 WT and mutant receptor models, we generated initial binding poses for the ligands CYM-5520 and S1P as follows. Ligands were prepared using ligprep Schrodinger Inc.) to generate ionization states (pH = 7) and stereoisomers resulting in a single representation for both S1P and CYM-5520. Ligands were initially docked into the receptor structures using the Induced Fit Docking (IFD) (Ref: Schrodinger Inc.) protocol with default settings. The IFD protocol includes a constraint receptor minimization step followed by initial flexible Glide docking of the ligand using a softened potential to generate an ensemble of poses. For each pose, the nearby receptor structure is then refined using Prime. Each ligand is then re-docked (using Glide) into its corresponding optimized low-energy receptor structure and ranked by Glide score. For S1P, we required two hydrogen bond interactions of polar receptor side chains known to interact with S1P (R120/108, E121/109, R292/K269; S1PR1/S1PR2). For CYM-5520 no constrains were used. The best pose with highest IFD score obtained for each ligand was again subjected to MD simulation (3–5 ns production runs) for further optimization of the protein ligand complex. The MD protocol includes a multi-step procedure of minimizations and short MD runs followed by the production MD simulation. The same parameter and settings as described above were used. Poses were stable during the production MD runs. The final frames of these simulations were then used for docking of ligands after constraint (impref) minimization (Maestro, Schrodinger Inc.). Ligands were re-docked using Glide SP and XP with default potential and other settings. The best pose of the ligand was selected based on the Glide scores, known interactions (e.g. head group) and visual inspection. MM-GB/SA implemented in Prime was performed to calculate the relative binding free energies for the studied ligands. Receptor flexibility cutoff was set to 4Å around the ligand. 2D ligand-receptor interaction diagrams were generated in Maestro and 3D plots were produced using PyMol.
To evaluate the hydrophobic interaction of ligand and receptor, the structural interaction fingerprint (SiFT) of the binding was calculated post docking. The resulting fingerprint was visualized in a matrix as heat map, sorted by similarity and clustered considering only the hydrophobic amino acid residues. The presence of interaction is shown as red and the absence green.
3 Results
3.1 S1PR2 HTS and uHTS Screening
We developed a HTS compatible assay for S1PR2 with beta-lactamase reporter readout. In order to identify the most appropriate reporter for detecting S1PR2 activation we compared the signal generated by transiently transfecting NFAT-bla or CRE-bla CHO cells with either pcDNA3.1 human S1pr2 or LacZ constructs. Stimulation with 1 micromolar S1P resulted in a reproducible 1.4 fold increase in the blue/green readout for the S1PR2 transfected cells and no increase for the control transfections. A stable cell line expressing S1PR2 in CRE-bla CHO was generated and conditions for HTS and ultra HTS (uHTS) optimized.18 Two complementary screening sets were probed, the Maybridge HitFinder and NIH MLSMR library. The Maybridge S1PR2 agonist screen was run in 384 well format, with 10 micromolar test compound. The screen had acceptable assay statistics (Z’ave =0.63; S/B 6.7). For the Maybridge screen a cutoff of greater than 40% of control was selected and 57 compounds were cherry picked and retested in the primary and parental CRE-bla cell line counterscreening assays in triplicate at 5 micromolar concentration. Compounds that were confirmed active in the primary assay and inactive in the counterscreen were purchased as powders for further testing. From the Maybridge screen only one compound, XAX-164 (Table 1), was active in the S1PR2 CRE-bla assay and inactive in the counterscreen assay. Dose response from a powder sample determined an EC50 of 1.3 μM in the S1PR2 CRE-bla reporter assay and absence of activity in the CRE-bla parental cell line counterscreen assay.
Table 1.
Confirmed S1PR2 agonists from the Maybridge and MLMSR Screens
| Name | Structure | IC50 S1PR2 (μM) | IC50 CRE-bla Counterscreen |
|---|---|---|---|
| 1 (CYM-5482) |
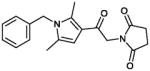
|
1.0 | Not Active |
| 2 (XAX-164) |
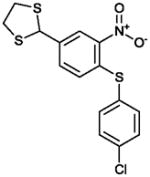
|
1.3 | Not Active |
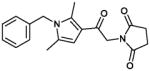
In the MLSCN S1PR2 uHTS campaign (deposited in PubChem AID 729), 96,881 compounds were tested in 1,536 well format at 5 micromolar. The 61 (of 64) available compounds with activation greater than 50% were tested in dose response against the S1PR2 Agonist (PubChem AID 854) and parental cell line counterscreen (PubChem AID 843) dose response assays. Only 2 compounds were active in the S1PR2 dose response assay, and inactive in the CRE-bla dose response counterscreen. The MLSMR compound MLS000049871 was inactive in the CRE-bla counterscreen, but was active in PubChem AID 662, an assay designed to find CRE activators, and was therefore not considered a viable lead. We resynthesized the remaining compound, CYM-5482 (1) (Table 1) and confirmed S1PR2 agonist activity with an EC50 of 1.0 μM.
3.2 Synthesis of 1, 10 and analogs
The structural integrity and biological activity of the original hit (1) were confirmed by re-synthesizing the title compound (Scheme 1). Reaction of commercially available chloroketone I with succinamide using DIPEA as a base provided the hit 1. Similarly, condensation of chloroketone I with 2-pyrrolidone using KOtBu as base furnished derivative 9. The synthesis of 1 and 9 is shown in Scheme 1. Condensation of 2-pyridone derivatives III–VI with chloroketones I and II, using DIPEA as base, provided the final products 10–14 (Scheme 2).
Scheme 1.
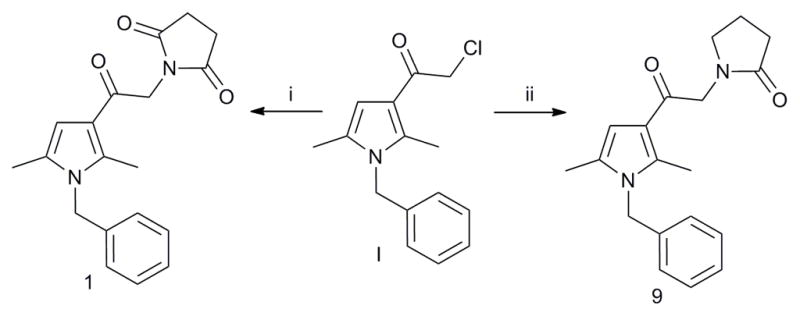
Reagents and conditions: (i) I (1 equiv.), succinamide (2 equiv.), DIPEA (2 equiv.), DMF, mw, 130 °C, 40 min, 19%; (ii) I (1 equiv.), 2-pyrrolidone (2 equiv.), KOtBu (0.5 equiv.), THF, 0 °C to rt, overnight, 60%.
Scheme 2.
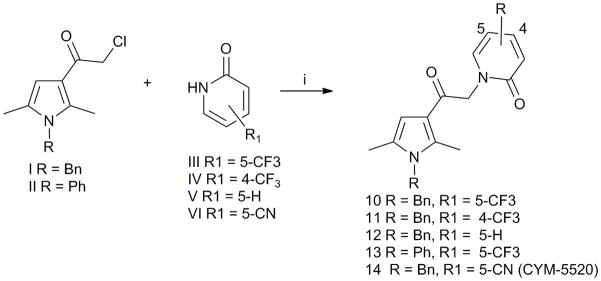
Reagents and conditions: (i) I or II (1 equiv.), III–VI (2 equiv.), DIPEA (2 equiv.), DMF, 70 °C, 48h (27–46%).
3.3 Structure Activity Analysis of Analogues
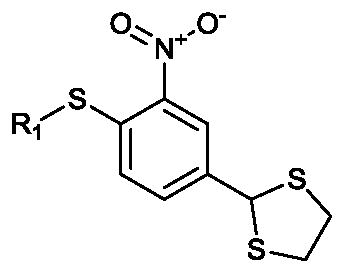
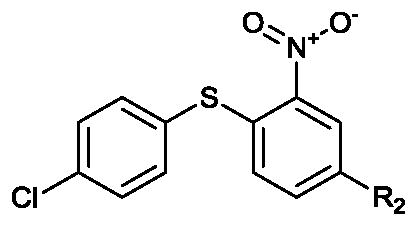
Six compounds similar to XAX-164 were purchased to explore what functional groups were required for S1PR2 activation (Table 2). The three compounds with 4-substituted phenyl groups in the R1 region are active. The methyl substitutedXAX-162 (EC50 0.55 micromolar) and bromide 4 (EC50 0.82 micromolar derivatives are modestly more potent than XAX-164. Substitution of the phenyl moiety with 6-(trifluoromethyl)pyridin-2-yl (5) or cyclohexy (6) results in loss of activity. Replacement of the 1,3-dithiolane group (R2) in 2 with either carboxyl (7) or chloride (8) results in a loss of S1PR2 activation. GPCR ligands for adrenergic and 5-HT receptors containing 1,3-dithiolanes have been reported.19
Table 2.
SAR by purchase for the Maybridge Hit XAX-164
| |||
|---|---|---|---|
| Name | R1 | EC50 S1PR2 (μM) | Max Activity (% of 1 μM S1P) |
| 2 (XAX-164) | 4-clorophenyl | 1.3 | 80% |
| 3 (XAX-162) | 4-methylphenyl | 0.55 | 130% |
| 4 (XAX-166) | 4-bromophenyl | 0.82 | 80% |
| 5 (XAX-159) | 6-(trifluoromethyl)pyridin-2-yl | >10 | 30% |
| 6 (XAX-160) | Cyclohexyl | >10 | 20% |
| |||
|---|---|---|---|
| Name | R2 | EC50 S1PR2 (μM) | Max Activity (%of 1 μM S1P) |
| 7 (BTB06747) | Carboxyl | >10 | 0% |
| 8 (CD05115) | Cl | >10 | 0% |
58 compounds structurally related to CYM-5482 were purchased and evaluated in the S1PR2 and counterscreen dose response assays (PubChem AIDs 872 and 874). Several compounds were active and are shown in Table S3 along with some of the inactive analogues. Interestingly, amongst the commercial analogs the 5-(trifluoromethyl)pyridin-2-one (10) was found slightly more potent than 1. Based upon these results, we then synthesized selected compounds (Table 3) in order to improve potency while maintaining selectivity. First we removed the labile functional group from 1, however the pyrrolidone (9) is inactive. Next we explored the SAR around 10. The 4-(trifluoromethyl)pyridin-2-one (11), the unsubstituted pyridine-2-one (12) and the N-phenyl (13) are completely inactive. Interestingly, changing the trifluoromethyl for a cyanide group (14, CYM5520) leads to a 1.6-fold increase in potency and inactivity in the CHO CRE-bla counterscreen.
Table 3.
Synthesized SAR compounds
| Name | Structure | EC50 S1PR2 (μM) | Max Activity (%of 1 μM S1P) |
|---|---|---|---|
| 1 (CYM-5482)α |
|
1.03 | 95% |
| 9 (CYM-5477) |
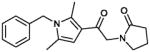
|
>10 | 35% |
| 10 (CYM-5478) |
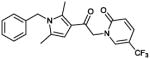
|
0.78 | 100% |
| 11 (CYM-5491) |
|
>10 | 9% |
| 12 (CYM-5481) |
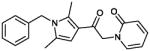
|
>10 | 10% |
| 13 (CYM-5473) |
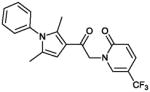
|
>10 | 0% |
| 14 (CYM-5520) |
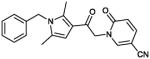
|
0.48 | 100% |
Resynthesized original hit.
3.4 Compound Selectivity
CYM-5520 and XAX-162 are highly selective for S1PR2 and were inactive in the S1PR1, S1PR3, S1PR4 and S1PR5 agonist assays (Supplemental table S4). We further characterized CYM-5520 in the PanLabs HitProfiling Screen of 29 receptors and transporters confirming selectivity for S1PR2 over other molecular targets with no assay inhibition greater than 20%. (Supplemental table S3)
3.5 Competition with the S1PR2 Antagonist JTE-013
JTE-013 is an antagonist of S1PR2.20 To compare S1P and CYM-5520 binding to S1PR2 we evaluated dose response curves of either S1P or CYM-5520 against several concentrations of JTE-013 in the S1PR2 CRE-bla reporter assay (Figure 2). The S1P concentration response curve shifts to the right in response to JTE-013, consistent with competitive antagonism. Linear regression of the Schild plot yields a Ki of 20 nM. With increasing JTE-013, the CYM-5520 dose response curves both shift to the right and the magnitude of the response is diminished. Both XAX-164 and XAX-162 have a similar response as CYM-5520 in the presence of increasing concentrations of JTE-013 (Supplemental figure S1). This type of agonist inhibition is best described with the noncompetitive binding model.21
Figure 2.
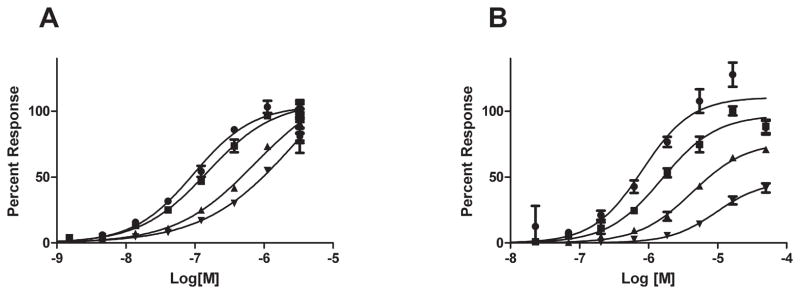
JTE-013 inhibition of agonists in the S1PR2-CRE-bla assay. Dose response of S1P (A) or CYM5520 (B) on S1PR2- Cre-bla CHO cells was challenged with increasing concentrations of JTE-013 (● DMSO control, ■ 10 nM JTE-013, ▲ 30 nM JTE-013, ▼ 90 nM JTE-013).
3.6 Radioligand binding competition to S1PR2
The competitive binding of the ligands used in this study for the S1PR2 receptor were evaluated with radiolabeled S1P. Both S1P and JTE-013 demonstrate dose-dependent inhibition of binding of 33P-S1P to S1PR2. The unlabeled S1P displaces the radiolabeled S1P with an IC50 of 25 nM. JTE-013 has an IC50 of 53 nM in this assay. In contrast, CYM-5520 does not block radiolabeled S1P binding (Figure 4). CYM-5520 was tested at a smaller range of higher concentrations because, in pilot experiments, we observed that it was not competitive with S1P binding. These results demonstrate that CYM-5520 is not competitive with S1P.
Figure 4.
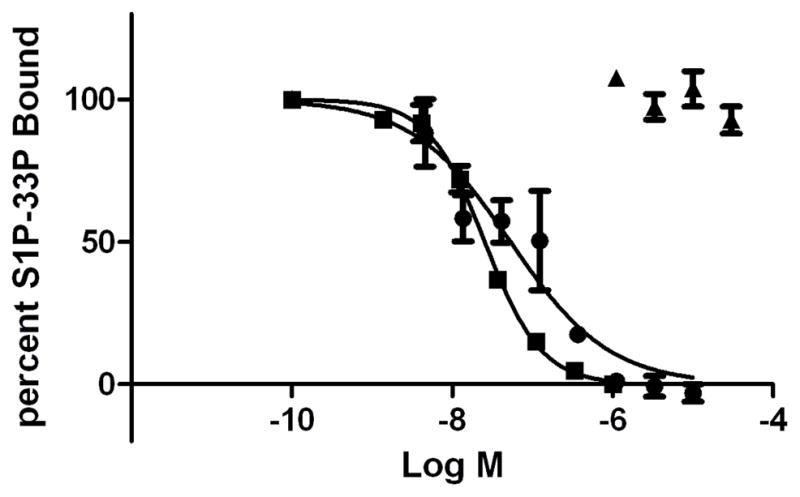
Radiolabeled 33P-S1P competition binding study. The graph shows the average of replicate samples. Results are representative of 3 independent experiments which was only tested one time. S1P (■), JTE-013, (■), CYM-5520 (▲)
3.7 CYM-5520 is an agonist for both wild type and headgroup mutant S1PR2 receptor
In S1PR1, the side chains from Arginine 120, Glutamic acid 121 and Arginine 292 form salt bonds with the phosphate and ammonium moieties of the zwitterionic S1P headgroup. These interactions are required for receptor binding of S1P to the receptor 22. Homologous polar residues responsible for S1P headgroup binding in S1PR2 were identified by alignment with the S1PR1 amino acid sequence. The residues in S1PR2 are Arginine 108, Glutamic acid 109 and Lysine 269. A S1pr2 cDNA was generated by overlapping PCR mutagenesis in which all three of these residues were mutated to alanine to generate the triple mutant (TM) construct. Stable cell lines with a single copy of either wild type (WT) or TM S1PR2-GFP fusion proteins were generated by targeted, single site integration. The WT S1PR2-GFP and TM S1PR2-GFP cell lines were then used to evaluate the ligand-stmulated cAMP response. We measured intracellular cAMP with a genetically encoded biosensor, based upon a single chain luciferase-cAMP fusion construct, which upon binding cAMP changes conformation to higher luciferase activity 23. S1P(EC50 10 nM) and CYM-5520 (EC50 1.6 μM) are full agonists for wild type S1PR2 (Figure 3A). Stimulation of cells expressing the triple mutant S1PR2 with S1P did not elicit a rise in luciferase activity, whereas the CYM-5520 was an agonist with an EC50 of 1.5 μM (Figure 3B).
Figure 3.
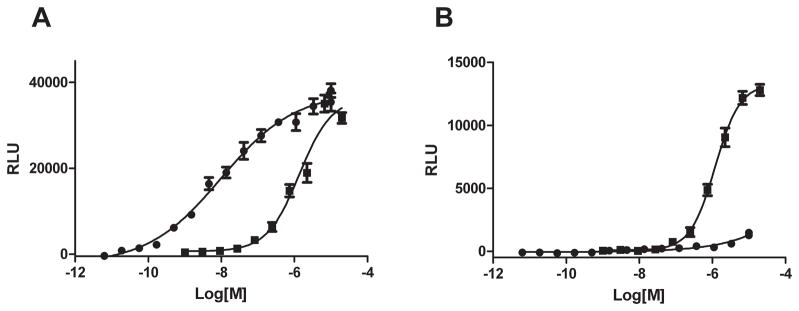
The effect of S1P and CYM5520 on intracellular cAMP levels in S1PR2 wild type- and triple mutant-expressed cells. Dose response of S1P (●) or CYM5520 (■) on intracellular content of cAMP in wild type- (A) and triple mutant- (B) S1PR2 expressed CHO cells was measured with luciferase activity.
3.8 Molecular Modeling
3.8.1 Computational Modeling Building
Because CYM-5520 is an allosteric agonist we modeled ligand binding and examined the binding pocket in detail to better understand how our observations could be explained. The questions examined were whether CYM-5520 and S1P co-binding in the optimized model was reasonable, and what hydrophobic contacts are important for CYM-5520 binding. A homology model was produced for S1PR2 by using the S1PR1 structure as obtained from PDB (3V2W, resolution 3.35Å) and the Uniprot sequence of S1PR2_Human (accession O95136). The primary aligment of the amino acids sequences used to build the model is in the supplemental materials Figure S3. The alignment score of S1PR1-S1PR2 was 0.016 and RMSD (C-alpha atoms of the aligned chains) was 0.625. RMSD is explained by the identity of two proteins of 51% and a 3% gap in the structures. The S1PR2 receptor model showed that disulfide bonds in the extracellular loops EL2 and EL3 were aligned with the corresponding S1PR1 disulfide bonds. Importantly, the S1P binding region of S1PR2 was well aligned with S1PR1 (compare supporting material) with a RMSD (alpha-C) of 0.322. The receptor structures were optimized by minimization and molecular dynamics. Optimized structures were used further for docking studies.
3.8.2 Docking study of S1PR2 with S1P and CYM-5520
Ligand-receptor binding models were generated as described in Methods. The final ligand-receptor complexes were ranked by glide score, emodel, and MM GB/SA energies, based on known interactions. The energetically most favorable and comparable poses representpossible binding modes in the S1PR1 and S1PR2 receptors. The docking scores are reported in Supplementary modeling table S5. In S1PR2, the S1P head group interacts with R108 and E109 while the hydrocarbon alkyl tail interacts in the hydrophobic pocket.(Figure 5A) CYM-5520 sits in the hydrophobic pocket close to F250 (Figure 5B) with no apparent head group interaction. In the presence of S1P, CYM-5520 moves away from F86 closer to the hydrophobic region formed by F250 and W256. Co-docking of S1P and CYM-5520 suggested that the receptor pocket could accommodate CYM-5520 in the hydrophobic pocket close to W246 and F250 while the S1P tail adjusts in the space around it (Figure 5C). Molecular dynamics simulations did not show any change in the binding pattern. The corresponding interaction diagrams for individual and co-docked poses showing all of the residues in close contact with the ligands are shown in Figure 6.
Figure 5.
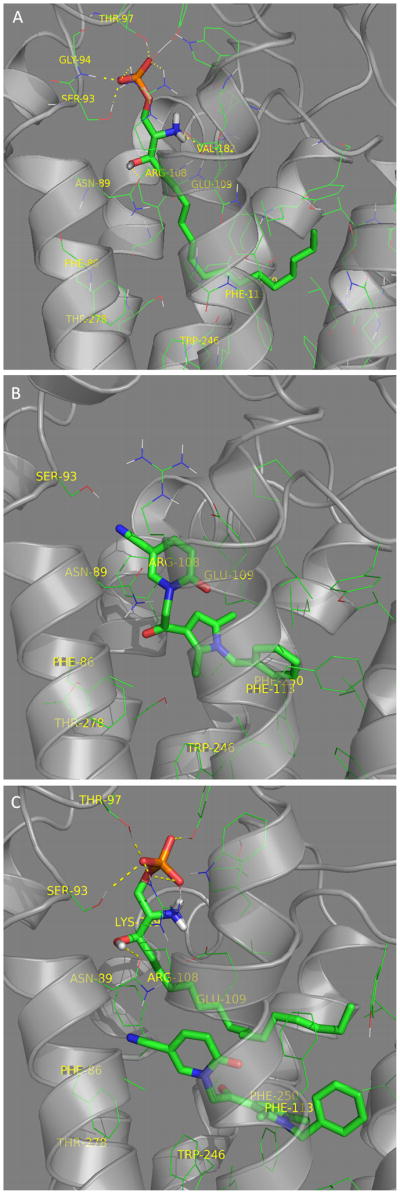
S1PR2 structure model. The ligands S1P (Panel A) and CYM-5520 (Panel B) are shown docked in the ligand binding pocket. S1P and CYM-5520 are co-docked in Panel C.
Figure 6.

Ligand interaction diagrams in the S1PR2 wild type pocket for (A) S1P, (B) CYM-5520, or in the S1PR2 co-docked WT pocket for (C) S1P and (D) CYM-5520. Amino acid type (sphere color):red - acidic (charged negative), purple - basic (charged positive), green – hydrophobic, turquoise – polar and gray - other (Gly). Ligand exposure: yellow - ligand atom exposed to solvent, Interaction type: solid pink - H-bond to protein backbone amd dotted pink - H-bond to protein site chain.
The SiFT analysis of the CYM-5520 ligand over several valid poses of S1PR2 post docking suggests important amino-acid residues that take part in hydrophobic interactions. The majority of interactions shown in the heat map matrix are conserved across the different poses. The SiFT analysis complements the ligand interaction map shown for CYM-5520.(Figure 6B)
4 Discussion and Conclusions
Chemical probes that elucidate interactions within and outside of the orthosteric binding pockets of GPCRs provide important insights both into mechanisms and potentially into interactions that make the pocket more pharmaceutically tractable. Detailed analysis of the recent liganded S1PR1 crystal structure 24 has provided insights into the S1PR1-S1PR5 ligand specificity. In the S1P receptor family the orthosteric ligand binding pocket is highly conserved, but residues that form binding pockets for allosteric ligands are more diverse.25 Defining the orthosteric pocket of S1PR1 defines the gatekeeper residues for selectivity of pharmacological ligands between receptors, and these side-chain protrusions into the pocket define binding pocket shape and impact on the variable efficiency of small molecule discovery between subtypes.13 For example, L276 in S1PR1 and its replacement by F263 in S1PR3 provided both loss of function mutations L276F for S1PR1 26, while that same mutation induced a gain of function for selective ligands of S1PR3 to bind S1PR1.13 The selectivity of fingolimod-phosphate for S1PR1, S1PR3, S1PR4 andS1PR5 while having no activity of S1PR2 27 can be explained by the steric interference of the F274 aromatic side-chain in S1PR2 precluding the binding of fingolimod while that residue is a conserved leucine in the remaining four receptors. Notably, a close contact between CYM5-520 and F274 is predicted in the S1PR2 model (Figure 6B). This residue is leucine in the other S1P receptors and thus this interaction may be important for the specificity of CYM-5520 binding to S1PR2. The significant difference between S1PR1 and S1PR4 with regard to subtype selectivity is M124 in S1PR1 (Leucine in S1PR4). In contrast S1PR1 and S1PR5 have no obvious gatekeeper residue differences and this is reflected in the parallel structure-activity relationships seen between these two receptor subtypes. The importance of screening is to define diverse chemical scaffolds that are not limited by the very difficult physical properties of the physiological lysophospholipid ligands.1 The hydrophobic, strong zwitterions, and their synthetic analogs28–32 are very sparingly soluble, do not cross biological membranes, are not orally bioavailable and require some significant stabilization of the phosphate ester bond to limit excessive lability in vivo.1, 33 Furthermore, the complete conservation of the zwitterionic headgroup interactions (E121R122) that provide >3 logs of potency 27 for ligand binding, select for lysophospholipid-like scaffolds that discriminate poorly between receptor subtypes. Only the addition of aromatic systems in place of the long acyl chain, and especially with the elaboration of heterocycles provides a useful basis for the identification of probes of the binding pocket that are truly selective and specific.13, 34 Reaching beyond the orthosteric interactions enhance the possibilities of finding truly novel chemical space.
High throughput screening of two diversity libraries identified novel S1PR2 agonists. The diaryl-oxadiazole chemical space activity “hot spot” identified in the comparison of the S1PR1 and S1PR3 HTS campaigns does not extend to these S1PR2 agonists, suggesting that the receptor family has significant diversity in ligand binding. We made analogues leading to CYM-5520 because this series was considered more amenable to chemical optimization, in part due to the nitro group in XAX-164. CYM-5520 lacks any functional group with any similarity to the zwitterionic head group of S1P. For these two reasons we focused these ligand-receptor binding studies to the CYM-5520/S1PR2 ligand/receptor system. Noncompetitive antagonist inhibition, functional response with triple mutant S1PR2, and absence of radioligand binding completion revealed a consistent scenario in which CYM-5520 is an allosteric agonist that can co-bind in the S1PR2 receptor with S1P. Since S1PR2 selective agonists are not currently available, these compounds may serve as leads for synthetic efforts designed to produce more potent chemical tools with utility in the study of the physiological function(s) of S1PR2.
Supplementary Material
Acknowledgments
This work was supported by Research Grants from the National Institute of Health, Molecular Library Probe Production Center Grant U54 MH084512 (E.R., H.R. P.H.), and a Grant-in-Aid for Young Scientists (A-20688005) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (H.S.) We thank Jill Ferguson for her help with editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki H, Ishizaka N, Sakurai T, Kurokawa K, Goto K, Kumada M, Takuwa Y. Molecular Cloning of a Novel Putative G Protein-Coupled Receptor Expressed in the Cardiovascular System. Biochemical and Biophysical Research Communications. 1993;190(3):1104–1109. doi: 10.1006/bbrc.1993.1163. [DOI] [PubMed] [Google Scholar]
- 3.Gonda K, Okamoto H, Takuwa N, Yatomi Y, Okazaki H, Sakurai T, Kimura S, Sillard R, Harii K, Takuwa Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and -insensitive G-proteins to multiple signalling pathways. Biochem J. 1999;337(1):67–75. [PMC free article] [PubMed] [Google Scholar]
- 4.Blom T, Bergelin N, Meinander A, Lof C, Slotte JP, Eriksson J, Tornquist K. An autocrine sphingosine-1-phosphate signaling loop enhances NF-kappaB-activation and survival. BMC Cell Biology. 2010;11(1):45. doi: 10.1186/1471-2121-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takashima S-i, Sugimoto N, Takuwa N, Okamoto Y, Yoshioka K, Takamura M, Takata S, Kaneko S, Takuwa Y. G12/13 and Gq mediate S1P2-induced inhibition of Rac and migration in vascular smooth muscle in a manner dependent on Rho but not Rho kinase. Cardiovascular Research. 2008;79(4):689–697. doi: 10.1093/cvr/cvn118. [DOI] [PubMed] [Google Scholar]
- 6.Kono M, Belyantseva IA, Skoura A, Frolenkov GI, Starost MF, Dreier JL, Lidington D, Bolz S-S, Friedman TB, Hla T, et al. Deafness and Stria Vascularis Defects in S1P2 Receptor-null Mice. Journal of Biological Chemistry. 2007;282(14):10690–10696. doi: 10.1074/jbc.M700370200. [DOI] [PubMed] [Google Scholar]
- 7.Skoura A, Sanchez T, Claffey K, Mandala SM, Proia RL, Hla T. Essential role of sphingosine 1–phosphate receptor 2 in pathological angiogenesis of the mouse retina. The Journal of Clinical Investigation. 2007;117(9):2506–2516. doi: 10.1172/JCI31123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cattoretti G, Mandelbaum J, Lee N, Chaves AH, Mahler AM, Chadburn A, Dalla-Favera R, Pasqualucci L, MacLennan AJ. Targeted Disruption of the S1P2 Sphingosine 1-Phosphate Receptor Gene Leads to Diffuse Large B-Cell Lymphoma Formation. Cancer Research. 2009;69(22):8686–8692. doi: 10.1158/0008-5472.CAN-09-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teijaro John R, Walsh Kevin B, Cahalan S, Fremgen Daniel M, Roberts E, Scott F, Martinborough E, Peach R, Oldstone Michael BA, Rosen H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell. 2011;146(6):980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanna MG, Wang S-K, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng W-C, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 11.Osada MM, Yatomi YY, Ohmori TT, Ikeda HH, Ozaki YY. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochemical and biophysical research communications. 2002;299(3):483–487. doi: 10.1016/s0006-291x(02)02671-2. [DOI] [PubMed] [Google Scholar]
- 12.Kim KK, Kim YLY-L, Sacket SJSJ, Kim HLH-L, Han MM, Park DSDS, Lee BKBK, Lee WKWK, Ha HJH-J, Im DSD-S. Sphingosine 1-phosphate (S1P) induces shape change in rat C6 glioma cells through the S1P2 receptor: development of an agonist for S1P receptors. Journal of Pharmacy and Pharmacology. 2007;59(7):1035–1041. doi: 10.1211/jpp.59.7.0017. [DOI] [PubMed] [Google Scholar]
- 13.Schurer SC, Brown SJ, Gonzalez-Cabrera PJ, Schaeffer MT, Chapman J, Jo E, Chase P, Spicer T, Hodder P, Rosen H. Ligand-binding pocket shape differences between sphingosine 1-phosphate (S1P) receptors S1P1 and S1P3 determine efficiency of chemical probe identification by ultrahigh-throughput screening. ACS Chem Biol. 2008;3(8):486–498. doi: 10.1021/cb800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aiyar A, Xiang Y, Leis J. Site-Directed Mutagenesis Using Overlap Extension PCR. In: Trower M, editor. In Vitro Mutagenesis Protocols. Vol. 57. Humana Press; 1996. pp. 177–191. [DOI] [PubMed] [Google Scholar]
- 15.Lieu PT, Machleidt T, Thyagarajan B, Fontes A, Frey E, Fuerstenau-Sharp M, Thompson DV, Swamilingiah GM, Derebail SS, Piper D, et al. Generation of Site-Specific Retargeting Platform Cell Lines for Drug Discovery Using phiC31 and R4 Integrases. Journal of Biomolecular Screening. 2009;14(10):1207–1215. doi: 10.1177/1087057109348941. [DOI] [PubMed] [Google Scholar]
- 16.Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. Crystal Structure of a Lipid G Protein–Coupled Receptor. Science. 2012;335(6070):851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); November 11–17 2006; Tampa, Florida, USA. 2006. [Google Scholar]
- 18.Kunapuli P, Ransom R, Murphy KL, Pettibone D, Kerby J, Grimwood S, Zuck P, Hodder P, Lacson R, Hoffman I, et al. Development of an intact cell reporter gene [beta]-lactamase assay for G protein-coupled receptors for high-throughput screening. Analytical Biochemistry. 2003;314(1):16–29. doi: 10.1016/s0003-2697(02)00587-0. [DOI] [PubMed] [Google Scholar]
- 19.Sorbi C, Franchini S, Tait A, Prandi A, Gallesi R, Angeli P, Marucci G, Pirona L, Poggesi E, Brasili L. 1,3-Dioxolane-Based Ligands as Rigid Analogues of Naftopidil: Structure–Affinity/Activity Relationships at α1 and 5-HT1A Receptors. Chem Med Chem. 2009;4(3):393–399. doi: 10.1002/cmdc.200800277. [DOI] [PubMed] [Google Scholar]
- 20.Osada M, Yatomi Y, Ohmori T, Ikeda H, Ozaki Y. Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochemical and Biophysical Research Communications. 2002;299(3):483–487. doi: 10.1016/s0006-291x(02)02671-2. [DOI] [PubMed] [Google Scholar]
- 21.Kenakin T. Pharmacological analysis of drug-receptor interaction. New York: Raven Press; 1987. [Google Scholar]
- 22.Parrill AL, Wang D, Bautista DL, Van Brocklyn JR, Lorincz Z, Fischer DJ, Baker DL, Liliom K, Spiegel S, Tigyi G. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J Biol Chem. 2000;275(50):39379–39384. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- 23.Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. Novel Genetically Encoded Biosensors Using Firefly Luciferase. ACS Chemical Biology. 2008;3(6):346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- 24.Hanson M, Roth C, Jo E, Griffith M, Scott F, Reinhart G, Desale H, Clemons B, Cahalan SM, Schurer SC, et al. Crystal Structure of a Lipid G-protein Coupled Receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MBA. Sphingosine-1-Phosphate and Its Receptors: Structure, Signaling, and Influence. Annual Review of Biochemistry. 2013;82(1) doi: 10.1146/annurev-biochem-062411-130916. null. [DOI] [PubMed] [Google Scholar]
- 26.Deng Q, Clemas JA, Chrebet G, Fischer P, Hale JJ, Li Z, Mills SG, Bergstrom J, Mandala S, Mosley R, et al. Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol Pharmacol. 2007;71(3):724–735. doi: 10.1124/mol.106.029223. [DOI] [PubMed] [Google Scholar]
- 27.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 28.Awad AS, Ye H, Huang LP, Li L, Foss FW, Macdonald TL, Lynch KR, Okusa MD. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. American Journal of Physiology-Renal Physiology. 2006;290(6):F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 29.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. The Journal of biological chemistry. 2005;280(11):9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 30.Foss FW, Jr, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, Macdonald TL. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorganic & medicinal chemistry. 2007;15 (2):663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR, Lin CY, Hla T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. The Journal of biological chemistry. 2007;282(12):9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- 32.Snyder AH, Foss FW, Davis MD, MacDonald TL, Lynch KR. Sphingosine 1-phosphate receptor antagonists and lymphocyte trafficking. Acta Pharmacologica Sinica. 2006;27:404–404. [Google Scholar]
- 33.Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–441. doi: 10.1038/nchembio804. [DOI] [PubMed] [Google Scholar]
- 34.Jo E, Bhhatarai B, Repetto E, Guerrero M, Riley S, Brown SJ, Kohno Y, Roberts E, Schürer SC, Rosen H. Novel Selective Allosteric and Bitopic Ligands for the S1P3 Receptor. ACS Chemical Biology. 2012;7(12):1975–1983. doi: 10.1021/cb300392z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.