

Summary
Cholera toxin (CT), a virulence factor elaborated by Vibrio cholerae, is sufficient to induce the severe diarrhea characteristic of cholera. The enzymatic moiety of CT (CtxA) increases cAMP synthesis in intestinal epithelial cells, leading to chloride ion (Cl−) efflux through the CFTR Cl− channel. To preserve electroneutrality and osmotic balance, sodium ions and water also flow into the intestinal lumen via a paracellular route. We find that CtxA-driven cAMP increase also inhibits Rab11/exocyst-mediated trafficking of host proteins including E-cadherin and Notch signaling components to cell-cell junctions in Drosophila, human intestinal epithelial cells, and ligated mouse ileal loops, thereby disrupting barrier function. Additionally, CtxA induces junctional damage, weight loss, and dye leakage in the Drosophila gut, contributing to lethality from live V. cholerae infection, all of which can be rescued by Rab11 over-expression. These barrier-disrupting effects of CtxA may act in parallel with Cl− secretion to drive the pathophysiology of cholera.
Keywords: Cholera toxin, Drosophila, intestinal epithelium, CACO-2 cells, T84 cells, exocyst, Rab11, Sec15, Cadherin, Notch, Delta, Jagged, ZO-1, Claudin-2, electron microscopy
Introduction
Vibrio cholerae (V.c.) produces the virulence factor cholera toxin (CT), which alone can cause the severe watery diarrhea pathognomonic of cholera ((De Haan and Hirst, 2004; Sack et al., 2004). CT binds to the GM1 ganglioside receptor of intestinal epithelial cells, and is then endocytosed and transported in a retrograde fashion to the endoplasmic reticulum, whereupon the enzymatically active A subunit (CtxA) is translocated into the cytoplasm. There CtxA binds the host co-factor GTP-ARF6 and transfers ADP-ribose from NAD to the alpha subunit of the stimulating G-protein (Gsα) to activate Gsα, which in turn stimulates host adenylate cyclases (AC) at the plasma membrane causing a pathological rise in cAMP concentration.
cAMP promotes fluid secretion from crypt intestinal cells primarily by activating cAMP-dependent protein kinase A (PKA), which phosphorylates several ion channels and transporters including the linchpin CFTR chloride ion channel (Cheng et al., 1991; Picciotto et al., 1992), basolateral K+ ion channels, and an ATP-dependent Na+/K+/Cl− co-transporter. These combined effects of PKA provoke massive secretion of Cl− ions into the intestinal lumen. Na+ ions flow into the lumen via a paracellular pathway to balance the charge of Cl− ions, thus resulting in a net efflux of NaCl into the gut. Luminal flow of electrolytes produces an osmotic gradient entraining compensatory water efflux, resulting in large volume fluid loss (up to 10–20 liters per day). This model of CtxA action is supported by a wealth of experimental evidence, a definitive example being the failure of purified CT to induce fluid secretion in the small intestine of CFTR−/− null mutant mice (Gabriel et al., 1994).
Recently, we found that edema factor (EF), a highly active adenylate cyclase (Leppla, 1982) produced by Bacillus anthracis (B.a.), disrupts endocytic trafficking of adhesion and signaling proteins to adherens junctions (AJs, Fig. 1A) (Guichard et al., 2010). This study revealed that EF reduces levels of Rab11, a small GTPase residing in late recycling endosomes which binds to Sec15, a component of the exocyst complex, to tether recycling vesicles to the plasma membrane (Fig. 1B, reviewed in (Heider and Munson, 2012). Inhibition of Rab11 leads to decreased junctional accumulation of Sec15 and cargo proteins including cadherins (Langevin et al., 2005; Murthy and Schwarz, 2004; Murthy et al., 2010) and Notch pathway components such as the Delta ligand (Guichard et al., 2010; Jafar-Nejad et al., 2005) in flies and in human vascular endothelial cells (Guichard et al., 2010).
Figure 1. Diagram of cell-cell junctions.
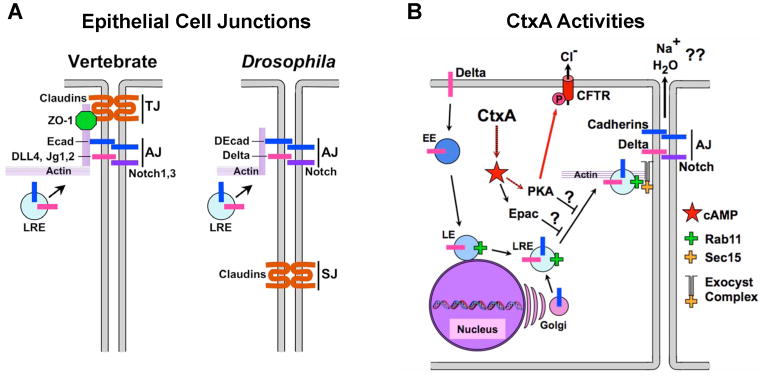
A) Schematic diagram of epithelial cell-cell junctions in vertebrates (left) and invertebrates (right). TJ = tight junction; AJ = adherens junction, SJ = septate junction (the functional equivalent of the TJ in invertebrates). B) Effect of CtxA and high-level cAMP production in epithelial cells. Notch ligands (e.g., Dl) are endocytosed and Rab11+ late recycling endosomes (LREs) fuse with Golgi vesicles containing newly synthesized protein cargo (e.g., E-cad). LREs are tethered to the exocyst complex at the plasma membrane via an interaction between Rab11 and Sec15 to initiate delivery of adhesion proteins (e.g., Ecad) and signaling components (e.g., Dl) to the AJ. CtxA leads to overproduction of cAMP to promote PKA mediated Cl− secretion via the CFTR ion channel. CtxA also blocks exocyst-mediated trafficking via the PKA and Epac cAMP effectors to disrupt cell junctions (this study). Fig. 1 is related to Supp. Fig. 1.
Here, we show that CtxA also disrupts Rab11-dependent protein trafficking to cell junctions in Drosophila wing and intestinal epithelial cells, in human intestinal epithelial cell lines, and in vivo in ligated murine ileal loops. CtxA also disrupts intestinal barrier integrity in Drosophila, and contributes to the lethality of live V.c. infection. Importantly, all of these effects of CtxA can be reversed by over-expression of Rab11. These previously undescribed effects of CtxA, acting in conjunction with its known induction of Cl− ion secretion, may contribute to the pathophysiology of severe cholera.
Results
CtxA disrupts exocyst-mediated junctional trafficking in Drosophila epithelial cells
CtxA activates Gsα pathways in the early Drosophila embryo (Morize et al., 1998) and wing (Katanayeva et al., 2010). Also, flies infected with V.c. die in a CtxA-dependent fashion through a process involving host cAMP-regulated ion channels (Blow et al., 2005). To explore CtxA activity in Drosophila, we expressed CtxA directly within the cytoplasm using the heterologous GAL4/UAS gene expression system (Brand and Perrimon, 1993), bypassing receptor binding and endocytic steps involved in host cell entry. In the well-characterized wing developmental system, strong expression of CtxA produced adult phenotypes similar to those observed in Notch mutants, consisting of thickened veins (Fig. 2B; compare to wild-type (WT) in panel A) and notches along the wing edge (margin) (Supp. Fig. 1B, arrow, compare to mild N−/+ phenotype in Supp. Fig. 1A). Furthermore, CtxA reduced expression of the Notch target gene cut (Fig. 2E, compare to 2D) along the wing margin primordium. Consistent with CtxA acting via the expected Gsα-mediated activation of endogenous AC in the wing, co-expressing CtxA with either of two Drosophila Gsα subunits caused wing phenotypes that were much stronger than those produced by CtxA alone (Supp. Fig. 1G–L). Also, expression of a constitutively active form of one of these Gsα subunits (Gsα60A) mimicked the effect of CtxA (Katanayeva et al., 2010). Reciprocally, RNAi knock-down of genes encoding any of three Gsα subunits (Supp. Fig. 1M–R) or the AC rutabaga (Supp. Fig. 1S, T) markedly suppressed CtxA phenotypes.
Figure 2. inhibits Notch signaling and Rab11 activity in Drosophila.

A–C, G–I) Drosophila wings of the indicated genotypes. Longitudinal veins = L2–L5, wing margin =M. D–F) Expression of the Notch target gene cut (detected by anti-Cut staining) along the margin in third instar larval imaginal discs of the indicated genotypes. J, L, N, P) WT wing discs, and K, M, O, Q) wing discs expressing CtxA under the control of the wingGAL4 driver stained for expression of exocyst (Rab11, Sec15-GFP) and AJ (Delta, DECad) components. Larvae were raised at 25°C for all panels except (P, Q) = raised at 29°C for 3hrs prior to dissection. Insets in panels J–Q are Z-sections. Insets in (N, O) are deeper horizontal sections. Arrows in panels in (N, O) indicate the two parallel rows of cells giving rise to the dorsal (magenta) and ventral (white) components of the wing margin. The wingGAL4 driver is expressed more strongly on the dorsal surface, consistent with the effects of CtxA expression being more pronounced on the dorsal component of the margin (O). Arrowheads in M indicate ectopic basal vesicles. Fig. 2 is related to Supp. Fig. 2.
Genetic epistasis experiments confirmed the Notch inhibitory activity of CtxA. For example, an activated allele of Notch (N*) fully suppressed the effect of CtxA (Supp. Fig. 1 E, compare to panels A, D) indicating that CtxA acts upstream of the Notch receptor. Conversely, CtxA reversed the vein-loss phenotype caused by ectopic expression of the Delta ligand in the second wing vein primordium (Supp. Fig. 1F), revealing that it acts downstream of ligand production. In addition, the thickened veins of CtxA-expressing wings were significantly enhanced in Notch−/+ heterozygotes (Supp. Fig. 1C).
CtxA-induced wing phenotypes are highly similar to those produced by a dominant-negative (DN) version of Rab11 (Fig. 2C, compare to 2B), a small GTPase involved in recycling endocytic vesicles to adherens junctions. Consistent with their closely allied adult wing phenotypes, CtxA and DN-Rab11 both reduced expression of the Notch target gene cut along the presumptive wing margin in imaginal discs (Fig. 2D–F). CtxA and DN-Rab11 acted synergistically when co-expressed (Fig. 2I, compare to 2B, C), while co-expression of WT-Rab11 (which on its own had no visible effect - Fig. 2G) with CtxA almost fully rescued the toxin phenotype (Fig. 2H, compare to 2B). Moreover, CtxA significantly reduced apical levels of Rab11 (Fig. 2J, K) and resulted in basal mis-localization of this GTPase (Fig. 2K, arrowhead in inset). CtxA also greatly reduced the levels of a GFP-tagged form of Sec15 (Fig. 2M), the exocyst binding partner of Rab11, which normally appears within large round structures near the apical cell surface (Fig. 2L). In CtxA-expressing cells most remaining Sec15-GFP vesicles appeared irregular in shape and were mislocalized more basally (Fig. 2M, arrowheads in inset). These alterations in Rab11/Sec15 levels and distribution were associated with expected defects in endocytic recycling of cargo such as the Notch ligand Delta (Fig. 2, N, O) and the cell adhesion molecule DECad (Fig. 2P, Q) to AJs. Reflecting the potent Rab11 rescue of the adult CtxA wing phenotype (Fig. 2H), apical Sec15-GFP vesicles and junctional levels of Delta and DECad were nearly fully restored by co-expression of Rab11 with CtxA (Supp. Fig. 2C–H). We conclude that CtxA inhibits exocyst-mediated trafficking of DECad and Delta to cell junctions in Drosophila wing epithelial cells.
CT disrupts cell-cell junctions and Notch signaling in human intestinal epithelial cells
We next asked whether CT treatment of human intestinal epithelial cells could disrupt trafficking to junctions as CtxA expression did in flies. We treated newly confluent cultures of CACO-2 or T84 cells with purified CT (CtxA/B) and examined the localization of Rab11, Sec15, and candidate cargo proteins at cell-cell junctions (Fig. 3). Well-defined AJs form in untreated CACO-2 cells, which are delineated by apically restricted E-cadherin (Ecad) staining (Fig. 3A, C). In these cells, Rab11 (Fig. 3E, G) and Sec15 (Fig. 3I, K) accumulate in a grainy cytoplasmic pool and are enriched within a narrow apical band co-localizing with the AJ (arrows in Fig. 3E, G - Rab11; and 3I, K - Sec15).
Figure 3. CT disrupts localization of exocyst and AJ components in human intestinal epithelial cells.

Expression of exocyst and AJ components in recently confluent monolayers of CACO-2 cells (A–L, O, P) and T84 (M, N) cells either untreated or CT-treated (three times over a 28 hour period) and fixed in methanol. A–D) Ecad viewed from above (A, B) or in cross section (C, D). A prominent cytoplasmic pool of Ecad vesicles is present only in untreated cells (A, asterisks). Ecad is mislocalized along the entire apical-basal axis of CT treated cells (C, D: brackets). E–L) The same field of cells triple stained for Rab11 (green), Sec15 (green), and Ecad (red). Arrows highlight narrow bands of Rab11 or Sec15 membrane-adjacent staining, which co-labels with Ecad (G, K center panels) and are separated from a pool of cytoplasmic staining by a subcortical zone virtually devoid of labeling (brackets in G, K). In contrast, CT-treated cells display a clear gap between Rab11/Sec15 staining from neighboring cells that corresponds to the position of Ecad staining (H, right panel, arrowheads). O, P) Localization of the EPAC effector Rap1 to points of cell-cell contact in a subset of cells (double arrow) is largely disrupted in CT-treated cells, which have more labeled small cytoplasmic vesicles (arrow). Fig. 3 is related to Supp. Fig. 3.
Reduced junctional levels of these components and altered distributions of their protein cargo were evident after 6h of CT treatment (not shown) and became more pronounced in cells treated three times over the course of 28h (henceforth our standard regimen). The distribution of core AJ component Ecad was dramatically altered in CT-treated cells leading to gaps in staining (Fig. 3B, arrows; compare to 3A) and loss of its apical restriction, with staining extending along the entire apical-basal axis (Fig. 3D, compare to 3C). In addition, a cytoplasmic pool of Ecad present in untreated cells (Fig. 3A, asterisks) virtually disappeared in CT-treated cells (Fig. 3B). CT also abolished junctional localization of Rab11 (Fig. 3F, H - compare to 3E, G) and Sec15 (Fig. 3J, L - compare to 3I, K) and caused ectopic accumulation of these proteins in subcortical regions (Fig. 3F, J - compare to 3E, I), which are zones virtually devoid of Rab11 and Sec15 label in untreated CACO-2 cells (brackets in Fig. 3G, K). Similarly in T84 cells, Sec15 staining at the cell surface, which in some cells was very pronounced (Fig. 3M), was greatly diminished by CT treatment (Fig. 3N), and the remaining Sec15 staining was observed in scattered cytoplasmic vesicles.
Transmission electron microscopy (EM) of CT-treated CACO-2 cells revealed prominent varicose gaps between cells and disorganized convoluted AJs. (Fig. 4A, B), which were paralleled by broadened and less uniform subcortical F-actin staining (Fig. 4C, D - arrows indicate tricellular varicosities in 4D - see also Supp. Fig. 3C, D). However, CT did not appreciably alter the apical meshwork of tubulin fibers (Supp. Fig. 4A–D), indicating that the cytoskeletal architecture was not globally disrupted.
Figure 4. CT disrupts localization of TJ components in intestinal epithelial cells.

Expression of TJ components in untreated (A, C, E, G, I, K, M) and CT-treated (B, D, F, H, J, L, N; 3X over 28 hours) recently confluent CACO-2 cells fixed in paraformaldehyde except for K, L (methanol fixed). A, B) Electron micrographs of untreated CACO-2 cells (A), and CT-treated cells (B). Prominent gaps (arrow) and convoluted borders are visible between cells, which can span a significant fraction of the apical-basal axis. Apical junctions (arrowheads) also tend to be less regular in CT-treated cells and microvilli (mv) are reduced. CT treatment disrupts regularly spaced cortical F-actin fibers (C), resulting in broader and disorganized phalloidin staining (D, arrows). Continuous staining of the TJ protein Claudin-2 (E), is interrupted in CT-treated cells (F, arrows). Regular ZO-1 staining (G) becomes irregular in CT-treated cells, punctuated by varicose accumulations (H, arrow), and ZO-1 staining accumulates in the cytoplasm, which in some cells forms a cap over the nucleus (H, asterisks). I, J) Z-section of ZO-1 stains. Bracket = apical region, arrow = ectopic basal ZO-1. The registration of the TJ and AJ (K) is uncoupled in CT-treated cells (L, arrows). M, N) ZO-1 and Rab11 co-localize in a subset of large vesicles adjacent to the plasma membrane (M, arrows) which is lost following CT treatment (N). Fig. 4 is related to Supp. Fig. 4.
We also examined the structure of TJs following CT treatment. In untreated CACO-2 cells, the TJ adhesion protein claudin-2 (Cld2), which serves as a major paracellular cation and water pore in the intestinal epithelium (Rosenthal et al., 2010), is localized to a narrow apical band (Fig. 4E). However, in CT-treated cells, Cld2 staining followed a convoluted path with abnormal cytoplasmic accumulations (Fig. 4F, arrows), and overall Cld2 levels were markedly increased compared to untreated cells (compare insets in Fig. 4E, F). Similarly, normally continuous staining of the TJ adaptor protein ZO-1 (Fig. 4G, K) assumed a tortuous path and accumulated in large patches along the borders of CT-treated cells (Fig. 4H, L). While CT treatment resulted in a net increase in ZO-1 staining, junctional levels of ZO-1 remained normal and restricted to the apical region of the cell (Fig. 4I, J; Supp. Fig. 4C, D), contrasting with mis-localization of the AJ protein Ecad that spanned the apical-basal axis (Fig. 3C, D). The most prominent increase in ZO-1 levels was in a basally located cytoplasmic pool (Supp. Fig. 4D) and in a bright disk above the nucleus in some CT-treated cells (Fig. 4H, asterisks). CT also disrupted the normal close alignment of AJ and TJ components. In untreated cells, these two junctions are closely stacked atop of one another (Fig. 4K, red apical ZO-1 and green AJ-level Ecad staining), a feature lost in CT-treated cultures. In some cases, the TJ path ran entirely out of register with that of the underlying AJ border (Fig. 4L, arrows). These effects of CT on the TJ were most pronounced in newly confluent cells, but also evident in mature confluent monolayers (Supp. Fig. 4E–H). Paralleling effects observed with ZO-1, CT induced an irregular pattern of occludin staining at cell borders as well as ectopic foci of cytoplasmic accumulation (Supp. Fig. 4I–L). Finally, CT disrupted a vesicular association of Rab11 with ZO-1. In untreated CACO-2 cells, a subset of large Rab11 vesicles co-localized with ZO-1 just below the level of the TJ (Fig. 4M, arrows), suggesting that Rab11-dependent endocytic recycling aids in trafficking this adaptor protein to cell-cell junctions. Such co-localization of Rab11 with ZO-1 was virtually abolished in CT-treated cells (Fig. 4N). Similarly in T84 cells, Rab11 and ZO-1 co-localized in large vesicles (Supp. Fig. 4O), and this association was greatly reduced by CT treatment (Supp. Fig. 4P). As in CACO-2 cells, high levels of ZO-1 also accumulated ectopically in basal regions of CT-treated T84 cells (Supp. Fig. 4R, arrows, compare to Supp. Fig. 4Q).
Since CtxA reduced Delta trafficking to the AJ and inhibited Notch signaling in Drosophila, we also examined the distribution of Notch pathway components in CACO-2 cells. Notch ligands Delta-like 4 (Dll4) and Jagged-2 (Jg2), and receptor Notch-3 (N3) are primarily detectable in punctate vesicles located in close proximity to the plasma membrane (Fig. 5A, C, E). In contrast, all Notch components exhibited a diffuse spongy cytoplasmic staining pattern in CT-treated cells (Fig. 5B, D, F). The CT effect was particularly pronounced in a subset of newly confluent CACO-2 cells, which formed scattered circular cup-like structures with rims delineated by F-actin (Supp. Fig. 3G–J). In untreated cells, Notch pathway components accumulated along the inner lip of these cups, forming rings of high level staining (Supp. Fig. 5A, C, E). CT treatment eliminated this staining and disrupted the radial symmetry of these cup-like structures (Supp. Fig. 5B, D, F). Consistent with its effects on localization of Notch pathway components, CT greatly reduced levels of signaling via the Notch1 receptor. Following ligand binding and receptor proteolysis, an activated cleavage product of the receptor (N1*) translocates to the nucleus and acts as a transcriptional cofactor to regulate expression of Notch target genes (Fortini, 2009). In untreated cells the great majority of N1* staining was confined to the nucleus (Fig. 5G), indicative of constitutive signaling via this receptor. Following CT treatment, nuclear N1* staining was greatly reduced (Fig. 5H), indicating strong inhibition of this pathway.
Figure 5. CT disrupts localization of Notch pathway components and signaling.

Recently confluent monolayers of CACO-2 cells, untreated or treated with CT (3X over 28 hours), were fixed with paraformaldehyde and stained for expression of the Notch pathway components as indicated. N1* = activated Notch1. The equalization of nuclear and cytoplasmic N1* staining observed here is also observed in cells treated for only 6 hours with CT in which the overall levels of staining have not yet been greatly diminished. Fig. 5 is related to Supp. Fig. 5.
cAMP exerts its effects through two known effectors, protein kinase A (PKA) and Epac (a guanine nucleotide exchange factor that activates the small GTPase Rap1). We asked whether these two major cAMP effector branches mediated the junction-disrupting effects of CtxA. In CACO-2 cells, inhibitors specific for PKA (H89) or Epac (ESI-09) markedly reduced CT effects (Supp. Fig. 5G–J) while cAMP analogs that selectively activate PKA (6Bnz: Supp. Fig. 5K) or Epac (8Cpt: Supp. Fig. 5L) mimicked CT-induced junction disruption assessed by ZO1 staining at TJs and Ecad staining at AJs (not shown). Consistent with CT acting on the Epac cAMP-effector branch, CT treated cells displayed reduced subcortical localization of the Epac mediator Rap1 (Fig. 3O, P). Paralleling these human cell results, expression of activated forms of PKA (PKA*) or Rap1 (Rap1*) in the Drosophila wing epithelium mimicked the effect of CtxA, including adult wing phenotypes (Supp. Fig. 2I–K) and reduced levels of Rab11 (Supp. Fig. 2L–N), Sec15-GFP (Supp. Fig. 2O–Q), and Delta (Supp. Fig. 2R–T).
CT reduces levels of exocyst components in ligated murine ileal loops
We next examined whether CT reduced levels of endocytic recycling components and disrupted cell-cell junctions in vivo employing the ligated murine ileal loop model. In anesthetized mice, two segments of the small intestine (ileum) were ligated with bowel clamps. CT was then injected into one of the segments and control saline into the other. The ligated intestines were returned to the body cavity, the incisions sutured, and the animals kept anesthetized for an additional 5 h before sacrifice. While control ileal loops appeared similar to adjacent non-ligated segments of the intestine (Fig. 6A), CT-treated segments were markedly inflated (Fig. 6B, and quantified in Methods), indicative of the classic fluid secretion associated with CT intoxication.
Figure 6. CT disrupts localization of exocyst components in vivo in ligated ileal loops.

Ligated ileal loops injected with saline (A) or CT (B) and incubated for 5 h. Rab11 staining in control loops (C, E) resembles that of non-treated animals (not shown), localizing to a narrow apical region (arrows) in epithelial cells of both the villus (C) and crypt (E), which is virtually abolished in of CT-injected loops (D, F). Crypts in CT-injected loops are also less regularly shaped than in controls (E, F). Apical Sec15 staining in the crypts of control loops (G) is greatly reduced in CT-injected loops (H). Regular F-actin staining in untreated control loops (I) becomes irregular and discontinuous (arrows) in CT-treated loops (J). Apical E-cadherin (Ecad) staining (arrows) is tightly aligned between neighboring epithelial cells in control injected loops (K), but often forks apically in CT-treated loops (L, arrows) revealing partial separation between adjacent cells. A cytoplasmic pool of Ecad in untreated epithelial cells (K, asterisks) is virtually abolished by CT treatment (L, asterisks indicate cytoplasmic void in staining). Albumin staining in control crypts, which is excluded from the lumen (M) accumulates at high levels along the luminal face of the crypt epithelium (arrow) as well as within the lumen itself (asterisk) in CT-treated injected loops (N). Fig. 6 is related to Supp. Fig. 6.
In uninjected mice or control ileal loops injected with saline, strong Rab11 staining was detected as an uninterrupted apical band running along the luminal epithelial surface of the crypt-to-villus axis (Fig. 6C, E), consistent with previous descriptions (Goldenring et al., 1996; Silvis et al., 2009). In tangential sections at the level of the crypts, Rab11 staining appeared as circular luminal rings (Fig. 6E). Sec15 staining was primarily confined to large cytoplasmic vesicles in the crypts (Fig. 6G), with much lower levels detected in the villus. In CT-injected loops, the staining for these endocytic trafficking components was dramatically reduced. Apical Rab11 staining virtually disappeared along the entire crypt-villus axis (Fig. 6D, F), and cross-sections of the crypts were less circular than in controls (Fig. 6F, arrows). Similarly, Sec15 staining was greatly reduced in a subset of crypts (Fig. 6H) that received high levels of CT (Supp. Fig. 6), which were also irregularly shaped. Paralleling the effects we observed in CACO-2 cells (Fig. 3C, D), CT induced a redistribution of Ecad in vivo. In untreated ileal loops, Ecad was concentrated apically at AJs (Fig. 6K, arrows) and accumulated in a prominent cytoplasmic pool (Fig. 6K, asterisks). In contrast, in CT-treated loops, Ecad staining expanded to occupy the entire apical-basal axis with a concomitant reduction of the cytoplasmic pool (Fig. 6L, asterisks). In addition, Ecad staining often forked apically in CT-treated loops (arrows in Fig. 6L, compare to 6K) revealing a clear separation between adjacent cells, evident also by apical gaps in F-actin staining (arrows in Fig. 6J, compare to smooth apical surface contour in 6I). Leakage of albumin from the vasculature into the intestinal lumen has been reported in human cholera patients (in the rice stool) as well as in CT-treated rabbits (De and Chatterje, 1953). Similarly, we found elevated levels of albumin staining accumulated adjacent to the luminal surface of some CT-treated crypts as well as within the lumen itself (Fig. 6 M, N). We conclude that CT reduces endocytic trafficking in vivo in the small intestine causing junctional defects similar to those observed in cell culture and leading to leakage of fluid and protein into the intestinal lumen.
Elevated Rab11 protects against CtxA-dependent V. cholerae infection in Drosophila
The Drosophila midgut epithelium shares many structural and regulatory mechanisms with the mammalian small intestine, and has been widely used as model for basic cell biological functions including innate immunity, response to damage, and stem cell homeostasis (reviewed in (Apidianakis and Rahme, 2011). Furthermore, oral infection of Drosophila with V.c. results in CT-dependent weight loss and lethality (Blow et al., 2005). We therefore used this system to examine the role of Rab11 in maintaining intestinal barrier function.
Targeted CtxA expression in the Drosophila midgut resulted in emaciated flies (Fig. 7B, compare to 7A) with reduced body weight (Fig. 7D), and co-expressing WT-Rab11 with CtxA counteracted these toxic effects (Fig. 7C, D). In addition, adult-specific expression of Ctx in the midgut reduced viability, an effect also rescued by Rab11 over-expression (Supp. Fig. 7A). Conversely, reduction in Rab11 levels (i.e., heterozygosity for either of two rab11- loss-of-function alleles: see Materials and Methods) resulted in midgut CtxA expression becoming 100% lethal (data not shown). Midgut expression CtxA also reduced apical levels of Rab11 (Fig. 7E), Sec15 (Fig. 7I, J), the AJ markers Ecad (Fig. 7L, M) and alpha-catenin (Supp. Fig. 7D–G), and the septate junction marker Polycheatoid (Pyd) - the Drosophila ortholog of ZO-1 (Supp. Fig. 7C–E). Ultrastructural analysis revealed that, similar to CT-treated CACO-2 cells, CtxA expression in the Drosophila midgut caused significant gaps and convoluted borders between intestinal epithelial cells (Fig. 7O, P; Supp. Fig. 7H–P). Over-expression of Rab11 rescued these CtxA-induced junctional defects in the midgut, restoring apical staining of Sec15-GFP (Fig. 7K) and Ecad (Fig. 7N), and reducing the size of gaps between enterocytes evident at the EM level (Fig. 7Q). In addition, weight loss caused by midgut expression of CtxA could be rescued by RNAi knock-down of Gsα60A or inhibition of the SK K+ channel with clotrimazole (Supp. Fig. 7B), both essential for CtxA-dependent weight loss and lethality in flies infected with V.c. (Blow et al., 2005). Thus, CtxA acts by very similar mechanisms in Drosophila intestinal and wing epithelial cells to disrupt Rab11-dependent junctional trafficking.
Figure 7. Rab11 rescues CtxA-dependent weight-loss and junction disruption in Drosophila.

A–D, F–H) Flies were fed a 5% sucrose plus dye solution. E, I–S) Flies were fed standard solid fly food. Adult NP1-GAL4/tubGAL80ts control flies (A) appear WT. Modest midgut expression of CtxA with this driver in flies raised at 22°C (RT), a permissive temperature for GAL80 that reduces but does not eliminate NP1-GAL4 activity, results in emaciated flies (B, arrows), which is counteracted by co-expression with Rab11 (C). Hindgut specific expression of CtxA also results in a similar emaciated phenotype (not shown). D) Average weights of NP1/GAL80ts control flies and flies expressing CtxA, CtxA+WT-Rab11, or WT-Rab11 alone in the gut (error bars = St. Dev). ** p = 0.0017 for the comparison of NP1/G80ts controls versus NP1/G80ts>CtxA flies; *** p = 0.0008 for the comparison of NP1/G80ts>Ctx versus NP1/G80ts>Ctx+Rab11. E) Apical Rab11 staining in the midgut of an NP1/+ control fly bracket in upper panel) is reduced by CtxA expression (asterisk in lower panel). F–H) Flies were fed with a sucrose dye solution, which remains strictly confined to the intestine in WT flies (F), but leaks into the body cavity of “smurfing” flies expressing CtxA (G) or DN-Rab11 (H) in the midgut. I–Q) Junction disrupting effects of CtxA can be rescued by co-expression with Rab11. Note: the DECad-Tomato construct is expressed from the endogenous DECad promoter. Sec15 = Sec15-GFP which by an anti-Sec15 antibody. R) Infection of flies with a Δctxa strain of V. cholerae (V.c., dashed red curve) results in decreased lethality compared to infection WT V.c. (solid red curve: p< 0.0001) and midgut specific expression of CtxA (with the NP1 GAL4 driver) restores lethality (dashed green curve: p< 0.0001) to the V.c. Δctxa strain. S) Rab11 rescues CtxA-dependent junction disruption caused by infection of flies with V.c.. DECad = DECad-Tomato. T) Summary model. Fig. 7 is related to Supp. Fig. 7.
Barrier disruption in the gut can also be assessed using a leakage assay in which flies are fed a solution of sugar water containing a colored food dye (Rera et al., 2011). Dye accumulation is restricted to the intestine in 100% of young WT flies, but if the epithelial barrier is breached, dye accesses internal body cavities and appendages, producing blue flies (“smurfing”)(Rera et al., 2011). In contrast to WT or NP1/+ control flies (Fig. 7F), midgut-specific expression of CtxA resulting in a significant fraction (≈ 20%) of flies smurfing (Fig. 7G). Expression of DN-Rab11 in enterocytes (Fig. 7H) also caused dye leakage at frequencies comparable to CtxA treatment, indicating that inhibition of endocytic recycling is sufficient to induce barrier disruption in the gut. Unlike CtxA, however, DN-Rab11 did not cause weight loss (data not shown), suggesting that a critical component of CtxA-dependent fluid leakage and weight loss are mediated by a Rab11-independent mechanism, such as cAMP-regulated ion channel activation (Blow et al., 2005).
As previously shown for exogenous CT provided during live infection of Drosophila with V.c. (Blow et al., 2005), gut-specific expression of CtxA restored lethality upon infection with a V.c. Δctxa mutant and also accelerated death caused by infection with the WT strain (Fig. 7R). Consistent with our model, over-expression of WT-Rab11 increased survival of flies infected with V.c. (Fig. 7R). Paralleling the effects of directly expressing CtxA in the midgut (e.g., in NP1>Ctx flies), V.c. infection of flies resulted in ctxA-dependent down-regulation of junctional Ecad levels (Fig. 7S), once again rescued by over-expression of Rab11 (Fig. 7S). Cumulatively, these findings implicate Rab11-dependent endocytic recycling in maintaining normal intestinal barrier function and indicate that boosting this process offers protection against CT-dependent effects of V.c. infection.
Discussion
In the current study, we find that CtxA causes emaciation, disorganized junctions, and barrier disruption when expressed in the Drosophila midgut and contributes to lethality during infection of flies with V.c.. Similarly, CT treatment produced apical gaps between epithelial cells in murine ileal loops, and histochemical staining and ultrastructural examination of CT-intoxicated human CACO-2 cells and Drosophila intestinal epithelial cells reveal parallel pathologies consisting of inter-cellular lacunae and convoluted borders between cells virtually identical to those observed in duodenal biopsies from human cholera patients (Mathan et al., 1995). Also, increased epithelial dye permeability has been reported in rats, mice, and rabbits following CT exposure (Magnusson et al., 1985; Ohishi and Odagiri, 1984; Triadafilopoulos et al., 1989), and serum albumin was found in the lumen of CT-treated rabbit ileal loops and in the stool of some cholera patients (De and Chatterje, 1953), which we too observed in CT-treated murine ileal loops. Furthermore, subcutaneous injection of CT in guinea pigs and rabbits leads to vascular effusion (Craig, 1965), which has been used as a criterion for assessing virulence of V.c. isolates (Sasmal et al., 1995).
Both CT and EF inhibit junction formation and barrier function at least in part by inhibiting Rab11-dependent endocytic recycling. Thus, over-expression of WT-Rab11 in the gut rescued CT-induced weight-loss, junctional organization, and intestinal barrier integrity in Drosophila as well as CT-dependent sensitivity to V.c. infection. Interestingly, gut-specific inhibition of Rab11 alone did not result in weight-loss, suggesting that the activation of ion channels is Rab11-independent (Blow et al., 2005). Cumulatively, these data provide strong evidence for a junction destabilizing effect of CT, which may act in concert with known toxin effects to stimulate ion conductances.
Potential role of junction disruption by CtxA with regard to its effects on ion secretion
A large body of work has established that CT-dependent cAMP production activates PKA, which then phosphorylates and increases the conductance of the apically localized CFTR, a Cl− ion transporter concentrated in epithelial cells of crypts in the small intestine (Jakab et al., 2011; Strong et al., 1994). The resulting Cl− flux is accompanied by efflux of Na+ ions and water into the intestinal lumen to preserve charge and osmotic balance, respectively. The exact path by which Na+ and water flow across the epithelium is not certain, but it is generally assumed that at least part of this flux occurs via the paracellular route (Barrett, 2006; Fromter and Diamond, 1972; Pappenheimer and Reiss, 1987). The ability of CT to inhibit endocytic recycling and disrupt epithelial junctions reported here could contribute to fluid secretion in several ways. First, by disrupting the highly regulated organization of the junctions, the paracellular flow of cations and water might be facilitated in the crypts where CT strongly down-regulated Rab11 and Sec15 and expression of the CFTR Cl− channel is maximal. Our observation that serum albumin accumulates within the lumen of crypts in CT-treated ileal loops is consistent with such compromised junctional barrier integrity. Second, elevated levels of Cld2, a paracellular channel selective for cations and water (Rosenthal et al., 2010), could contribute to increasing Na+ conductances, as suggested by a recent study in which Notch inhibition in CACO-2 cells lead to elevated levels of Cld2 and increased epithelial conductances (Dahan et al., 2011). CT may also destabilize junctions in the villus as it reduced Rab11 levels along the entire villus, altered expression of Ecad, and induced apical gaps.
CT inhibition of exocyst-mediated recycling could also alter the dynamics of ion channel turnover or targeting to the apical and basal membranes. Indeed, Rab11 has been implicated in recycling of CFTR in the crypt (Silvis et al., 2009). The degree to which such potential perturbations in protein trafficking of ion channels might contribute to the overall effect of CT in the intestinal epithelium merits further examination.
In addition, the exocyst is involved in secretion of various factors and therefore its inhibition by CT could alter release of cytokines from immune cells or endocrine intestinal epithelial cells, which normally contribute to regulating fluid balance via the enteric nervous system and submucosal immune cells (Lundgren, 2002). Indeed, Rab11 has recently been shown to be required for secretion of the permeability signals TNFα and IFNγ from natural killer T-cells (Reefman et al., 2010).
Disruption of cell-cell junctions by cAMP-producing toxins
The junction disrupting effects of CtxA in Drosophila epithelial cells and in human intestinal cell lines resemble those caused by anthrax EF toxin in human vascular endothelial cells (Guichard et al., 2010), consistent with these toxins acting via the common mechanism of cAMP overproduction. EF toxin decreased TER (Ebrahimi et al., 2011), increased dye permeability in HBMEC cells, and subepidermal and pulmonary vascular effusion caused by injection of B.a. bacteria were found to be EF-dependent (Guichard et al., 2010). Since ET also induces the formation of transcellular tunnels in vascular endothelial cells (Maddugoda et al., 2011), it will be interesting to determine if this effect is also exocyst-dependent. In the current study we observed junctional defects mimicking those caused by CT in CACO-2 cells treated with cAMP analogs specific for the primary cAMP effectors PKA and Epac, consistent with a prior report of similar, albeit weaker effects, in cells treated with the broad spectrum cAMP analog 8Br-cAMP (Boucher et al., 2005). Likewise in Drosophila, expression of activated forms of either PKA or Rap1 (the small GTPase activated by Epac) caused wing phenotypes similar to those caused by CT and down-regulated junctional Rab11, Sec15-GFP, and Delta. Conversely, the effect of CT in CACO-2 cells was attenuated by specific inhibitors of either PKA or Epac, further supporting a role of both of these cAMP pathways in the junction disrupting effects of CT.
One interesting aspect of CT action is that it has different effects on the AJ (e.g., reduction in levels of Rab11, Sec15, and Notch components and apico-basal mis-localization of Ecad) versus TJ (e.g., increased junctional levels of ZO-1 and Cld2, which remain apically restricted). CT treatment also uncoupled these two junctional complexes, suggesting that it disrupts a process that normally ensures their close alignment. Consistent with our observations that co-localization of a subpool of Rab11+ vesicles with ZO-1 is disrupted by CT treatment in CACO-2 or T84 cells, the exocyst has been proposed to mediate direct delivery of proteins to the TJ (Hazelett et al., 2011).
Determining how the various effects of cAMP overproduction are integrated to alter barrier integrity and assessing the full contribution of the junctional defects we describe here to the pathogenesis of cholera are important questions to address in future studies.
Experimental Procedures
Drosophila genetics
The UAS-CtxA transgene (see below) was stably transformed into the Drosophila genome. Flies carrying the UAS-CtxA construct were then crossed to various GAL4 driver stocks to drive toxin expression in specific cells. Timing of GAL4 activity was limited by a temperature sensitive form of GAL80, which inhibits GAL4 function below 29°C.
Transgenic fly stocks include: UAS-CtxA1 UAS-Flp/TM6Tb, UAS-Sec15-GFP/CyO (from H. Bellen), UAS-Rab11-YFPwt, wingG4 = MS1096-GAL4, prdG4 = paired-GAL4, L2G4 = E-GAL4, NP1-GAL4, the rab11- loss-of-function alleles rab11[93Bi] and rab11[j2D1], N−= N55e11 and N* = NAx-1 (available at the Bloomington Drosophila Stock Center or by request), UAS-Pyd-GFP (from A. Fanning), and DECad-tomato (from Y. Hong).
Construction of the UAS-CtxA transgene
A cDNA was PCR amplified from V.c. chromosomal DNA encoding the enzymatically active CtxA1 moiety of CT was inserted into the pUASt vector between the EcoR1 and Not1 sites of the polylinker (details available upon request). The PCR primers used deleted CtxA the signal peptide, introduced a Drosophila Kozak sequence followed by an ATG start codon, and inserted a STOP codon at the junction between the A1 and A2 subunits of CtxA. After confirming the sequence of the cloned cDNA, CtxA1 sequences were excised with Kpn1 and Spe1 and inserted into the pUASt vector.
Infection of Drosophila with V. cholerae
MO10, a V.c. O139 clinical isolate, and a CT subunit B deletion strain (Δctxa) were used for Drosophila infections. Flies were infected orally as described (Wang et al., 2010). Briefly, thirty male flies were divided among three vials that contained a plug saturated with a 1:10 dilution of an overnight culture. Numbers of dead flies in each vial were recorded at least once each day. Survival curves were constructed, and log-rank analysis was used to determine statistical significance.
Dye ingestion assay
Flies 4–8 days old were starved for 2 h in empty vials, then transferred to vials with a Kimwipe soaked with 1.2 ml of a 5% sucrose solution containing green (Yellow 5 and Blue1, Shilling) food colorant (4 drops per 10 ml of sucrose solution). Flies were left on the sucrose dye solution and examined twice daily for presence of dye in the abdomen and entire body cavity.
Immunofluorescence
Drosophila wing imaginal discs and midgut
Immunostaining was performed according to standard protocol (e.g., (Cook et al., 2004) using the following antibodies: anti-Dl (clone C594.9B from Developmental Studies Hybridoma Bank - DSHB, 1/500), rat anti-Drosophila Rab11 antibody (from R. Cohen, 1/500) or mouse anti-Rab11 (BD Transduction laboratories, #610656, 1/200), guinea pig anti-Sec15 (from H. Bellen, 1/1000), mouse anti-Cut (DSHB, clone 2B10-c, 1/100), rat anti-DECAD (DSHB, 1/500), rat anti D-CAT1 (DSHB, 1/500), and rabbit anti-GFP (Invitrogen #A11122, 1/500).
Intestinal cell lines
Human CACO-2 or T-84 intestinal epithelial cells were fixed in 4% paraformaldehyde/1X Phosphate Buffered Saline (PBS) for 30′ at RT or in methanol for 10′ at −20°C (only for Ecad, Sec15, Rab11 stains in Fig. 3 A–L, Ecad/ZO-1 stains of Fig. 4K, L, and occludin stains), washed in PBT (PBS+0.1% TritonX100), blocked in 1% BSA-PBT and processed for staining according to standard protocols. Antibodies were diluted in 1% BSA-PBT. Junctional anti E-cad staining was performed with methanol fixation. Antibodies used include: rabbit anti-Rab11 (Invitrogen, # 715300, 1/200); Goat anti-Sec15 (F-15, Santa Cruz, sc-34366, 1/50); rabbit anti-DLL4 (Lifespan, LS-C19035, 1/150); Goat anti-Jg2 (N-19, sc-34475); goat anti-N3 (M-20, Santa Cruz, sc-7424, 1/100); rabbit anti-activated Notch1 (Abcam, ab83253, 1/500); mouse anti-Ecad (BD, #610404), 1/50); mouse anti-ZO-1 (Invitrogen, #339100, 1/100), rabbit anti-claudin-2 (Invitrogen, 51–6100, 1/100); rabbit anti-occludin (Invitrogen, 71–1500, 1/100); and mouse anti-Rap1 (BD #610195, 1/100).
Cell culture and CT treatment
CACO-2 cells were grown in DME/F12 50/50 (Cellgro CN 10-092-cv) medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), Nu-Serum (BD #355500), and a penicillin/streptomycin mix for ≈ 1 week (recently confluent cultures) or for ≈ 3 weeks (fully confluent cultures). T84 cells were grown in the same medium with antibiotics, supplemented with 5% newborn calf serum (NCS, no heat inactivation). For CT treatment, cells were grown in 24-well plates on glass coverslips to sub-confluence. CT (100 ng/ml Sigma, C8052, diluted in 500 μl of medium) or 500μl of control medium was added 3X at 12 h intervals, and cells were fixed 4 h after the last treatment. Cells were also treated with pharmacological agents following the temporal protocol used for 3X CT treatment including: 6-Bnz (BioLog, Cat. B 009-10, 100uM); 8-Cpt (Tocris, # 1645, 5uM); H-89 (Sigma-Aldrich, # B1427, 10uM); ESI-09 (BioLog, # B 133, 10uM).
Ligated intestinal loop assay
A midline laparotomy was performed on two 5 week old male C57BL6 anesthetized mice, and two ligated intestinal loops (≈ 2 cm) were prepared in the jejunum of each animal using two double clamps of the intestine. 100 μl Hanks’ balanced salt solution (HBSS) containing Ca++ and Mg++ and 1μg of CT holotoxin, was injected into one loop obliquely with a 29-gauge needle and HBSS alone was injected into the second loop. Wounds were stitched, and animals were kept anesthetized until euthanization 5 h after surgery. Loops were excised, measured, and weighed before and after luminal content removal for fluid quantification. The fold increase in extruded fluid in CT versus control loops was calculated per unit length of intestine = fluid secretion (Mouse 1: control fluid secretion (mg/mm) = 1.03; CT-induced fluid secretion (mg/mm) = 3.88; Mouse 2: control fluid secretion (mg/mm): 0.85; CT-induced fluid secretion (mg/mm) = 5.13: SE = 1.0, p = 0.03; average = 4.9 fold increase ± 1.13 SEM). Specimens of intestinal loop tissue, were collected, flash frozen in liquid nitrogen and stored at −80°C. One section of each loop was placed in a plastic mold filled with OCT compound (Tissue-Tek, Sakura Finetek, USA), and quickly frozen in 2-methylbutane (Fischer Scientific, USA) with dry ice for immunofluorescence studies. All animal experiments were approved by the UCSD Animal Care Program.
Ileal tissue sectioning and staining
5 μm sections of fixed intestinal tissue were cut in a cryostat at −18°C. Slides were air dried at RT for 30 minutes after sectioning, blocked with 1% BSA (Sigma #A4503) in PBST washing buffer (PBS, 0.1% Tween20) for 10 min, washed 3 times with PBST, fixed for 30 min in Formaldehyde-Fresh Solution (Fisher #SF94-4), and washed 3X in PBST. Primary antibodies were incubated overnight (4°C), slides were washed 3X in PBST, incubated with secondary antibodies for 30 minutes (RT), and washed 3X in PBST. Antibodies used were: goat anti-Sec15 (F-15, Santa Cruz, sc-34366, 1/10); rabbit anti-Rab11 (Invitrogen, # 715300, 1/250); rabbit anti-E-cad (Cell Signaling, #3195S, 1/250); goat anti-CtxB (Calbiochem #227040, 1/10); and rabbit anti-ms Albumin (Abcam, ab19196, 1/200). F-actin was detected using AlexaFluor 488 Phalloidin (Invitrogen A12379, 1/100).
Confocal Microscopy
Stained Drosophila tissues, sections of murine ileum, and human cell lines (CACO-2, T84) were examined on Leica SP2 or SP5 scanning confocal microscopes. Specific settings for the various experiments varied, but these settings were identical between compared samples (e.g., Supp. Fig. 2A–D).
Electron Microscopy
CACO-2 cells grown on transwell filters or Drosophila intestines were fixed in modified Karnovsky’s fixative (2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M sodium cacodylate buffer, pH 7.4) for 4 h, postfixed in 1% osmium tetroxide in 0.15 M cacodylate buffer for 1 h and stained en bloc in 2% uranyl acetate for 1 h. Samples were dehydrated in ethanol, embedded in Durcupan epoxy resin (Sigma-Aldrich), sectioned at 50- 60 nm, and picked up on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5 minutes and Sato’s lead stain for 1 min. Grids were viewed using a Tecnai G2 Spirit BioTWIN transmission electron microscope equipped with an Eagle 4k HS digital camera (FEI, Hilsboro, OR).
Supplementary Material
Highlights.
Cholera toxin disrupts Rab11/exocyst-mediated protein trafficking to cell junctions
Cholera toxin inhibits Notch signaling in Drosophila and in human intestinal cells
Elevating Rab11 rescues Ctx-induced weight loss and junctional defects in the gut
Elevating Rab11 rescues Ctx-dependent lethality of V. cholerae infection in flies
Acknowledgments
We thank Nathalie Franc for generous access to her confocal microscope and helpful comments on the manuscript, Nissi Varki and Liwen Deng for help with preparing and analyzing sections of murine ileal loops, Dr. Marilyn Farquhar for use of the electron microscopy facility, Ying Jones for EM sample preparation, Bill McGinnis, Steve Wasserman, Emily Troemel, Karla Satchell, and members of the Bier and Nizet labs for helpful discussions, and Allan Fanning and Y. Hong for kindly providing respectively the UAS-Pyd-GFP and DECad-tom fly stocks. B.A. is a postdoctoral fellow in the UCSD/SDSU IRACDA Program (GM06852). This study was supported by National Institutes of Health (NIH) R01 grants AI070654 (E.B.) and AI057153 (V.N.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apidianakis Y, Rahme LG. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis Model Mech. 2011;4:21–30. doi: 10.1242/dmm.003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett KE. Chapter 5: Water and electrolyte absorption and secretion. In: Malley J, Naglieri C, editors. Gastrointestinal Physiology. McGraw-Hill; 2006. pp. 59–100. [Google Scholar]
- Blow NS, Salomon RN, Garrity K, Reveillaud I, Kopin A, Jackson FR, Watnick PI. Vibrio cholerae infection of Drosophila melanogaster mimics the human disease cholera. PLoS Pathog. 2005;1:e8. doi: 10.1371/journal.ppat.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher MJ, Laprise P, Rivard N. Cyclic AMP-dependent protein kinase A negatively modulates adherens junction integrity and differentiation of intestinal epithelial cells. J Cell Physiol. 2005;202:178–190. doi: 10.1002/jcp.20104. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Rich DP, Marshall J, Gregory RJ, Welsh MJ, Smith AE. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Cook O, Biehs B, Bier E. brinker and optomotor-blind act coordinately to initiate development of the L5 wing vein primordium in Drosophila. Development. 2004;131:2113–2124. doi: 10.1242/dev.01100. [DOI] [PubMed] [Google Scholar]
- Craig JP. A permeability factor (toxin) found in cholera stools and culture filtrates and its neutralization by convalescent cholera sera. Nature. 1965;207:614–616. doi: 10.1038/207614a0. [DOI] [PubMed] [Google Scholar]
- Dahan S, Rabinowitz KM, Martin AP, Berin MC, Unkeless JC, Mayer L. Notch-1 signaling regulates intestinal epithelial barrier function through Interaction with CD4(+) T-cells, in mice and humans. Gastroenterology. 2011 doi: 10.1053/j.gastro.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan L, Hirst TR. Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms. Mol Membr Biol. 2004;21:77–92. doi: 10.1080/09687680410001663267. [DOI] [PubMed] [Google Scholar]
- De SN, Chatterje DN. An experimental study of the mechanism of action of Vibrio cholerae on the intestinal mucous membrane. J Pathol Bacteriol. 1953;66:559–562. doi: 10.1002/path.1700660228. [DOI] [PubMed] [Google Scholar]
- Ebrahimi CM, Sheen TR, Renken CW, Gottlieb RA, Doran KS. Contribution of lethal toxin and edema toxin to the pathogenesis of anthrax meningitis. Infect Immun. 2011;79:2510–2518. doi: 10.1128/IAI.00006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Fromter E, Diamond J. Route of passive ion permeation in epithelia. Nat New Biol. 1972;235:9–13. doi: 10.1038/newbio235009a0. [DOI] [PubMed] [Google Scholar]
- Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994;266:107–109. doi: 10.1126/science.7524148. [DOI] [PubMed] [Google Scholar]
- Goldenring JR, Smith J, Vaughan HD, Cameron P, Hawkins W, Navarre J. Rab11 is an apically located small GTP-binding protein in epithelial tissues. Am J Physiol. 1996;270:G515–525. doi: 10.1152/ajpgi.1996.270.3.G515. [DOI] [PubMed] [Google Scholar]
- Guichard A, McGillivray SM, Cruz-Moreno B, van Sorge NM, Nizet V, Bier E. Anthrax toxins cooperatively inhibit endocytic recycling by the Rab11/Sec15 exocyst. Nature. 2010;467:854–858. doi: 10.1038/nature09446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelett CC, Sheff D, Yeaman C. RalA and RalB differentially regulate development of epithelial tight junctions. Mol Biol Cell. 2011;22:4787–4800. doi: 10.1091/mbc.E11-07-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, Munson M. Exorcising the exocyst complex. Traffic. 2012;13:898–907. doi: 10.1111/j.1600-0854.2012.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafar-Nejad H, Andrews HK, Acar M, Bayat V, Wirtz-Peitz F, Mehta SQ, Knoblich JA, Bellen HJ. Sec15, a component of the exocyst, promotes Notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev Cell. 2005;9:351–363. doi: 10.1016/j.devcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Jakab RL, Collaco AM, Ameen NA. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt-villus axis in the intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G82–98. doi: 10.1152/ajpgi.00245.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katanayeva N, Kopein D, Portmann R, Hess D, Katanaev VL. Competing activities of heterotrimeric G proteins in Drosophila wing maturation. PLoS One. 2010;5:e12331. doi: 10.1371/journal.pone.0012331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin J, Morgan MJ, Sibarita JB, Aresta S, Murthy M, Schwarz T, Camonis J, Bellaiche Y. Drosophila exocyst components Sec5, Sec6, and Sec15 regulate DE-Cadherin trafficking from recycling endosomes to the plasma membrane. Dev Cell. 2005;9:365–376. doi: 10.1016/j.devcel.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren O. Enteric nerves and diarrhoea. Pharmacol Toxicol. 2002;90:109–120. doi: 10.1034/j.1600-0773.2002.900301.x. [DOI] [PubMed] [Google Scholar]
- Maddugoda MP, Stefani C, Gonzalez-Rodriguez D, Saarikangas J, Torrino S, Janel S, Munro P, Doye A, Prodon F, Aurrand-Lions M, et al. cAMP Signaling by Anthrax Edema Toxin Induces Transendothelial Cell Tunnels, which Are Resealed by MIM via Arp2/3-Driven Actin Polymerization. Cell Host Microbe. 2011;10:464–474. doi: 10.1016/j.chom.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Magnusson KE, Kihlstrom E, Sundqvist T. Effect of cholera toxin on rat intestinal permeability assessed with fluorescent dextran 3000. FEMS Microbiol Letters. 1985;29:15–18. [Google Scholar]
- Mathan MM, Chandy G, Mathan VI. Ultrastructural changes in the upper small intestinal mucosa in patients with cholera. Gastroenterology. 1995;109:422–430. doi: 10.1016/0016-5085(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Morize P, Christiansen AE, Costa M, Parks S, Wieschaus E. Hyperactivation of the folded gastrulation pathway induces specific cell shape changes. Development. 1998;125:589–597. doi: 10.1242/dev.125.4.589. [DOI] [PubMed] [Google Scholar]
- Murthy M, Schwarz TL. The exocyst component Sec5 is required for membrane traffic and polarity in the Drosophila ovary. Development. 2004;131:377–388. doi: 10.1242/dev.00931. [DOI] [PubMed] [Google Scholar]
- Murthy M, Teodoro RO, Miller TP, Schwarz TL. Sec5, a member of the exocyst complex, mediates Drosophila embryo cellularization. Development. 2010;137:2773–2783. doi: 10.1242/dev.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohishi I, Odagiri Y. Histopathological effect of botulinum C2 toxin on mouse intestines. Infect Immun. 1984;43:54–58. doi: 10.1128/iai.43.1.54-58.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappenheimer JR, Reiss KZ. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J Membr Biol. 1987;100:123–136. doi: 10.1007/BF02209145. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, Nairn AC. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- Reefman E, Kay JG, Wood SM, Offenhauser C, Brown DL, Roy S, Stanley AC, Low PC, Manderson AP, Stow JL. Cytokine secretion is distinct from secretion of cytotoxic granules in NK cells. J Immunol. 2010;184:4852–4862. doi: 10.4049/jimmunol.0803954. [DOI] [PubMed] [Google Scholar]
- Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T, Jr, Jones DL, Walker DW. Modulation of Longevity and Tissue Homeostasis by the Drosophila PGC-1 Homolog. Cell Metab. 2011;14:623–634. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Milatz S, Krug SM, Oelrich B, Schulzke JD, Amasheh S, Gunzel D, Fromm M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J Cell Sci. 2010;123:1913–1921. doi: 10.1242/jcs.060665. [DOI] [PubMed] [Google Scholar]
- Sack DA, Sack RB, Nair GB, Siddique AK. Cholera. Lancet. 2004;363:223–233. doi: 10.1016/s0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- Sasmal D, Guhathakurta B, Ghosh AN, Pal CR, Datta A. Studies on adhesion, haemagglutination and other biological properties of Vibrio cholerae O139. FEMS Immunol Med Microbiol. 1995;10:199–205. doi: 10.1111/j.1574-695X.1995.tb00034.x. [DOI] [PubMed] [Google Scholar]
- Silvis MR, Bertrand CA, Ameen N, Golin-Bisello F, Butterworth MB, Frizzell RA, Bradbury NA. Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol Biol Cell. 2009;20:2337–2350. doi: 10.1091/mbc.E08-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong TV, Boehm K, Collins FS. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest. 1994;93:347–354. doi: 10.1172/JCI116966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triadafilopoulos G, Pothoulakis C, Weiss R, Giampaolo C, Lamont JT. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology. 1989;97:1186–1192. doi: 10.1016/0016-5085(89)91689-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.