

Abstract
The Janus kinases (or Jak kinases) mediate cytokine and growth factor signal transduction. Acquired or inherited Jak mutations can result in dysregulation of Jak-mediated signal transduction and can be critical to disease acquisition in neoplasias including acute myeloid, acute lymphoblastic and acute megakaryoblastic leukemias, and in rare X-linked severe combined immunodeficiency. The discovery of an acquired Jak2 point mutation, V617F, in significant numbers of patients with classical myeloproliferative disorders has increased the interest in development of Jak2-specific tyrosine kinase inhibitors and consequently there are now over 20 publically available structures of Jak kinase domains that describe all four family members, Jak1, Jak2, Jak3, and Tyk2. Here we review the recent advances in understanding the druggable structure and function of the Jak family, with a focus on the structural biology of the Jak kinase domain. We will discuss how these advances impact the development of Jak-targeted therapeutics.
Keywords: Jak kinase, Crystal structure, CP-690, 550, Jak2-V617F, Kinase inhibitor
Overview of cytokine signaling through Jak kinases
The Janus kinases (Jak) are a family of four non-receptor tyrosine kinases whose central role is to respond to cytokine or growth factor receptor activation [1]. Signals from more than 38 cytokines are transduced through four Jak family members and seven STATs [2]. Three of the four Jak family members: Jak1, Jak2, and tyrosine kinase 2 (Tyk2) are ubiquitously expressed in vertebrates, while the fourth, Jak3, is mostly limited to hematopoietic cells [1, 3]. To transduce cytokine signaling, a series of conformational changes occur in the receptor-Jak complex upon extracellular ligand binding. This results in trans-activation of the receptor-associated Jaks followed by phosphorylation of receptor tail tyrosine sites. The Signal Transducers and Activators of Transcription (STAT) are then recruited to the receptor tail, become phosphorylated and translocate to the nucleus to regulate transcription [4] (Fig. 1A). The Jak-STAT signaling cascade is tightly controlled, and even conservative acquired point mutations that alter regulation of the pathway can have critical effects on cell proliferation.
Figure 1. Structural descriptions of Jak signaling in the Jak-STAT pathway.
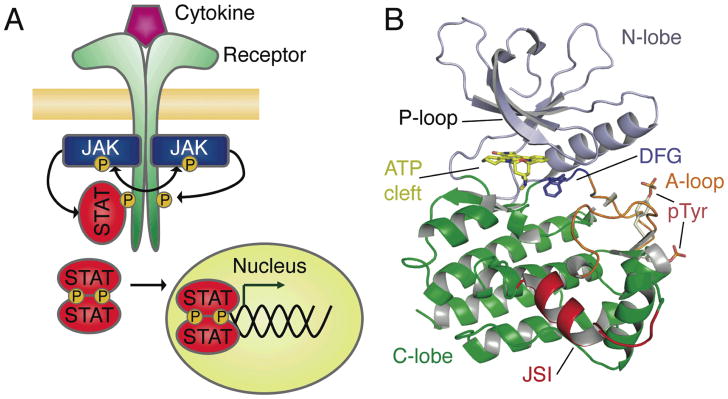
A) The Jak-STAT signaling pathway. Cytokine signaling is initiated upon ligand binding to receptor, this facilitates Jak kinase activation by trans-autophosphorylation. Jaks then phosphorylate the receptor tails creating docking sites for STAT molecules, which bind through their SH2 domains. STATs become phosphorylated by Jaks and form dimers that are able to translocate to the nucleus and regulate gene expression. B) Crystal structures of all Jak kinase domains have been determined in the active state. As is typical for kinase domains, these structures contain a predominantly β-sheet N-lobe (blue) that is conformationally flexible, facilitating activation regulation and ATP/ADP binding and release. The nucleotide-binding glycine-rich loop (P-loop) is indicated. The activation loop (orange) is extended in all of these structures, and is phosphorylated in almost all. The DFG is found in its ‘in’ conformation in all of these structures. The C-lobe of the kinase domain (green) is the most structurally consistent region, with the exception of the Jak-specific insert (red). All structural figures created with Pymol (www.pymol.org).
The domain structure of the Jak kinases suggests seven evolutionarily conserved Jak Homology (JH) regions that are predicted to fold into four domains. At the amino-terminus the JH7 to JH3 regions are predicted to fold as FERM (band four-point-one, ezrin, radixin, and moesin) and SH2 (Src Homology-2) domains [1, 5]. Although the extreme N-terminal JH domains (JH7 and JH6) are required for cytokine binding, the whole region (JH7-JH3) may be important for normal association with cytokine receptor tails by interacting with three regions named Box 1, interbox, and Box 2 [6–12]. The Jak SH2 domain has incomplete conservation of the β5 arginine conserved in all SH2 domains that bind phosphotyrosine [13] suggesting that phosphotyrosine binding for this domain is not required for normal Jak function [14, 15].
At the carboxy-terminus of the Jak domain structure, the JH2 and JH1 regions fold into the pseudokinase domain and the catalytic tyrosine kinase domain respectively. The pseudokinase domain, while predicted to share the same fold as catalytically active kinases, lacks key residues that are required for phosphotransfer. These include residues within the ATP-binding glycine-rich loop (GxGxxG is GxGxxT in the Jak pseudokinases), the HRD motif that contains the catalytic aspartic acid (HRD is HGN in Jak pseudokinases), and the DFG motif important for binding magnesium and ATP orientation (DFG is DPG in Jak pseudokinases) [16]. These alterations in Jak pseudokinase domains suggest likely catalytic inactivity. Instead of possessing catalytic activity the Jak pseudokinase domain directly regulates catalytic activity of the tyrosine kinase domain [17–21]. The kinase domain (JH1) is responsible for tyrosine phosphorylation of Jak substrates and is the only region of the Jak family that has been investigated by atomic-level structural biology techniques; crystal structures of kinase domains for all four Jaks have been determined [22–32] with all demonstrating a classic bilobed fold characteristic of a protein kinase (Fig. 1B) [33].
The Jak kinases are regulated by layers of enzymatic and non-enzymatic mechanisms. First, as is often seen for kinase regulation [34], the Jak kinase domain is regulated by phosphorylation of the activation loop (Fig. 1B), this is associated with the catalytically competent kinase conformation and is distinct from the inactive kinase conformation. All current Jak kinase domain crystal structures are determined in the active state conformation [22–32]. Second, the pseudokinase domain directly modulates Jak catalytic activity [19, 20, 35] and the FERM domain plays a role in maintenance of an active state [36]. Third, the suppressor of cytokine signaling (SOCS) family and tyrosine phosphatases directly regulate Jak activity. The SOCS family target the kinase-receptor complex for proteasomal degradation and can directly inhibit kinase activity through pseudosubstrate interactions by a ‘kinase inhibitory region’ at the N-terminus of SOCS1 and SOCS3 [37]. Phosphatase-mediated regulation of cytokine signaling is achieved by dephosphorylation of receptor tyrosine residues and tyrosines within the kinase activation loop [38, 39] important for kinase conformational switching between inactive and active states [33]. The multiple layers of regulation for these kinases indicate physiological importance, and multiple disorders have been described in which altered Jak activity is observed.
Dysregulation of Jak signaling in disease
Dysregulation of Jak catalytic activity can manifest as either a reduction or an increase in kinase activity, and is observed in pathogeneses including immunodeficiency, inflammatory diseases, hematological defects, autoimmune and myeloproliferative disorders, and susceptibility to infection [40]. Altered Jak regulation occurs by multiple mechanisms, including: gene translocations, somatic or inherited point mutations, receptor mutations, and alterations in the activity of Jak regulators such as SOCS or phosphatases.
Translocations
Hyperactivated Jak2 has been observed in acute leukemia and atypical CML as the protein product of t(9;12)(p24;p13) gene translocation. This translocation fuses the helix-loop-helix oligomerization domain of the TEL transcription factor with C-terminal Jak regions that include the kinase domain, facilitating kinase trans-activation in a cytokine independent fashion [41, 42]. Similar translocations occur that result in BCR-Jak2 [43] and PCM1-Jak2 [44, 45] in chronic and acute leukemias.
Activating point mutations
In 2005, multiple groups simultaneously reported the presence of an activating mutation within the Jak2 pseudokinase domain, V617F, that is highly correlated with the classical myeloproliferative disorders [46–50]. This somatic mutation is present in almost all polycythemia vera cases, and is found in the majority of primary myelofibrosis and essential thrombocythemia cases [51]. The corresponding mutation in Jak1, V658F, has been found in acute lymphoblastic leukemia (ALL) patients [52, 53]. Although the precise mechanisms of kinase activation are not known, and await atomic-level structural investigation, deregulation of kinase autoinhibition seems to be achievable by mutation to phenylalanine of the analogous valine in Jak1 and Tyk2 [21]. Co-evolution of residue M592 and another, as yet unknown, region of the protein in Jak3 may account for the inability to activate Jak3 by mutation to phenylalanine. On the whole, however, these studies suggest conserved mechanisms for the conformational regulation of the four Jak family members.
Other Jak activating mutations have been found in all domains of Jak3 and drive acute megakaryoblastic leukemia [54, 55]. Similarly, mutations were found throughout Jak1 and are present in approximately 18% of cases of adult acute lymphoblastic leukemia [56]. For Jak1 and Jak3, several of these point mutations seen in patients confer cytokine independent kinase activity and cell proliferation [54, 56].
Inactivating point mutations
Inactivating mutations in Jak3 are associated with X-linked severe combined immunodeficiency (X-SCID) variant, and account for between 7 and 14% of SCID cases [57, 58]. The majority of these inherited mutations result in Jak3 truncation, however, point mutations throughout the kinase can adversely affect receptor binding, intracellular trafficking, and kinase activity [58]. Inactivating mutations in Tyk2 have also been linked to an immunodeficiency syndrome [59].
Mutations in receptors and regulators associated with Jak signaling
Regulation of Jak signaling cascades can also be altered by somatic point mutations in cytokine receptors and alterations in expression of regulators such as the SOCS family. Examples of these changes in Jak regulation are observed in somatic point mutations in the thrombopoietin receptor, MPL, have been shown to alter Jak activity and are associated with Jak2-V617F-negative myeloproliferative disorders [60, 61] and in many human cancers by hypermethylation of SOCS genes [37]. A SOCS3 SH2 domain point mutation has also recently been associated with myeloproliferative disorders [62].
Targeted inhibition of Jak activity
The linkage of these pathologies with altered Jak catalytic activity has increased interest in development of Jak-specific inhibitors to treat classical myeloproliferative disorders and cancers, or to be used as immunosuppressants. Indeed, there are now multiple Jak2-and Jak3-targeted kinase inhibitors in clinical trials (e.g. INCB018424, CEP-701, AZD1480, XL019, TG101348, CYT387, CP-690,550, and VX-509). Jak2-targeted inhibitors are currently undergoing trials for the treatment of myeloproliferative disorders, and Jak3 inhibitors as immunosuppressants for organ transplants and rheumatoid arthritis.
Structural biology plays an important role in rational inhibitor design, and consequently, over the last few years crystal structures for all of the enzymatically active Jak kinase domains have been determined in complex with various ATP-competitive inhibitors [22–32] (Table 1)(Fig. 2). In all of these Jak kinase domain structures a small molecule inhibitor binds in the ATP-cleft juxtaposed between a β-sheet-rich N-lobe and α-helix-rich C-lobe. All of these structures were determined in the active state, with a DFG-in conformation [63] and an extended activation loop. Analysis of the Jak crystal structures shows that the major conformational differences are centered on the glycine rich loop (P-loop) and the activation loop [26–28]. Moreover, two pan-Jak small molecule inhibitors have been structurally investigated with all four Jak family members; CMP6 a Jak inhibitor developed by Merck [64] but not used in the clinic, and CP-690,550 an inhibitor designed by Pfizer to target Jak3 that is currently in Phase III clinical trials [65]. These structural studies therefore provide a relatively comprehensive foundation to understand the conformational diversity of the active-state Jak kinases and potential methods to design small-molecule specificity.
Table 1. Current public domain Jak kinase crystal structures.
Summary of the public domain Jak kinase crystal structures [22–31]. Inhibitors are labeled by their chemical formula or documented name.
| PDB ID | Jak kinase | Reported resolution limit (Å) | Inhibitor/ATP analogue | DFG- in/out | Activation loop | Phosphorylat ion status | Space Group | Cell (a, b, c) (Å) | Publication |
|---|---|---|---|---|---|---|---|---|---|
| 3EYG | Jak1 | 1.9 | CP-690,550 | DFG-in | Extended | pYpY | C2221 | 45, 88, 146 | Williams, et al., 2009 |
| 3EYH | Jak1 | 2.0 | CMP6 | DFG-in | Extended | pYpY | C2221 | 45, 88, 145 | Williams, et al., 2009 |
| 2B7A | Jak2 | 2.0 | CMP6 | DFG-in | Extended | pYpY | P41 | 111, 111, 71 | Lucet et al., 2006 |
| 2W1I | Jak2 | 2.6 | AT9283 | DFG-in | Extended | pYpY | C21 | 103, 93, 123 | Howard, et al., 2009 |
| 3E62 | Jak2 | 1.92 | 5-BROMO-1H-INDAZOL- 3-AMINE | DFG-in | Extended | pYpY | C2221 | 93, 102, 68 | Antonysamy et al., 2009 |
| 3E63 | Jak2 | 1.9 | 5-PHENYL-1H-INDAZOL- 3-AMINE | DFG-in | Extended | pYpY | C2221 | 93,103, 68 | Antonysamy et al., 2009 |
| 3E64 | Jak2 | 1.8 | 4-(3-AMINO-1H- INDAZOL-5-YL)-N-TERT- BUTYLBENZENESULFO NAMIDE | DFG-in | Extended | pYpY | C2221 | 93, 103, 68 | Antonysamy et al., 2009 |
| 3FUP | Jak2 | 2.4 | CP-690,550 | DFG-in | Extended | pYpY | P41 | 110, 110, 70 | Williams, et al., 2009 |
| 3IO7 | Jak2 | 2.6 | (3S)-1-[6-(2- AMINOPYRAZOLO[1,5- A]PYRIMIDIN-3- YL)PYRIMIDIN-4-YL]-N,N- DIETHYLPIPERIDINE-3- CARBOXAMIDE | DFG-in | Extended | pYpY | C2221 | 94, 102, 67 | Ledeboer et al., 2009 |
| 3IOK | Jak2 | 2.1 | 3-(6-[[(1S)-1-(4- FLUOROPHENYL)ETHYL]AMINO]PYRIMIDIN-4- YL)PYRAZOLO[1,5- A]PYRIMIDIN-2-AMINE | DFG-in | Extended | pYpY | C2221 | 94, 103, 68 | Ledeboer et al., 2009 |
| 3JY9 | Jak2 | 2.1 | (3S)-3-(4- HYDROXYPHENYL)-1,5- DIHYDRO-1,5,12- TRIAZABENZO[4,5]CYCL OOCTA[1,2,3-CD]INDEN- 4(3H)-ONE | DFG-in | Extended | pYpY | C2221 | 95, 101, 68 | Wang et al., 2009 |
| 3KCK | Jak2 | 2.2 | 3-CHLORO-4-(4H-3,4,7- TRIAZADIBENZO[CD,F]A ZULEN-6-YL)PHENOL | DFG-in | Extended | pYpY | C2221 | 95, 101, 68 | Wang et al., 2010 |
| 3KRR | Jak2 | 1.8 | NVP-BSK805 | DFG-in | Extended | pYpY | C2221 | 93, 103, 69 | Baffert et al., 2010 |
| 3LPB | Jak2 | 2.0 | N-METHYL-4-[3-(3,4,5- TRIMETHOXYPHENYL)Q UINOXALIN-5- YL]BENZENESULFONAM IDE | DFG-in | Extended | pYpY | P41 | 112, 112, 71 | Pissot- Soldermann et al., 2010 |
| 1YVJ | Jak3 | 2.55 | AFN941 | DFG-in | Extended | pYpY | P212121 | 46, 54, 119 | Boggon et al., 2005 |
| 3LXK | Jak3 | 2.0 | CP-690,550 | DFG-in | Extended | YY | P212121 | 48, 75, 88 | Chrencik et al., 2010 |
| 3LXL | Jak3 | 1.74 | CMP6 | DFG-in | Extended | YY | P212121 | 47, 75, 89 | Chrencik et al., 2010 |
| 3LXN | Tyk2 | 2.5 | CP-690,550 | DFG-in | Extended | pYY | P212121 | 36, 74, 106 | Chrencik et al., 2010 |
| 3LXP | Tyk2 | 1.6 | CMP6 | DFG-in | Extended | pYY | P212121 | 38, 76, 97 | Chrencik et al., 2010 |
Figure 2. ATP-binding clefts of Jak kinase domain structures.

Comparison of the ATP-binding clefts and bound small molecules for all Jak family kinase domain crystal structures. In these structures the kinase nucleotide-binding glycine-rich loop (P-loop) has been trapped in multiple conformations, describing its flexibility. Additionally, in four of the Jak2 structures (3JY9, 3KCK, 3IOK, 3IO7) the P-loop is not visible in the electron density, indicating significant flexibility within these crystals. Jak1 is colored cyan, Jak2 in green, Jak3 in orange and Tyk2 in magenta. The protein data bank accession code is indicated for each structure. If two molecules exist in the crystal structure with different conformations these are indicated by parentheses.
So how is Jak specificity achieved? Certain small molecules have been demonstrated to specifically target one Jak over other family members [15]. The molecular basis for this is unclear as the Jak family members are very homologous, especially true in the region of the ATP-binding site, where only two residues are divergent within 6 Å of the ATP-binding site between the Jak family members. In Jak1, Jak2, and Tyk2 these residues are serine and glycine, while in Jak3 they are cysteine and alanine [28]. To investigate whether these Cys-Ser or Gly-Ala differences were the reason for differential Jak2 and Jak3 specificity achieved by CP-690,550 and CMP6, Chrencik et al. mutated Jak2 residues Ser936 and Gly993 to the corresponding Jak3 amino acids (Cys and Ala respectively). They found that small molecule specificity for the mutant Jak2 was not affected [28]. This suggests that specificity for CP-690,550 and CMP6 is derived outside of the immediate hydrogen-bonding and van der Waals profile of the ATP-binding cleft [28]. Indeed, analysis of the crystal structures suggests that the mode of binding for CP-690,550 and CMP6 is not affected by these Cys-Ser or Gly-Ala differences.
The work of Chrencik et al. suggests that there are other structural differences affecting small molecule inhibitor specificity for the Jaks. One possible mechanism is that sequence differences outside of the active site can alter the conformational dynamics of the kinases themselves. This is illustrated by analysis of the glycine-rich P-loop in the determined crystal structures (Fig. 2). The P-loop is conformationally flexible in order to accommodate adenosine binding and ATP/ADP entry and release, there are a couple of features, however, in the structures of Jak1 and Tyk2 that suggest differences in the flexibility of this loop between Jak family members. First, a salt bridge between the lysine residue at the tip of strand β3 (Lys911 in Jak1 and Lys933 in Tyk2) and a glutamate within the P-loop (Glu883 in Jak1 and Glu905 in Tyk2) is not seen in Jak2 or Jak3 structures, which have a glutamine residue in place of the lysine (Gln885 in Jak2 and Gln858 in Jak3) [26]. Second, at the tip of the P-loop of Jak1 and Tyk2 a histidine residue interacts with a C-lobe aspartate (Asp1023 in Tyk2 and Asp1003 in Jak1). These salt bridges could provide some stability for a collapsed conformation of the P-loop [26, 28] and are potentially important for the design of small molecule inhibitors selective for specific Jak family members as they may indicate structural conformations that can be rationally targeted. Small molecule specificity differences between the Jaks may therefore be driven by Jak accessibility to specific kinase domain conformations. There is consequently a need to understand the extent of conformational flexibility and structural space accessed by these kinase domains.
Determination of kinase domain crystal structures in multiple crystal forms, i.e. with different space groups and unit cell parameters, can provide a comprehensive map of accessible structural space. Structures in multiple crystal forms can provide an understanding of the protein’s conformational dynamics and extent of conformational space accessible. For kinases this information can aid in understanding specificity, small molecule docking and rational drug discovery. It is well established that kinases are conformationally flexible molecules, and that a protein crystal ‘traps’ the molecule in a defined structural population, it is therefore preferential to determine an array of crystal structures for a particular kinase domain target of interest. This is particularly true when molecular modeling is being utilized as crystal packing can ‘trap’ the kinase in a single population that may not be optimal for a docking of the small molecule of interest. Illustrating this, molecular modeling of four CP-690,550 enantiomers to the crystal structure of Jak3 [66] utilized the only available Jak3 structure at the time, bound to the staurosporine analogue AFN941 [31]. The molecular basis for CP-690,550 binding was predicted, however, when the structure of Jak3 bound to CP-690,550 was determined the small molecule was found to be bound in a divergent fashion [28]. This seems to be because the conformation of Jak3 in the AFN941-bound crystal was not optimal to model CP-690,550 binding. This kind of problem can be mitigated by utilization of molecular dynamics simulations for kinase conformational changes [67], a technique that would be particularly powerful if referenced using multiple crystal structures. The current structural descriptions, especially for Jak2 (Table 1) provide such a referencing framework.
Structure-directed drug discovery has, however, been used successfully in some cases to surmount the significant structural and sequential similarity between the Jak family members [15]. These efforts include the design of TG101209 [68] and TG101348 [69] which were generated by structure-based design using Jak2 and Jak3 crystal structures to target Jak2. TG101209 showed a 28-fold selectivity over Jak3 kinase with an IC50 of 6 nM [68] and has proven effective for the treatment of a Jak2-V617F induced hematopoietic disorder in mice. Similarly, TG101348 inhibits proliferation of a human and mouse cell lines expressing human JAK2V617F [69] and when tested on a murine model, TG101348 was able to treat a JAK2-V617F-induced MPD. In silico drug discovery has also been conducted using the Jak3 kinase domain structure. The Jak3 crystal structure was used to create a Jak2 homology model and conduct virtual screening using an in silico screen of the National Cancer Institute compound library [70]. This technique discovered a Jak2 inhibitor able to suppress the pathological growth of bone marrow cells from a PV patient by more than 50% [70]. A Jak3 homology model was also used to discover WHI-P131 [71, 72] and computational modeling of conformational changes has been used to aid the design of targeted specificity [67]. Significantly, while the current published structural studies for the Jak kinases have focused on the active state, the inactive state Jak remains unexplored. Inactive conformations of kinases are potential therapeutic targets, and have been targeted by multiple small molecule inhibitors, e.g. imatinib [73] and lapatinib [74]. Structures of inactive state Jak kinase domains may provide an alternate target for structure-directed drug discovery.
Further considerations for the structure and druggable surfaces of the Jak kinases
Jak kinase activity is regulated by interactions with the pseudokinase and FERM domains [19, 20, 75]. In the long-term, these interactions may provide alternative targets for dysregulated Jak activity. The best studied of these regulatory interactions is the inhibitory role of the pseudokinase domain on kinase catalytic activity. In vitro and in vivo data suggest direct regulation by pseudokinase interactions with the kinase domain. The molecular basis for this is unknown, but structural modeling [76], based on a crystal structure of the FGF receptor tyrosine kinase domain [77] suggests an asymmetric dimer. This model, however, has significant similarity to the FGFR crystal structure it was based on and does not correspond to mapped surfaces of activating Jak residues [15]. Interestingly, a structural map of the pseudokinase domain Jak-activating mutations shows that these mutations are concentrated at the predicted N-lobe, the structural reason for this is yet to be determined [75]. Additionally, the ability to bind ATP may be conserved in the Jak pseudokinase domains, as has been seen for other pseudokinases [78], although ATP-binding may not cause conformational changes in certain pseudokinases [78]. A structural level understanding of pseudokinase regulation of kinase activity can potentially facilitate novel approaches for Jak kinase inhibitor design.
There are other potentially interesting sites of intra- and inter-molecular interaction within the Jak family that may yield alternative drug targets. For example, in the Jaks a novel insertion not seen in other tyrosine kinases (‘FG-Helix’ in [31], Jak-specific insertion, JSI, in [79] and ‘Lip’ in [27]) is conserved in vertebrate Jak sequences, suggesting participation in intra- or intermolecular interactions. This conformationally flexible insertion (Fig. 3) has been suggested to play a role in regulation of the kinase. A recent report suggests that mutagenesis and deletion studies in the JSI region can reduce or abrogate Jak autophosphorylation [79], it will therefore be interesting to discover the molecular mechanism of how kinase activity is affected by this Jak-specific kinase domain insertion and whether this can guide alternative Jak-targeted small molecule or peptide mimetic inhibitors. Other intermolecular interactions are better established to be direct regulators of Jak activity. The SOCS family members SOCS1, and SOCS3 directly regulate Jak activity by interaction of a 12 residue region termed the kinase inhibitory region (KIR) with the Jak kinase domain [37]. The mechanism of this regulation is probably by substrate mimicry, but is not yet described at the atomic level. Use of a SOCS1 KIR peptide mimetic has been documented to down-regulate Jak activity, raising the potential of SOCS-KIR mimetics as Jak-specific inhibitors [80, 81].
Figure 3. Conformational flexibility of the Jak-specific insertion.

Structural alignment of all public domain Jak kinase crystal structures. The Jak family kinase domain C-lobe is conformationally conserved in all published crystal structures, however, flexibility is observed in the Jak-specific insertion (JSI). Other areas of conformational diversity predominantly lie in N-lobe and activation loop. Jak1 is colored cyan, Jak2 in green, Jak3 in orange and Tyk2 in magenta.
Conclusions
Recent advances in structural biology descriptions of the Jak kinase family have provided over 20 publically available kinase domain crystal structures. These structures have been determined in complex with multiple small molecule inhibitors and ATP analogues, providing a relatively large public-domain description of Jak structural space. Although these structures are restricted to the active, DFG-in, state, the current publicly accessible Jak kinase domain structures (Table 1) provide a relatively comprehensive library for structure-directed kinase inhibitor design that has already played a role in discovery of some small molecule Jak inhibitors [68, 69]. There is still, however, much to be learned from determination of further Jak kinase domain crystal forms. Structural differences between crystal forms may be influenced by crystal packing effects and inhibitor-induced conformation, still, all of these structures represent accessible protein conformations. Knowledge of the conformational space that a kinase can adopt should allow exploitation of structure-directed inhibitor design targeted at a specific orientation of the active state. Consequently, the solution of a single kinase domain crystal structure is only a starting point to understand and discover improved inhibitors by a rational structure-directed approach; further crystal structures will increase the knowledge of conformational states accessible to the Jak kinase family and facilitate in silico rational design of improved inhibitors.
There is still much we do not yet understand about the molecular-level basis for Jak-STAT signaling. At the kinase level, we do not yet know what conformation the inactive-state Jak kinase domain assumes, and therefore how to target the inactive state kinase. Also, Jak kinase activity regulation by the pseudokinase domain is yet to be elucidated; although there are tantalizing clues to this regulation mechanism a better understanding awaits crystallographic analysis. Likewise, our understanding of the atomic-level and global conformations of the full-length Jak proteins, and how they are regulated by inter-domain interactions between the kinase and FERM domain, for example, are still not described. These questions will undoubtedly be rapidly addressed by the multiple structural biology groups working in the field and may provide a molecular-level basis for further development of Jak inhibitors.
Acknowledgments
This work is funded by NIAID, R01 AI075133 and Diversity Supplement R01 AI075133-S1. We apologize to colleagues whose work was not cited due to space constraints.
References
- 1.Yamaoka K, Saharinen P, Pesu M, Holt VE, 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks) Genome Biol. 2004;5(12):253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol. 2007;44(10):2497–506. doi: 10.1016/j.molimm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 3.Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11(2):69–74. doi: 10.1016/s0168-9525(00)89000-9. [DOI] [PubMed] [Google Scholar]
- 4.Darnell JJ, Kerr I, Stark G. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 5.Girault JA, Labesse G, Mornon JP, Callebaut I. The N-termini of FAK and JAKs contain divergent band 4. 1 domains. Trends Biochem Sci. 1999;24(2):54–7. doi: 10.1016/s0968-0004(98)01331-0. [DOI] [PubMed] [Google Scholar]
- 6.Usacheva A, Sandoval R, Domanski P, Kotenko SV, Nelms K, Goldsmith MA, et al. Contribution of the Box 1 and Box 2 motifs of cytokine receptors to Jak1 association and activation. J Biol Chem. 2002;277(50):48220–6. doi: 10.1074/jbc.M205757200. [DOI] [PubMed] [Google Scholar]
- 7.Haan C, Hermanns HM, Heinrich PC, Behrmann I. A single amino acid substitution (Trp(666)-->Ala) in the interbox1/2 region of the interleukin-6 signal transducer gp130 abrogates binding of JAK1, and dominantly impairs signal transduction. Biochem J. 2000;349(Pt 1):261–6. doi: 10.1042/0264-6021:3490261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cacalano NA, Migone TS, Bazan F, Hanson EP, Chen M, Candotti F, et al. Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. Embo J. 1999;18(6):1549–58. doi: 10.1093/emboj/18.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen M, Cheng A, Chen YQ, Hymel A, Hanson EP, Kimmel L, et al. The amino terminus of JAK3 is necessary and sufficient for binding to the common gamma chain and confers the ability to transmit interleukin 2-mediated signals. Proc Natl Acad Sci U S A. 1997;94(13):6910–5. doi: 10.1073/pnas.94.13.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pesu M, Candotti F, Husa M, Hofmann SR, Notarangelo LD, O’Shea JJ. Jak3, severe combined immunodeficiency, and a new class of immunosuppressive drugs. Immunol Rev. 2005;203:127–42. doi: 10.1111/j.0105-2896.2005.00220.x. [DOI] [PubMed] [Google Scholar]
- 11.Radtke S, Jorissen A, de Leur HS, Heinrich PC, Behrmann I. Three dileucine-like motifs within the interbox1/2 region of the human oncostatin M receptor prevent efficient surface expression in the absence of an associated Janus kinase. J Biol Chem. 2006;281(7):4024–34. doi: 10.1074/jbc.M511779200. [DOI] [PubMed] [Google Scholar]
- 12.Haan C, Heinrich PC, Behrmann I. Structural requirements of the interleukin-6 signal transducer gp130 for its interaction with Janus kinase 1: the receptor is crucial for kinase activation. Biochem J. 2002;361(Pt 1):105–11. doi: 10.1042/0264-6021:3610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radtke S, Haan S, Jorissen A, Hermanns HM, Diefenbach S, Smyczek T, et al. The Jak1 SH2 domain does not fulfill a classical SH2 function in Jak/STAT signaling but plays a structural role for receptor interaction and up-regulation of receptor surface expression. J Biol Chem. 2005;280(27):25760–8. doi: 10.1074/jbc.M500822200. [DOI] [PubMed] [Google Scholar]
- 14.Haan C, Kreis S, Margue C, Behrmann I. Jaks and cytokine receptors--an intimate relationship. Biochem Pharmacol. 2006;72(11):1538–46. doi: 10.1016/j.bcp.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Haan C, Behrmann I, Haan S. Perspectives for the use of structural information and chemical genetics to develop inhibitors of Janus kinases. J Cell Mol Med. 2010;14(3):504–27. doi: 10.1111/j.1582-4934.2010.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16(9):443–52. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Chen M, Cheng A, Candotti F, Zhou YJ, Hymel A, Fasth A, et al. Complex effects of naturally occurring mutations in the JAK3 pseudokinase domain: evidence for interactions between the kinase and pseudokinase domains. Mol Cell Biol. 2000;20(3):947–56. doi: 10.1128/mcb.20.3.947-956.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ihle JN, Gilliland DG. Jak2: normal function and role in hematopoietic disorders. Curr Opin Genet Dev. 2007;17(1):8–14. doi: 10.1016/j.gde.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–63. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 20.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–95. doi: 10.1128/mcb.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staerk J, Kallin A, Demoulin JB, Vainchenker W, Constantinescu SN. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280(51):41893–9. doi: 10.1074/jbc.C500358200. [DOI] [PubMed] [Google Scholar]
- 22.Pissot-Soldermann C, Gerspacher M, Furet P, Gaul C, Holzer P, McCarthy C, et al. Discovery and SAR of potent, orally available 2,8-diaryl-quinoxalines as a new class of JAK2 inhibitors. Bioorg Med Chem Lett. 2010;20(8):2609–13. doi: 10.1016/j.bmcl.2010.02.056. [DOI] [PubMed] [Google Scholar]
- 23.Wang T, Duffy J, Wang J, Halas S, Salituro F, Pierce A, et al. Janus kinase 2 inhibitors. Synthesis and characterization of a novel polycyclic azaindole. J Med Chem. 2009;52(24):7938–41. doi: 10.1021/jm901383u. [DOI] [PubMed] [Google Scholar]
- 24.Wang T, Ledeboer M, Duffy J, Pierce A, Zuccola H, Block E, et al. A novel chemotype of kinase inhibitors: Discovery of 3,4-ring fused 7-azaindoles and deazapurines as potent JAK2 inhibitors. Bioorg Med Chem Lett. 2010;20(1):153–6. doi: 10.1016/j.bmcl.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Ledeboer M, Pierce A, Duffy J, Gao H, Messersmith D, Salituro F, et al. 2-Aminopyrazolo[1,5-a]pyrimidines as potent and selective inhibitors of JAK2. Bioorg Med Chem Lett. 2009;19(23):6529–33. doi: 10.1016/j.bmcl.2009.10.053. [DOI] [PubMed] [Google Scholar]
- 26.Williams NK, Bamert RS, Patel O, Wang C, Walden PM, Wilks AF, et al. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol. 2009;387(1):219–32. doi: 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 27.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107(1):176–83. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 28.Chrencik JE, Patny A, Leung IK, Korniski B, Emmons TL, Hall T, et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J Mol Biol. 2010;400(3):413–33. doi: 10.1016/j.jmb.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 29.Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, et al. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J Med Chem. 2009;52(2):379–88. doi: 10.1021/jm800984v. [DOI] [PubMed] [Google Scholar]
- 30.Baffert F, Regnier CH, De Pover A, Pissot-Soldermann C, Tavares GA, Blasco F, et al. Potent and selective inhibition of polycythemia by the quinoxaline JAK2 inhibitor NVP-BSK805. Mol Cancer Ther. 2010;9(7):1945–55. doi: 10.1158/1535-7163.MCT-10-0053. [DOI] [PubMed] [Google Scholar]
- 31.Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106(3):996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonysamy S, Hirst G, Park F, Sprengeler P, Stappenbeck F, Steensma R, et al. Fragment-based discovery of JAK-2 inhibitors. Bioorg Med Chem Lett. 2009;19(1):279–82. doi: 10.1016/j.bmcl.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 33.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109(3):275–82. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 34.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23(48):7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 35.Saharinen P, Vihinen M, Silvennoinen O. Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol Biol Cell. 2003;14(4):1448–59. doi: 10.1091/mbc.E02-06-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou YJ, Chen M, Cusack NA, Kimmel LH, Magnuson KS, Boyd JG, et al. Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol Cell. 2001;8(5):959–69. doi: 10.1016/s1097-2765(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 37.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19(4):414–22. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourdeau A, Dube N, Tremblay ML. Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr Opin Cell Biol. 2005;17(2):203–9. doi: 10.1016/j.ceb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Zhou YJ, Hanson EP, Chen YQ, Magnuson K, Chen M, Swann PG, et al. Distinct tyrosine phosphorylation sites in JAK3 kinase domain positively and negatively regulate its enzymatic activity. Proc Natl Acad Sci U S A. 1997;94(25):13850–5. doi: 10.1073/pnas.94.25.13850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrajoli A, Faderl S, Ravandi F, Estrov Z. The JAK-STAT pathway: a therapeutic target in hematological malignancies. Curr Cancer Drug Targets. 2006;6(8):671–9. doi: 10.2174/156800906779010227. [DOI] [PubMed] [Google Scholar]
- 41.Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. 1997;278(5341):1309–12. doi: 10.1126/science.278.5341.1309. [DOI] [PubMed] [Google Scholar]
- 42.Peeters P, Raynaud SD, Cools J, Wlodarska I, Grosgeorge J, Philip P, et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood. 1997;90(7):2535–40. [PubMed] [Google Scholar]
- 43.Griesinger F, Hennig H, Hillmer F, Podleschny M, Steffens R, Pies A, et al. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11. 2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer. 2005;44(3):329–33. doi: 10.1002/gcc.20235. [DOI] [PubMed] [Google Scholar]
- 44.Bousquet M, Quelen C, De Mas V, Duchayne E, Roquefeuil B, Delsol G, et al. The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24(48):7248–52. doi: 10.1038/sj.onc.1208850. [DOI] [PubMed] [Google Scholar]
- 45.Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65(7):2662–7. doi: 10.1158/0008-5472.CAN-04-4263. [DOI] [PubMed] [Google Scholar]
- 46.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 47.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 48.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 49.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 50.Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem. 2005;280(24):22788–92. doi: 10.1074/jbc.C500138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190–8. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106(23):9414–8. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ, et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res. 2008;14(12):3716–21. doi: 10.1158/1078-0432.CCR-07-4839. [DOI] [PubMed] [Google Scholar]
- 54.Walters DK, Mercher T, Gu TL, O’Hare T, Tyner JW, Loriaux M, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10(1):65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Cornejo MG, Boggon TJ, Mercher T. JAK3: a two-faced player in hematological disorders. Int J Biochem Cell Biol. 2009;41(12):2376–9. doi: 10.1016/j.biocel.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205(4):751–8. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Notarangelo LD, Mella P, Jones A, de Saint Basile G, Savoldi G, Cranston T, et al. Mutations in severe combined immune deficiency (SCID) due to JAK3 deficiency. Hum Mutat. 2001;18(4):255–63. doi: 10.1002/humu.1188. [DOI] [PubMed] [Google Scholar]
- 58.O’Shea JJ, Husa M, Li D, Hofmann SR, Watford W, Roberts JL, et al. Jak3 and the pathogenesis of severe combined immunodeficiency. Mol Immunol. 2004;41(6–7):727–37. doi: 10.1016/j.molimm.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 59.Watford WT, O’Shea JJ. Human tyk2 kinase deficiency: another primary immunodeficiency syndrome. Immunity. 2006;25(5):695–7. doi: 10.1016/j.immuni.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 60.Kilpivaara O, Levine RL. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia. 2008;22(10):1813–7. doi: 10.1038/leu.2008.229. [DOI] [PubMed] [Google Scholar]
- 61.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suessmuth Y, Elliott J, Percy MJ, Inami M, Attal H, Harrison CN, et al. A new polycythaemia vera-associated SOCS3 SH2 mutant (SOCS3F136L) cannot regulate erythropoietin responses. Br J Haematol. 2009;147(4):450–8. doi: 10.1111/j.1365-2141.2009.07860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, et al. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 2006;4(5):e144. doi: 10.1371/journal.pbio.0040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, et al. Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett. 2002;12(8):1219–23. doi: 10.1016/s0960-894x(02)00106-3. [DOI] [PubMed] [Google Scholar]
- 65.Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302(5646):875–8. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- 66.Jiang JK, Ghoreschi K, Deflorian F, Chen Z, Perreira M, Pesu M, et al. Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperi din-1-yl)-3-oxopropanenitrile (CP-690,550) J Med Chem. 2008;51(24):8012–8. doi: 10.1021/jm801142b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang X, Hu Y, Yuan Z. Computational analyses of JAK1 kinase domain: subtle changes in the catalytic cleft influence inhibitor specificity. Biochem Biophys Res Commun. 2008;370(1):72–6. doi: 10.1016/j.bbrc.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 68.Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21(8):1658–68. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 69.Geron I, Abrahamsson A, Barroga C, Kavalerchik E, Gotlib J, Hood J, et al. Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors. Cancer Cell. 2008;13(4):321–30. doi: 10.1016/j.ccr.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 70.Kiss R, Polgár T, Kirabo A, Sayyah J, Figueroa N, List A, et al. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg Med Chem Lett. 2009;19(13):3598–601. doi: 10.1016/j.bmcl.2009.04.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sudbeck EA, Liu XP, Narla RK, Mahajan S, Ghosh S, Mao C, et al. Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents. Clin Cancer Res. 1999;5(6):1569–82. [PubMed] [Google Scholar]
- 72.Uckun F, Roers B, Waurzyniak B, Liu X, Cetkovic-Cvrlje M. Janus kinase 3 inhibitor WHI-P131/JANEX-1 prevents graft-versus-host disease but spares the graft-versus-leukemia function of the bone marrow allografts in a murine bone marrow transplantation model. Blood. 2002;99(11):4192–9. doi: 10.1182/blood.v99.11.4192. [DOI] [PubMed] [Google Scholar]
- 73.Schindler T, Bornmann W, Pellicena P, Miller W, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 74.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–9. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 75.Zhao L, Dong H, Zhang CC, Kinch L, Osawa M, Iacovino M, et al. A JAK2 interdomain linker relays Epo receptor engagement signals to kinase activation. J Biol Chem. 2009;284(39):26988–98. doi: 10.1074/jbc.M109.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lindauer K, Loerting T, Liedl KR, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 2001;14(1):27–37. doi: 10.1093/protein/14.1.27. [DOI] [PubMed] [Google Scholar]
- 77.Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86(4):577–87. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- 78.Fukuda K, Gupta S, Chen K, Wu C, Qin J. The pseudoactive site of ILK is essential for its binding to alpha-Parvin and localization to focal adhesions. Mol Cell. 2009;36(5):819–30. doi: 10.1016/j.molcel.2009.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haan C, Kroy DC, Wuller S, Sommer U, Nocker T, Rolvering C, et al. An unusual insertion in Jak2 is crucial for kinase activity and differentially affects cytokine responses. J Immunol. 2009;182(5):2969–77. doi: 10.4049/jimmunol.0800572. [DOI] [PubMed] [Google Scholar]
- 80.Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol. 2007;178(8):5058–68. doi: 10.4049/jimmunol.178.8.5058. [DOI] [PubMed] [Google Scholar]
- 81.Ahmed CM, Dabelic R, Waiboci LW, Jager LD, Heron LL, Johnson HM. SOCS-1 mimetics protect mice against lethal poxvirus infection: identification of a novel endogenous antiviral system. J Virol. 2009;83(3):1402–15. doi: 10.1128/JVI.01138-08. [DOI] [PMC free article] [PubMed] [Google Scholar]