

Abstract
During locomotion, visual cortical neurons fire at higher rates to visual stimuli than during immobility while maintaining orientation selectivity. The mechanisms underlying this change in gain are not understood. We performed whole cell recordings from layer 2/3 and layer 4 visual cortical excitatory neurons as well as from parvalbumin-positive and somatostatin-positive inhibitory neurons in mice free to rest or run on a spherical treadmill. We found that the membrane potential of all cell types became more depolarized and (with the exception of somatostatin-positive interneurons) less variable during locomotion. Cholinergic input was essential for maintaining the unimodal membrane potential distribution during immobility, while noradrenergic input was necessary for the tonic depolarization associated with locomotion. Our results provide a mechanism for how neuromodulation controls the gain and signal-to-noise ratio of visual cortical neurons during changes in the state of vigilance.
Introduction
Cortical neuronal output, even in primary sensory areas, results from the interaction between sensory-driven and internally-generated activity1, 2. The characteristics of this spontaneous cortical activity, which depends on the behavioral state3, 4, have an important impact on the integration of incoming sensory information 5–7. In rodents, locomotion is associated with higher visually-evoked firing rates in visual cortex (V1) neurons 8–10. However the cellular mechanisms leading to distinct modes of visual information processing during immobility and locomotion remain unknown.
Several mechanisms such as a decrease in inhibitory drive 11–13, an increase in excitatory inputs 9, or an alteration of neuronal activity by neuromodulators14 could potentially modify the gain of sensory neurons. To determine which of these processes are critical for brain-state-dependent changes in information processing, it is essential to record the membrane potential (Vm) of the neurons and determine the subthreshold activity leading to the alteration of visual evoked activity. Using current-clamp whole cell recording of excitatory and inhibitory neurons in layer 2/3 (L2/3) and excitatory neurons in layer 4 (L4), visual stimulation and local pharmacological interventions in mice free to run or rest on a spherical treadmill, we showed that acetylcholine and norepinephrine control two distinct membrane potential (Vm) dynamics of V1 neurons during wakefulness. Acetylcholine was essential for maintaining the unimodal and broad distribution of Vm during quiescent periods, whereas norepinephrine was necessary to depolarize neurons during locomotion. The modification of Vm dynamics during locomotion enhanced the gain and the signal-to-noise ratio of the cortical neurons. These results provide a mechanism for how behavioral context modulates the collection and processing of sensory information by the cerebral cortex.
Results
L2/3 neuron activity during locomotion
To determine the cellular mechanisms underlying the increase of gain in V1 L2/3 neurons during locomotion, we performed in-vivo two-photon guided whole cell recordings from these neurons in head-fixed mice habituated to rest or run on a spherical treadmill (n= 53 neurons; Fig. 1a). Recordings were performed simultaneously with an electrocorticogram (ECoG) located in the vicinity (<1mm) of the recorded neuron. We first recorded the spontaneous activity (Fig. 1b) while the LCD monitor placed in front of the animal displayed an isoluminant gray screen. During locomotion, as previously described8, the V1 ECoG power spectrum demonstrated lower power at lower frequencies (typically <30 Hz) and higher power at higher frequencies (typically >30 Hz) than during stationary periods (Mann-Whitney U Test: p < 0.001; Fig. 1c; Supplementary Fig. 1). This change in ECoG frequency content was associated with a change in Vm dynamics as reflected in the neuronal Vm distribution (Fig. 1d). The Vm distribution, which was unimodal for almost all L2/3 neurons during immobility (n = 50 out of 53 neurons), was always unimodal, shifted toward more depolarized potentials, and narrower during locomotion (Fig. 1d). In agreement with this observation, the mean Vm was significantly more depolarized and the Vm less variable during locomotion than during stationary periods, but the mean firing rate did not change (Fig. 1e; Supplementary Table 1). The depolarization was not associated with a change in the membrane resistance or membrane time constant of the neurons (n= 10 neurons; Supplementary Fig. 2; Supplementary Table 2).
Figure 1.
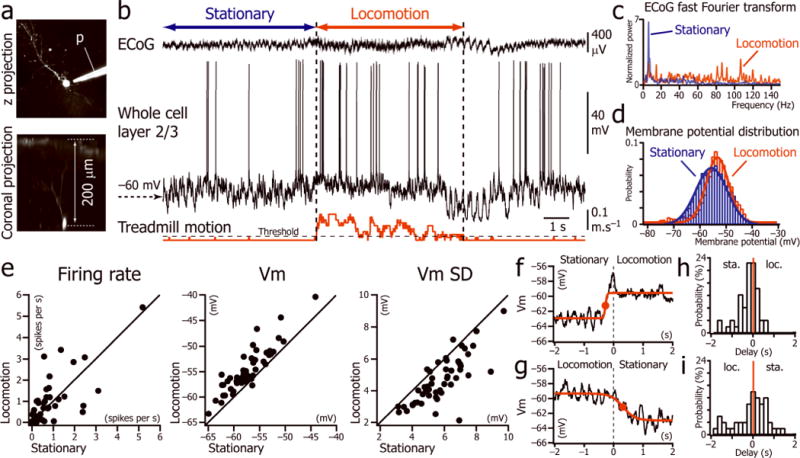
Spontaneous activity of L2/3 neurons during stationary and locomotion periods. (a) Two-photon images of a V1 L2/3 neuron labeled with Alexa-594 through the recording pipette (p). (b) Current-clamp recording of a L2/3 neuron (middle trace) simultaneously with V1 ECoG (top trace) and treadmill motion (bottom trace). Period of locomotion (between vertical dotted lines) defined as beginning when the velocity is greater than the first step of velocity detection (threshold) for more than 1s, and resuming when velocity is lower than threshold for more than 1s. (c) Fast Fourier transforms of the ECoG signal shown in bduring the periods labeled “stationary” and “locomotion”. (d) Distributions of the Vm fitted by Gaussian functions during the periods labeled “stationary” and “locomotion” in b. (e) Plots of the mean firing rate, the mean Vm and the Vm standard deviation (Vm SD) during stationary versus locomotion periods (n=53 neurons from 36 mice). (f, g) Vm averages triggered by the beginning (f) and the end (g) of 62 locomotion episodes in one neuron. Times at half rise (red dots)of the sigmoid fits were used to determine the delay between the Vm depolarization and the beginning and end of locomotion episodes. (h, i) Distribution of delays between depolarization and beginning of locomotion (loc.) (h); and repolarization and immobility (sta.) (i) for all the neurons (n= 53 from 36 mice).
We measured the temporal relationship between the change in Vm and locomotion by calculating the average of the Vm triggered by the beginning (Fig. 1f) and the end (Fig. 1g) of the locomotion periods, as measured by the movement of the spherical treadmill. This demonstrated that the onset of depolarization occurred slightly before locomotion onset (mean delay: −362 ± 481 ms; p < 0.001, one-sample signed rank test; Fig. 1h) while there was no significant delay between Vm repolarization and onset of immobility (mean delay: −315 ± 864 ms; p = 0.07, one-sample signed rank test; Fig. 1i).
To understand how locomotion-related changes in Vm dynamics alter the integrative properties of V1 L2/3 neurons during visual stimulation, we presented a series of sine-wave drifting gratings (6 orientations in both directions) interleaved with isoluminant gray screens while the animal was free to run or stay stationary (Fig. 2a–b). For 22 neurons, we obtained a complete orientation tuning curve of the Vm, the Vm SD, and the firing rate for both locomotion and immobility (Fig. 2c). The population orientation tuning curve showed that the tonic depolarization and decrease in Vm variability was associated with an increase in the gain of L2/3 excitatory neurons without a change in orientation tuning (OSI during immobility: 0.48 ± 0.19; OSI during locomotion: 0.54 ± 0.28; p =0.4; n=22 neurons; Fig. 2c,e,f). During presentations of drifting gratings of the preferred orientation, the Vm was significantly more depolarized and less variable during locomotion than during immobility (Fig. 2c–d; Supplementary Table 3). The firing rate evoked during locomotion by the drifting grating of the preferred orientation was about twice as high as the firing rate measured during immobility (Fig. 2c–d; Supplementary Table 3) whereas it remained unchanged for the orthogonal orientations (p = 1 and p = 0.7 for gratings oriented respectively −90o and +90o to the preferred orientation; Fig. 2c). Therefore, during locomotion, Vm depolarization and decreased Vm variability increased the gain of visual cortical neurons without altering orientation selectivity. While the Vm depolarization was additive in nature, it resulted in a multiplicative increase in firing rate to visual stimulation (Supplementary Fig. 3).
Figure 2.
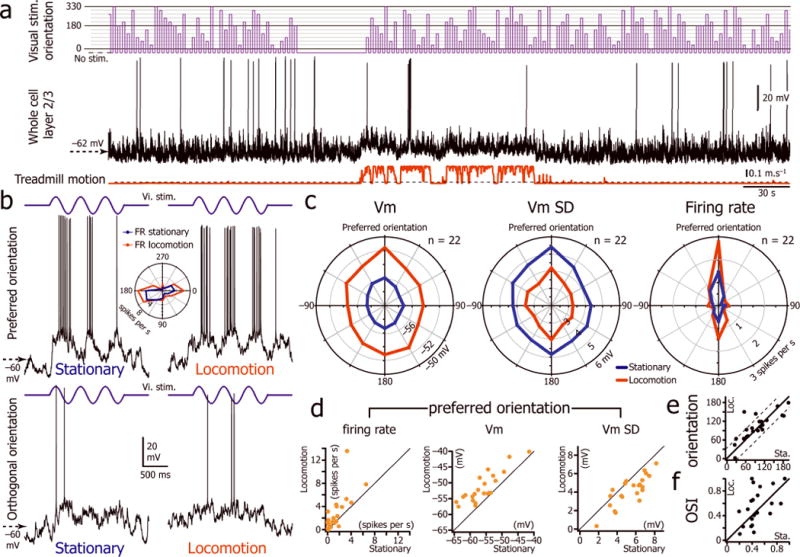
Locomotion is associated with an increase in the gain of L2/3 excitatory neurons. (a) V1 L2/3 whole cell recording (middle trace) during the presentation of drifting gratings of 12 different orientations (top trace) interleaved with the presentation of an isoluminant gray screen (“no stim.”) while the animal was free to run or rest on the spherical treadmill (bottom trace). (b) Vm changes (bottom traces) evoked in the same L2/3 neuron by three cycles of a drifting grating at 2 Hz (top traces, vi. stim.) of preferred orientation (180 degrees, top panels) and orthogonal orientation (270 degrees, bottom panels) during stationary (left) and locomotion (right) periods. Inset: orientation tuning curve of the neuron during immobility (blue) and locomotion (red). (c) Orientation tuning curve for the Vm, Vm SD and firing rate of the L2/3 neuronal population during immobility and locomotion (n=22 neurons from 18 mice). (d) Plots of firing rate, Vm and Vm SD measured for the preferred orientation during the stationary periods versus the locomotion periods (n=22 neurons from 18 mice). (e) Plot of the preferred orientation measured during immobility versus locomotion. Beyond the dashed lines the difference of orientation is greater than 30 degrees. (f) Plot of the orientation selectivity index (OSI) measured during immobility versus locomotion. OSI = 1 correspond to a perfectly oriented neuron.
L2/3 interneurons and L4 neurons during locomotion
We investigated the origin of the tonic depolarization and decreased Vm variability of L2/3 neurons during locomotion by recording the main excitatory and inhibitory inputs to these cells. As a tonic depolarization could potentially result from a decrease in inhibition, we recorded from parvalbumin-positive (PV+) and somatostatin-positive (SOM+) interneurons, the two types of interneuron providing the majority of inhibitory inputs to L2/3 excitatory neurons15. To specifically target PV+ and SOM+ interneurons, we used PV-Cre X Ai9 and SOM-Cre X Ai9 mice, where these neurons are specifically labeled with tdTomato16 (Supplementary Fig. 4).
We performed whole cell recordings from 9 PV+ neurons expressing tdTomato in PV-Cre x Ai9 mice (Fig. 3a; recording duration: mean = 26 ± 7 minutes). All of these neurons fired narrow action potentials at high rates (Fig. 3b,d; Supplementary Table 4) and a significant proportion of them (44%) exhibited a bimodal distribution of their membrane potential during immobility (Hartigan’s dip test <0.05; Fig. 3b,d). Similar to excitatory neurons, PV+ interneurons exhibited a more depolarized and less variable Vm during locomotion but, unlike excitatory neurons, spontaneous firing rates dramatically increased (Fig. 3c; Supplementary Table 1). Recordings during visual stimulation showed that PV+ interneurons had poor orientation selectivity (OSI = 0.20 ± 0.09; n= 9 neurons; Fig. 3e) and fired robustly for all orientations during immobility and locomotion (Fig. 3d–e). For the orientation for which the neurons had the highest firing rate during stationary periods, locomotion significantly depolarized PV+ neurons, decreased their Vm variability and increased their evoked firing rate (Fig. 3e; Supplementary Table 3).
Figure 3.
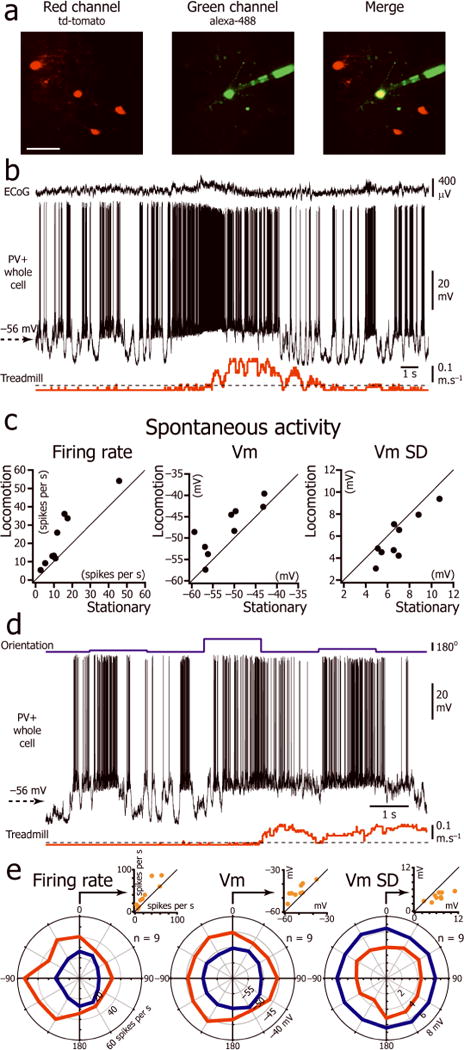
Effect of locomotion on Vm of L2/3 parvalbumin positive interneurons. (a) In vivo two-photon image of a neuron (yellow) injected with Alexa-488 (green) during the recording in a mouse expressing tdTomato (red) in PV+ neurons. Scale bar: 50 μm. (b) Spontaneous activity of a L2/3 PV+ interneuron during immobility and locomotion. (c) Plots of firing rate, Vm and Vm SD during immobility versus locomotion for 9 L2/3 PV+ interneurons (8 mice). (d) Vm of the neuron shown in c during the presentation of drifting gratings of three different orientations (top trace) interleaved with presentation of an isoluminant gray screen. (e) Orientation tuning curve of the L2/3 PV+ interneuron population for firing rate, Vm, and Vm SD during immobility and locomotion (n=9 neurons from 8 mice). The orientation “0” was assigned for each neuron to the orientation at which the stationary firing rate evoked by the visual stimulus was maximal. Insert: Plot of firing rate, Vm, and Vm SD during immobility versus locomotion for the orientation “0”.
SOM+ interneurons localized in L2/3 (recording duration: mean = 23 ± 13 minutes; n=10 neurons; Fig. 4a) were characterized by an action potential rise time intermediate between L2/3 excitatory neurons and PV+ interneurons (Supplementary Table 4). During locomotion, SOM+ interneurons were also more depolarized and fired at higher rates than during immobility (Fig. 4b–c; Supplementary Table 1), but we found no significant decrease in Vm SD in these neurons (Fig. 4c; Supplementary Table 1). Similar to PV+ neurons, SOM+ neurons showed poor orientation selectivity (OSI: 0.14 ± 0.13; n=10 neurons) and fired at high rates during both locomotion and immobility (Fig. 4b,d). For the orientation at which the neurons had the highest firing rate during stationary periods, Vm was significantly more depolarized and the evoked firing rate significantly increased (Fig. 4d; Supplementary Table 3).
Figure 4.
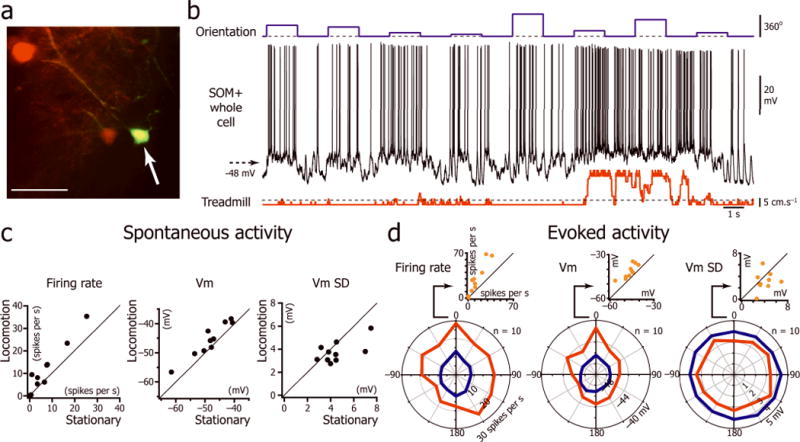
Effect of locomotion on the intracellular activity of L2/3 somatostatin positive interneurons. (a) In vivo two-photon imaging of a neuron (arrow) injected with Alexa-488 (green) during the intracellular recording in a mouse expressing tdTomato in SOM+ neurons (red). Scale bar: 50 μm. (b) Vm activity evoked by a series of drifting gratings interleaved with isoluminant gray screens (top trace) of a L2/3 SOM+ interneuron during immobility and locomotion. (c) Plots for 10 L2/3 SOM+ interneurons of the spontaneous firing rate, Vm and Vm SD during immobility versus locomotion (8 mice). (d) L2/3 SOM+ interneuron population orientation tuning curves for the firing rate, Vm, Vm SD during immobility and locomotion (n=10 neurons from 8 mice). The orientation “0” was assigned for each neuron to the orientation at which the stationary firing rate evoked by the visual stimulus was maximal. Insert: Plot of firing rate, Vm, and Vm SD during immobility versus locomotion for the orientation “0”.
As these results disproved the hypothesis that the tonic depolarization of L2/3 neurons during locomotion resulted from a decrease in PV+ and SOM+ inhibitory inputs, we asked whether the depolarization could result from an increase in L4 neuronal firing rate, as this constitutes one of the main excitatory inputs to L2/3 neurons. We performed whole cell recordings from 10 L4 neurons (depth: 383 ± 23 μm from the surface; range 350 to 426 μm; Fig. 5a). During presentations of an isoluminant gray screen, locomotion was associated with a tonic depolarization but did not alter the spontaneous firing rate of these cells (Fig. 5c; Supplementary Table 1), ruling out the possibility that the tonic depolarization of L2/3 neurons resulted from an increase of the feed-forward excitation from L4. During visual stimulation (Fig. 5b), the Vm significantly depolarized and the Vm SD significantly decreased (Fig. 5d,e; Supplementary Table 3). The firing rate evoked by the preferred orientation significantly increased (Supplementary Table 3) while the firing rate for the orthogonal orientations stayed unchanged (p = 0.3 and p = 0.1 for gratings oriented respectively −90o and +90oto the preferred orientation). Therefore, an increase in the feed-forward excitation from L4 did not mediate the tonic depolarization of L2/3 neurons during locomotion.
Figure 5.
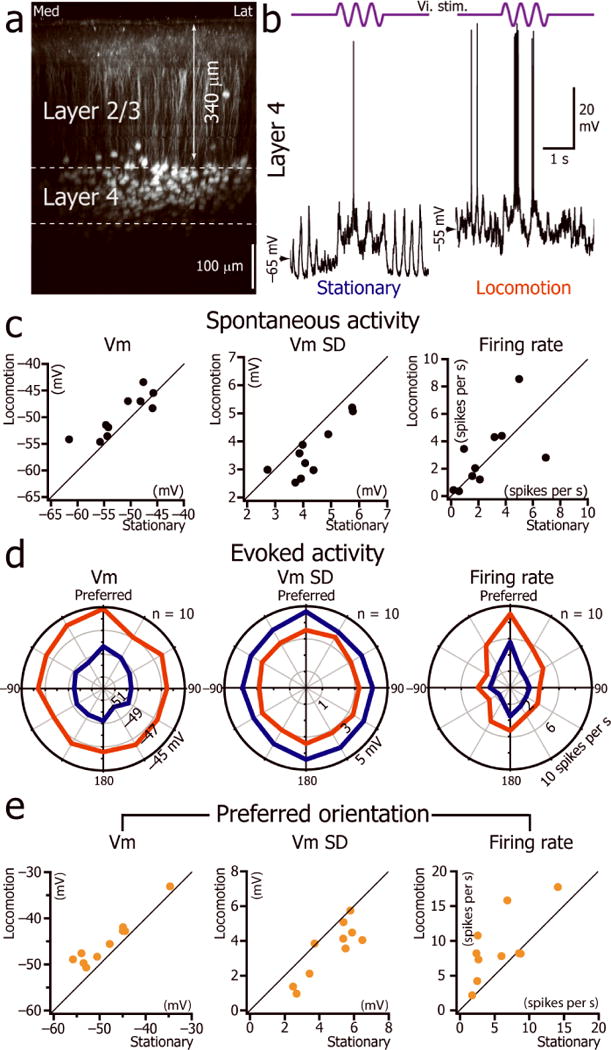
L4 neuron signal-to-noise ratio increases during locomotion. (a) Coronal view of V1 in a SCNN1a-Cre × Ai9 mouse expressing tdTomato in L4. (b) Current-clamp whole cell recordings from a V1 L4 neuron during the presentation of a 2Hz drifting grating of preferred orientation (top trace) when the animal was immobile (left) or during locomotion (right). (c) Plots of the spontaneous Vm, Vm SD and firing rate of L4 neurons during immobility versus locomotion (n= 10neurons from 10 mice). (d) L4 population orientation tuning curve for Vm, the Vm SD and firing rate during immobility and locomotion (n=10 neurons from 10 mice). (e) Plot of mean Vm, Vm SD and mean firing rate during immobility versus locomotion for the preferred orientation.
Cholinergic blockade during immobility and locomotion
As the depolarization and decreased Vm fluctuations of L2/3 excitatory neurons during locomotion could not be explained by increased excitation from L4 or decreased local inhibition, we explored the role of neuromodulators that had previously been shown to modulate the awake cortical brain state 14, 17–21. We first tested the role of cholinergic input, which plays an important role in arousal, electroencephalographic activation, attentional processing 1, 22, and enhances stimulus driven neuronal activity in the visual cortex 23–25. We recorded 6 L2/3 neurons before, during, and after local perfusion of 1 mM atropine and mecamylamine (muscarinic and nicotinic antagonists respectively) using a pipette whose tip was positioned within 250 μm of the patch pipette tip (Fig. 6a). After cholinergic blockade, the distribution of the Vm during stationary periods became bimodal (Hartigan’s dip test of unimodal distribution after local drug injection: p = 0.007 ± 0.012; range: 0 to 0.035; n=6; Fig. 6b–d). Moreover, local injection of cholinergic antagonists led to aberrant synchronization characterized by large amplitude ECoG spikes associated with neuronal paroxysmal depolarizations crowned with bursts of action potentials (Fig. 6e). However, cholinergic antagonists did not prevent the unimodal depolarization associated with locomotion (Vm depolarization during baseline: mean = 2.9 ± 1.7 mV; Vm depolarization under cholinergic blockade: mean = 4.3 ± 1.3 mV; p = 0.2; n=6 neurons; Fig. 6d,f,g and Supplementary Table 5; Hartigan’s dip test > 0.05). Therefore, while cholinergic input was essential for maintaining the unimodal distribution of the Vm during stationary periods, it did not mediate the tonic depolarization induced by locomotion. As cholinergic inputs maintain the unimodal Vm distribution during stationary states and have been shown to reduce the variability of evoked firing rates25, we tested if cholinergic blockade would increase the Vm SD measured during locomotion. However there was no significant change in the Vm SD during locomotion before and after cholinergic blockade (Supplementary Table 5; Mann-Whitney U test: p = 0.7) suggesting that cholinergic inputs do not play a role in decreasing the membrane potential variability during locomotion.
Figure 6.
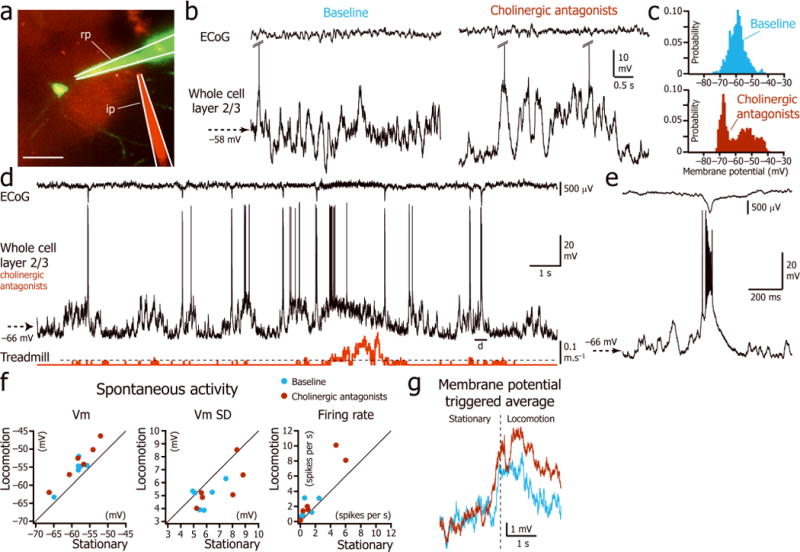
Effect of cholinergic antagonists on the L2/3 neuron Vm during stationary and locomotion periods. (a) Two-photon image of a L2/3 neuron labeled with Alexa-488 (green) through the recording pipette (rp) and the local drug injection via an injection pipette (ip) visualized by addition of Alexa-594 (red) to the drug vehicle. Scale bar: 50 μm. (b) L2/3 neuron spontaneous activity during immobility before (left) and after (right) local injection of cholinergic antagonists. (c) Distribution of the Vm for the two examples shown in b. (d) Vm (middle trace) and ECoG (top trace) during stationary and locomotion periods (bottom trace) after local injection of cholinergic antagonists atropine and mecamylamine. (e) Magnified view of the paroxysmal burst associated with the ECoG spike indicated in d. (f) Superimposition of the plots of the spontaneous Vm, Vm SD and firing rate during immobility versus locomotion during baseline (blue) and cholinergic blockade (maroon). (n = 6 neurons from 6 mice). (g) Superimposition of the averages of the Vm triggered by the beginning of locomotion (vertical dotted line) during baseline (blue; n = 57 episodes in 6 neurons) and cholinergic blockade (maroon; n= 144 episodes in 6 neurons).
Noradrenergic blockade during immobility and locomotion
As norepinephrine also plays a major role in arousal and attention20, 26, and the firing rate of locus coeruleus neurons increases during locomotion 27, we tested the role of noradrenergic input by performing local injections of α1, α2, and β noradrenergic receptor antagonists (prazosin, yohimbine, and propranolol respectively; 1mM) in V1. Noradrenergic blockade led to a hyperpolarization of the Vm of L2/3 neurons localized in the vicinity of the injection pipette (500 μm radius; stationary Vm during baseline vs. stationary Vm during norepinephrine blockade: p = 0.02, n=8 neurons; Fig. 7a,b and Supplementary Table 5). These injections dramatically decreased spontaneous Vm variability (stationary Vm SD during baseline vs. stationary Vm SD during norepinephrine blockade: p = 0.008, n=8 neurons; Fig. 7a,b and Supplementary Table 5) and firing rate (stationary firing rate during baseline vs. stationary firing rate during norepinephrine blockade: p = 0.008, n=8 neurons; Fig. 7a,b and Supplementary Table 5). These injections also reduced the amplitude of the mean depolarization induced by visual stimulation (Fig. 7c,e). Finally, this alteration of Vm dynamics was concomitant with the loss of the depolarization associated with locomotion (depolarization during noradrenergic blockade: 0.5 ± 0.7 mV; One-Sample Signed Rank Test: p = 0.08, n=8 neurons; depolarization during baseline: 2.9 ± 1.2mV; One-Sample Signed Rank Test: p =0.008; Fig. 7a,d).
Figure 7.
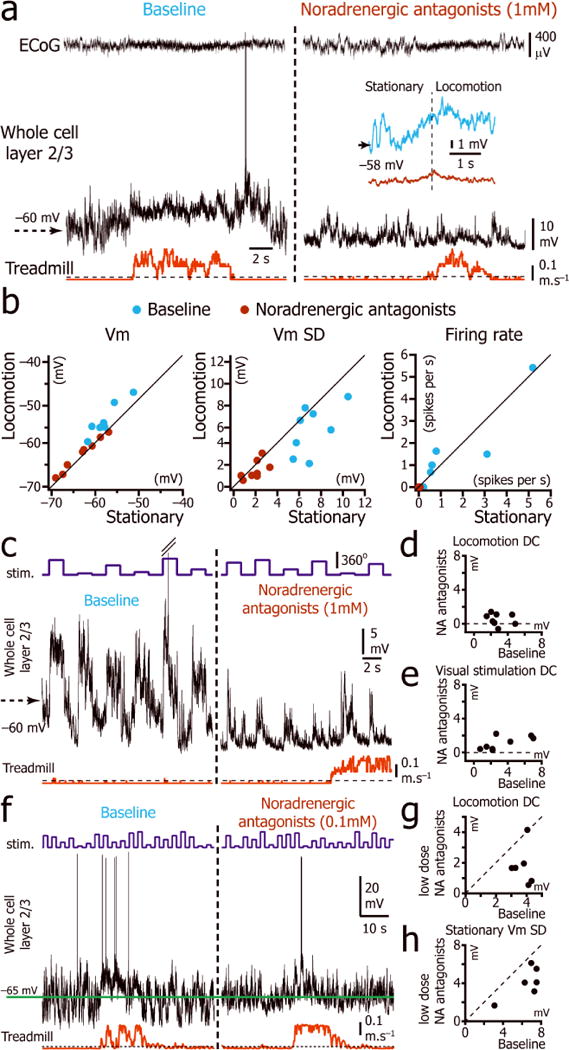
Effect of norepinephrine antagonists on the Vm of L2/3 neurons during stationary and locomotion periods. (a) Vm (middle trace) and ECoG (top trace) recordings during stationary and locomotion periods (bottom trace) before (left) and after (right) local injection of noradrenergic antagonistsprazosin, yohimbine, and propranolol. Inset: Average of the Vm triggered by the beginning of locomotion (vertical dotted line) during baseline (blue; n = 58 episodes in 8 neurons from 8 mice) and noradrenergic blockade (maroon; n= 79 episodes in 8 neurons from 8 mice). (b) Plot of the spontaneous baseline (blue) and noradrenergic blockade (maroon) Vm, Vm SD and firing rate during immobility versus locomotion. (c) Activity evoked by a series of drifting gratings (top trace) to a L2/3 neuron (middle trace) before (left) and after (right) local injection of noradrenergic antagonists. (d) Plot of the difference between the mean Vm during locomotion and the mean Vm during immobility, during baseline periods versus during noradrenergic (NA) blockade. (n = 8 neurons). (e) Plot of the mean depolarization during visual stimulation (all orientations) during baseline versus during noradrenergic blockade. (n = 8 neurons from 8 mice). (f) Vm recording (middle trace) during stationary and locomotion periods (bottom trace) while visual stimulation was presented (top trace) before (left) and after (right) local injection of low dose (0.1 mM) noradrenergic antagonists prazosin, yohimbine, and propranolol. Green horizontal line indicates the mean stationary Vm during baseline. (g) Plot of the difference between the locomotion Vm and stationary Vm during baseline versus partial noradrenergic blockade (n = 6 neurons from 6 mice). (h) Plot of the stationary Vm SD during baseline versus partial noradrenergic blockade (n = 6 neurons from 6 mice).
To rule out the possibility that the blockade of the tonic depolarization was due to a non-specific effect associated with high dose of antagonists, we tested if we could partially block the tonic depolarization associated with locomotion using low concentrations of prazosin, yohimbine, and propranolol (0.1 mM; Fig. 7f). Partial blockade of noradrenergic inputs significantly decreased the amplitude of the depolarization associated with locomotion by 51% (baseline: 3.8 ± 0.5 mV; partial blockade: 1.8 ± 1.3 mV; n = 6; Mann-Whitney U test: p = 0.04; Fig. 7g) showing that this effect is specific to noradrenergic blockade. Low dose noradrenergic blockade also decreased Vm fluctuations induced by spontaneous synaptic activity by 37% (stationary Vm SD during baseline: 6.4 ±1.7 mV; during partial blockade: 4.1± 1.6 mV; n = 6; Mann-Whitney U test: p = 0.03; Fig. 7h), without changing the mean stationary Vm (baseline: −61.4 ± 7.9 mV; during partial blockade: −62.3 ± 6.4 mV; n = 6; Mann-Whitney U test: p = 0.7; Fig. 7f). This led to a decrease of the spontaneous firing rate (baseline: 0.7±0.7spikes per s; during partial blockade: 0.1 ± 0.2 spikes per s; n = 6; Mann-Whitney U test: p = 0.03) and visually evoked firing rates (Supplementary Fig. 5). Therefore, low-dose noradrenergic blockade reduced tonic depolarization associated with locomotion indicating that the previous results with full blockade resulted from a specific antagonism of noradrenergic receptors.
Glutamatergic blockade during immobility and locomotion
To determine if the depolarization associated with locomotion was due to a direct effect of norepinephrine on V1 neurons or whether norepinephrine indirectly allowed the recruitment of short or long-range glutamatergic circuits, including for example excitatory inputs from the motor cortex 9, we recorded 6 V1 L2/3 neurons before and after local injection of AMPA and NMDA glutamate receptor antagonists CNQX and AP5 (1 mM). This injection quickly shut down spontaneous and visually evoked synaptic activity, leaving the cell hyperpolarized (stationary Vm: −74.3 ± 2.3 mV; Fig. 8a). However, even in absence of spontaneous and evoked synaptic activity, the tonic depolarization associated with locomotion was still present (Fig. 8b,c), although its amplitude was significantly reduced by 57% (depolarization amplitude during baseline 2.4 ± 0.6 mV; during glutamatergic blockade: 0.8 ± 0.5 mV, n= 6; Mann-Whitney U test: p = 0.009; Fig. 8d). These results show that a diminished tonic depolarization associated with locomotion is still present in the absence of glutamatergic inputs, indicating that glutamatergic inputs only amplify the direct depolarizing effect of norepinephrine on V1 neurons.
Figure 8. Effect of glutamatergic antagonists on the Vm of L2/3 neurons during stationary and locomotion periods.
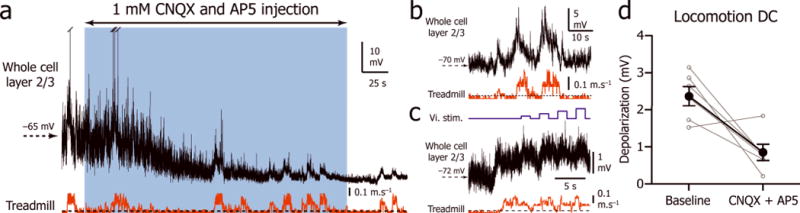
(a) Vm (top trace) recordings during stationary and locomotion periods (bottom trace) before (left), during (blue box) and after (right) local injection of 1mM AMPA and NMDA antagonists CNQX and AP5. (b) Tonic depolarization of the Vm (top trace) during locomotion episodes (bottom trace) during blockade of the spontaneous activity by CNQX and AP5 in another neuron. (c) Recording of the Vm (middle trace) of a third neuron during the transition between stationary and locomotion (bottom trace) while visual stimuli are presented (top trace). (d)) Plot of the difference between the locomotion Vm and stationary Vm during baseline and during glutamatergic blockade (n = 6 neurons from 6 mice). Solid circles indicate mean ±s.e.m. The decrease in locomotion depolarization during glutamatergic blockade is significant (Mann-Whitney U Test, p = 0.009).
Discussion
We showed that the membrane potential of L2/3 and L4 excitatory cortical neurons as well as L2/3 PV+ and SOM+ interneurons depolarized and became less variable (except for SOM+ interneurons) during locomotion. In L2/3 and L4 excitatory neurons, this depolarization (which can be considered as a summation; Supplementary Fig. 3) did not increase the spontaneous firing rate but did significantly enhance the gain (multiplicative transformation; Supplementary Fig. 3) of the V1 neurons, increasing signal-to-noise ratios. PV+ and SOM+ interneurons increased both their spontaneous and visually evoked firing rates during locomotion. Finally, while cholinergic input to the visual cortex was essential for maintaining the unimodal distribution of the membrane potential of L2/3 excitatory neurons during immobility, it was not necessary for inducing the tonic depolarization associated with locomotion. Noradrenergic input, on the other hand, was essential for the locomotion-related tonic depolarization.
Our work provides a mechanism for the enhanced signal-to-noise ratio of visual cortical neurons during locomotion 8–10. In L2/3 and L4 excitatory neurons, during spontaneous activity, the effects of the tonic depolarization on action potential generation is counteracted by the decrease in membrane potential variability, which lowers the probability that the membrane potential crosses the action potential threshold 28, 29. This phenomenon could maintain low spontaneous firing rates, while increasing the likelihood that visual input can drive L2/3 and L4 cortical neurons30. This mechanism could also play an important role in suppressing firing rates during stimulation at non-ideal orientations, and therefore maintaining orientation selectivity31. Similarly in the barrel cortex, whisking in head-restrained mice, which is associated with a tonic depolarization and a decrease in Vm variability, enhances the signal-to-noise ratio32. While the mechanisms underlying decreased variability of the membrane potential during locomotion are likely to be complex, it is highly plausible that they are induced at least in part by the massive increase in both perisomatic and dendritic inhibition on the cell via PV+ and SOM+ interneurons 33, 34.
It is unlikely that the tonic depolarization of L2/3 excitatory neurons results from an increase in feed-forward drive as neither L4 nor LGN spontaneous firing rates increase during locomotion8. It is also highly unlikely that the tonic depolarization was entirely due to excitatory inputs originating from motor cortex9 or other cortical or subcortical areas, as it persisted after glutamatergic blockade. We also show that both PV+ and SOM+ interneurons increase their firing during locomotion. It is possible that the V1 interneurons, which have been shown to decrease or cease firing during locomotion 8, belong to the minority of interneurons which are neither SOM+ nor PV+15. In the barrel cortex, GABAergic fast spiking and SOM+ interneurons tend to hyperpolarize and decrease their spontaneous action potential discharge, and ventrobasal thalamic neurons increase their firing rates during whisking35, 36. This suggests that the mechanisms of signal-to-noise ratio increase in the barrel cortex during whisking and in the primary visual cortex during locomotion may be different.
The results of our pharmacological experiments extend the conclusions of previous studies on the role of acetylcholine and norepinephrine on the membrane potential dynamics during wakefulness 18, 19, 21, 37. Acetylcholine has been shown to promote desynchronized activity 38, visual attention39, and to facilitate visual cortical responses mainly through the activation of muscarinic receptors40. We showed that a local injection of cholinergic antagonists is sufficient to allow the occurrence of locally generated UP and DOWN states41 characteristic of sleep and anesthesia3, 18, and of epileptic spikes42 associated with the paroxysmal synchronous ECoG events associated with local atropine cortical injections 43. These results differ from those obtained in rats transiently awakened from anesthesia18 possibly because in those animals, anincrease in noradrenergic drive could make up for the cholinergic deficit and keep the cell depolarized. The broad unimodal distribution of the membrane potential during immobility is likely to be controlled by both acetylcholine and norepinephrine as they both dramatically alter the membrane potential dynamics during stationary periods. Our finding that the activation of noradrenergic receptors is essential for the tonic depolarization during locomotion suggests that the increase in norepinephrine levels induced by the higher firing rates of locus coeruleus neurons during locomotion27 is essential for depolarizing L2/3 and L4 neurons, but may rely on glutamatergic inputs to fully depolarize the Vm. This hypothesis is supported by previous in-vitro studies that have shown that norepinephrine acting through α1-adrenergic receptors can tonically depolarize both excitatory44–47 and inhibitory 48 cortical neurons through a reduction of a leak potassium current. The similar input resistance of L2/3 neurons during immobility and locomotion may result from the counterbalanced closing of potassium channels induced by noradrenergic activation and opening of channels associated with GABAergic receptors.
Cognitive processes such as attention that are associated with a change in neuromodulation alter the gain of sensory tuning without affecting the shape of tuning curves49, 50. We propose that norepinephrine could enhance visual attention during locomotion by increasing the signal-to-noise ratio of excitatory neurons.
Online Methods
Surgery
All experimental procedures were approved by the University of California Los Angeles Office for Protection of Research Subjects and the Chancellor’s Animal Research Committee. 10 minutes after injection of a systemic analgesic (carprofen, 5mg.kg−1), adult male and female C57Bl6/J mice, SOM-Cre (JAX number 013044) X Ai9 (JAX number 007909), PV-Cre (JAX number 008069) X Ai9 (1 – 12 months of age), and Scnn1a-Tg3-Cre (JAX number 009613) X Ai9 were anesthetized with isoflurane (3–5% induction, 1.5% maintenance) and placed in a stereotaxic frame. Animals were kept at 37o C at all times using a Harvard Apparatus feedback-controlled heating pad. Pressure points and incision sites were injected with a local anesthetic (lidocaine 2%), and eyes were protected from desiccation using artificial tear ointment. The skin above the skull was incised, a custom-made light-weight metal head holder was implanted on the skull using Vetbond (3M), and a recording chamber was built using dental cement Ortho-Jet (Lang). Mice recovered from surgery for 5 days during which they were administered antibiotic (Amoxicillin: 0.25 mg.mL−1 in drinking water) through the water supply. After the recovery period, mice were habituated to head fixation on the spherical treadmill (at least 2 sessions of 20 minutes). The treadmill consisted of an 8 inch Styrofoam ball resting inside a Styrofoam hollow half-sphere (Graham Sweet) into which compressed air was blown. The air kept the spherical ball floating and allowed the mice to freely run or rest. On the day of the recording, mice were anesthetized with isoflurane. A circular craniotomy (diameter: 3 mm) was performed above V1 and a 3mm diameter coverslip drilled with a 500 μm diameter hole was placed over the dura such that the coverslip fit entirely in the craniotomy and was flush with the skull surface. For some experiments, the coverslip was drilled with two holes to allow the access of the local pharmacological injection pipette. The coverslip was maintained in place using Vetbond and dental cement and the recording chamber was filled with cortex buffer containing (in mM): NaCl 135, KCl 5, HEPES 5, CaCl2 1.8, and MgCl2 1. The head-bar was fixed to a post and the animal was placed on the spherical treadmill to recover from anesthesia. All recordings were performed at least 2 hours after the end of anesthesia.
Electrophysiological recordings
Long-tapered micropipettes made of borosilicate glass (1.5 mm OD, 0.86 ID; Sutter Instrument, Novato, CA USA) were pulled on Sutter Instruments P-97 pipette puller to a resistance of 3–7 MΩ, and filled with an internal solution containing (in mM): KGluc 115, KCl 20, HEPES 10, phosphocreatine 10, ATP-Mg14, GTP 0.3, Alexa-594 (for experiments with C57Bl/6 mice) or Alexa-488 (for interneuron and pharmacology experiments) 0.01–0.05. Pipettes under positive pressure (initial pressure 70 millibars then 20–30 millibars after advancing past the dura) were advanced into the cortex at 20–30 degrees from the horizontal while monitoring the ECoG to assure that the internal solution did not cause spreading depression. While pipettes were lowered into the brain, two-photon imaging was performed with a Sutter MOM microscope using a Ti-Sapphire Ultra-2 laser (Coherent) at 875 nm, and a 40× 0.8 NA Zeiss water-immersion objective. The objective was tilted at 30 degrees such that the objective lens was parallel to the dural surface. Laser power was kept below 70 mW at the sample. Images were acquired using Scanimage software 3.251. Images were processed using ImageJ software 52. Neuronal cell bodies could be clearly visualized as silhouettes on the background of neuropil stained by the Alexa dye. Pipettes were guided to the soma until the resistance of the micropipette increased. Negative pressure was then transiently applied to establish a 4–5 GΩ seal. 10 minutes after establishing the seal, further negative pressure was applied to obtain the whole cell configuration. Whole cell current-clamp recordings were performed using the bridge mode of an Axoclamp 2A amplifier (Molecular Devices), further amplified and low pass filtered at 5 KHz using a Warner Instruments amplifier (LPF 202A). Series of current pulses of small intensity (typically −100pA) were used to balance the bridge and compensate the pipette capacitance. Access resistance ranged between 30 and 150 MΩ but was typically below 60 MΩ. No DC current was injected during the recordings and the membrane potential was not corrected for liquid junction potentials (estimated to be about 10 mV). Recordings lasted typically 30 minutes (range 5 to 50 minutes). Recordings or parts of recordings with unstable membrane potential and/or action potentials < 35 mV were excluded from analysis. ECoG recordings were performed with an AC/DC differential amplifier (Model 3000, A-M system) and band pass filtered at 0.1–3,000 Hz. Analog signals were digitized at 10 kHz with a NIDAQ card (National Instruments) running under the WinEDR (Strathclyde University). In some cases, the cell’s anatomy was reconstructed by performing multiple overlapping image stacks, using the two-photon microscope. For the whole study, we assessed the depth of each recorded neuron by measuring the distance between the cell body and the dura using the two-photon microscope (Fig. 1a). We determined the location of the border between L4 and L2/3 by measuring the depth of the most superficial tdTomato-positive neurons in adult SCNN1a-Cre X Ai9 mice, a mouse line with selective labeling of L4 neurons. The transition between L4 and L2/3 was located 340 μm below the dura (range: 332 and 356 μm, n=3 mice; Fig. 5a).
Visual Presentation
A 40 cm diagonal LCD monitor was placed in the monocular visual field of the mouse at a distance of 30 cm, contralateral to the craniotomy. Custom-made software developed with Psychtoolbox in Matlab was used to display drifting sine wave gratings (series of 12 orientations spaced by 30 degrees randomly permuted, temporal frequency: 2 Hz, spatial frequency: 0.04 cycle per degree, contrast: 100%). The presentation of each orientation lasted 1.5 seconds and was followed by the presentation of a gray isoluminant screen for an additional 1.5 seconds. To synchronize data acquisition and visual presentation, a photodiode was attached to a corner of the monitor where a 2cm × 2cm square (masked so it would not be visible to the mouse) flipped from black to white at each screen refresh during drifting grating presentation. The photodiode signal was digitized simultaneously with the electrophysiological signal, and two analog signals coding for the spatial and temporal properties of the grating. The treadmill motion was measured every 25 ms (40 Hz) by an optical mouse whose signal was converted into two servo pulse analog signals (front-back and left-right) using an external PIC microcontroller, and acquired simultaneously with the electrophysiological data. The pre-compiled HEX file originally developed by Evan Dudzik for programming the PIC chip can be found at the following address: http://imakeprojects.com/Projects/seeing-eye-mouse/Gfx/downloads/Seeing-Eye-Mouse-v1.0-PIC16F628A.hex. Servo signals were analyzed post-hoc and the velocity in the cardinal directions was summed; this signal is referred to in the text and figures as treadmill motion.
Histology
Mice were anesthetized with Nembutal and perfused with 0.1 M phosphate buffer (PB) followed by 4% paraformaldehyde and kept in a refrigerator for 1 h before the brains were removed. Brains were post-fixed for 1 h in the same fixative, then rinsed with 0.1 M PB for 30 min × 3 changes and kept in 30% sucrose in PB untill they sank. Brains were embedded with optimal cutting temperature medium and cut with cryostat at 30 μm. 10 visual cortex sections were rinsed with 0.1 M Tris Buffered Saline (TBS). Non-specific combining sites were blocked with 10% normal goat serum in 0.1 M TBS containing 0.3% Triton X-100. Sections were then incubated in rabbit primary antiserum to either parvalbumin (1:8000; Swant) or somatostatin (1:5000; T-4103, Peninsula Laboratories) in TBS containing 0.1% NaN3 and 2% NGS for 7 nights. After a thorough rinse, sections were incubated in Alexa Fluor 488 conjugated goat anti-rabbit IgG for 4 hours, rinsed in TBS, mounted on glass slides and coverslipped with antifade medium.
Pharmacology
Cholinergic, noradrenergic and glutamatergic blockers were purchased from Sigma (Mecamylamine M9020; Yohimbine Y3125; Propranolol P0884; Prazosin P7791; CNQX C239; AP5 A169) except for atropine (American Regent), dissolved in warm cortex buffer, mixed with Alexa-488 and filtered. Blockers were ejected from a glass pipette similar to the patch electrode (5–7 MΩ) by applying gradually increasing pressure up to 100 millibars for several minutes. As no drug effect was ever found when the injection pipette was more than 750 μm away from the recording pipette, we estimate our maximal volume of ejection to be 1.8 mm3.
Data Analysis
Data analysis was performed using custom made routines in Igor Pro 6.0 (WaveMetrics). The initiation of a locomotion period was defined as the time at which the treadmill motion signal crossed the first level of velocity detection (indicated as threshold and/or dashed lines in the figures and corresponding to a velocity of 2 cm.s−1) and remained greater than threshold for at least one second. The end of a locomotion period was defined as the time at which the treadmill motion signal crossed the threshold and remained below this level for at least one second. The spontaneous firing rate was defined for each neuron as the number of action potentials divided by the total duration of the period of immobility or locomotion. Spontaneous mean Vm and Vm SD were measured as the average of the mean and standard deviation of a Vm trace for which spikes were replaced by the threshold value. Evoked FR, Vm and Vm SD were measured between the beginning and the end of the visual stimulus using the same methodology as for spontaneous activity. Two tuning curves, one for trials occurring during immobility, the other one for trials occurring during locomotion, were computed for the firing rate, Vm and Vm SD of each neuron. A population orientation tuning curve was computed by averaging the orientation tuning curve of cells having a complete tuning curve for both immobility and locomotion. Before averaging, the orientation tuning curve of each cell was circularly permuted such that the preferred orientation would be 0 degree.
Orientation and orientation selectivity index: The preferred orientation was computed as the angle φ of the mean vector of the orientation tuning curve F(θ) with φ = atan2(Im(V), Re(V)) and . Orientation selectivity index was measured as ||V|| 53. The preferred direction of the neuron was determined by using the circular mean of the orientation tuning curve.
Input resistance and action potential parameters: Apparent membrane input resistance was assessed using the mean membrane potential change induced by hyperpolarizing current pulses of 100 pA (250 ms duration, applied every 1.25 s). The difference of potential was measured between the mean potential 250 ms before the pulse and the mean potential during the second half of the pulse. The membrane time constant was given by the coefficient of the exponential decay fit of the hyperpolarization. Action potential amplitude and the rise time were calculated by averaging action potentials occurring during immobility and measuring the potential and time differences between the voltage threshold, measured as the membrane potential at which the second derivative of the membrane potential is maximal, and the peak of the spike waveform.
Frequency analysis: power spectra were computed as the absolute value of the Fast Fourier transform signal (obtained using a Hanning window) divided by N / (2*0.375) to satisfy Parseval’s Theorem (N represents the number of points of the ECoG signal bit). Spectra were then normalized by applying a 1/f correction. For each frequency band, the normalized power was calculated as the area under the power spectrum curve. For presentation only, the peak corresponding to 60 Hz was removed.
Statistics
Unless stated otherwise, statistical significance was calculated by Mann-Whitney U test using SigmaPlot 12.0 (Systat Software). One-Sample Signed Rank Test tested the hypothesis: the population median value is 0. Hartigan’s dip testswere performed to test for unimodal distribution using Nicholas Price’s routine for Matlab (http://www.nicprice.net/diptest/) on the membrane potential distribution of the whole spontaneous activity recorded during immobility and locomotion. No statistical methods were used to pre-determine sample sizes. The number of cells used for each test is denoted by ‘n’ in the text. Data collection and analysis were not performed blind to the conditions of the experiments. No randomization was used to collect and process data or to assign animals to the various experiments.
Supplementary Material
Acknowledgments
The authors acknowledge R. Gruver for technical assistance on visual stimulation; Z. Peng and C. Houser for histology; D. Contreras, M. Einstein, T. Indersmitten, M. Javaherian, C. Kaba, S. Mahon, A. Silva, and S. Singh for their thoughtful comments on the manuscript. This work was supported by the National Institutes of Health (KO8 NS0562101), the Whitehall Foundation (Grant 2012-05-83) and a Veterans Affairs Merit Review Award (1I01BX001524-01A1).
Footnotes
Author contributions P-O.P. and P.G. conceived and designed the experiments. P-O.P., J.F. and P.G. built the experimental setup. P-O.P. acquired and analyzed the experimental data. P-O.P. and P.G. wrote the manuscript.
References
- 1.Harris KD, Thiele A. Cortical state and attention. Nature reviews. Neuroscience. 2011;12:509–523. doi: 10.1038/nrn3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ringach DL. Spontaneous and driven cortical activity: implications for computation. Current opinion in neurobiology. 2009;19:439–444. doi: 10.1016/j.conb.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- 4.Buzsáki G, Draguhn A. Neuronal Oscillations in Cortical Networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- 5.Hasenstaub A, Sachdev RN, McCormick DA. State changes rapidly modulate cortical neuronal responsiveness. The Journal of neuroscience. 2007;27:9607–9622. doi: 10.1523/JNEUROSCI.2184-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crochet S, Petersen CCH. Correlating whisker behavior with membrane potential in barrel cortex of awake mice. Nature neuroscience. 2006;9:608–610. doi: 10.1038/nn1690. [DOI] [PubMed] [Google Scholar]
- 7.Bereshpolova Y, Amitai Y, Gusev AG, Stoelzel CR, Swadlow HA. Dendritic backpropagation and the state of the awake neocortex. The Journal of neuroscience. 2007;27:9392–9399. doi: 10.1523/JNEUROSCI.2218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niell CM, Stryker MP. Modulation of visual responses by behavioral state in mouse visual cortex. Neuron. 2010;65:472–479. doi: 10.1016/j.neuron.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keller Georg B, Bonhoeffer T, Hübener M. Sensorimotor Mismatch Signals in Primary Visual Cortex of the Behaving Mouse. Neuron. 2012;74:809–815. doi: 10.1016/j.neuron.2012.03.040. [DOI] [PubMed] [Google Scholar]
- 10.Szuts TA, et al. A wireless multi-channel neural amplifier for freely moving animals. Nature neuroscience. 2011;14:263–269. doi: 10.1038/nn.2730. [DOI] [PubMed] [Google Scholar]
- 11.Lee SH, et al. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson NR, Runyan CA, Wang FL, Sur M. Division and subtraction by distinct cortical inhibitory networks in vivo. Nature. 2012;488:343–348. doi: 10.1038/nature11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goard M, Dan Y. Basal forebrain activation enhances cortical coding of natural scenes. Nature neuroscience. 2009;12:1444–1449. doi: 10.1038/nn.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonchar Y, Wang Q, Burkhalter A. Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Frontiers in neuroanatomy. 2007;1:3. doi: 10.3389/neuro.05.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrero JL, et al. Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature. 2008;454:1110–1114. doi: 10.1038/nature07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constantinople CM, Bruno RM. Effects and mechanisms of wakefulness on local cortical networks. Neuron. 2011;69:1061–1068. doi: 10.1016/j.neuron.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steriade M, Amzica F, Nunez A. Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. Journal of neurophysiology. 1993;70:1385–1400. doi: 10.1152/jn.1993.70.4.1385. [DOI] [PubMed] [Google Scholar]
- 20.Berridge CW, Page ME, Valentino RJ, Foote SL. Effects of locus coeruleus inactivation on electroencephalographic activity in neocortex and hippocampus. Neuroscience. 1993;55:381–393. doi: 10.1016/0306-4522(93)90507-c. [DOI] [PubMed] [Google Scholar]
- 21.Metherate R, Cox C, Ashe J. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. The Journal of Neuroscience. 1992;12:4701–4711. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarter M, Bruno JP. Cortical cholinergic inputs mediating arousal, attentional processing and dreaming: differential afferent regulation of the basal forebrain by telencephalic and brainstem afferents. Neuroscience. 2000;95:933–952. doi: 10.1016/s0306-4522(99)00487-x. [DOI] [PubMed] [Google Scholar]
- 23.Sato H, Hata Y, Hagihara K, Tsumoto T. Effects of cholinergic depletion on neuron activities in the cat visual cortex. Journal of neurophysiology. 1987;58:781–794. doi: 10.1152/jn.1987.58.4.781. [DOI] [PubMed] [Google Scholar]
- 24.Muller CM, Singer W. Acetylcholine-induced inhibition in the cat visual cortex is mediated by a GABAergic mechanism. Brain research. 1989;487:335–342. doi: 10.1016/0006-8993(89)90837-8. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez R, Kallenbach U, Singer W, Munk MH. Stabilization of visual responses through cholinergic activation. Neuroscience. 2010;165:944–954. doi: 10.1016/j.neuroscience.2009.10.059. [DOI] [PubMed] [Google Scholar]
- 26.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain research. Brain research reviews. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 27.Foote SL, Aston-Jones G, Bloom FE. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:3033–3037. doi: 10.1073/pnas.77.5.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chance FS, Abbott LF, Reyes AD. Gain modulation from background synaptic input. Neuron. 2002;35:773–782. doi: 10.1016/s0896-6273(02)00820-6. [DOI] [PubMed] [Google Scholar]
- 29.Fellous JM, Rudolph M, Destexhe A, Sejnowski TJ. Synaptic background noise controls the input/output characteristics of single cells in an in vitro model of in vivo activity. Neuroscience. 2003;122:811–829. doi: 10.1016/j.neuroscience.2003.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haider B, Duque A, Hasenstaub AR, Yu Y, McCormick DA. Enhancement of visual responsiveness by spontaneous local network activity in vivo. Journal of neurophysiology. 2007;97:4186–4202. doi: 10.1152/jn.01114.2006. [DOI] [PubMed] [Google Scholar]
- 31.Anderson JS, Lampl I, Gillespie DC, Ferster D. The contribution of noise to contrast invariance of orientation tuning in cat visual cortex. Science. 2000;290:1968–1972. doi: 10.1126/science.290.5498.1968. [DOI] [PubMed] [Google Scholar]
- 32.Poulet JFA, Petersen CCH. Internal brain state regulates membrane potential synchrony in barrel cortex of behaving mice. Nature. 2008;454:881–885. doi: 10.1038/nature07150. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez-Vives MV, et al. Inhibitory modulation of cortical up states. Journal of neurophysiology. 2010;104:1314–1324. doi: 10.1152/jn.00178.2010. [DOI] [PubMed] [Google Scholar]
- 34.Hasenstaub A, et al. Inhibitory postsynaptic potentials carry synchronized frequency information in active cortical networks. Neuron. 2005;47:423–435. doi: 10.1016/j.neuron.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Gentet LJ, Avermann M, Matyas F, Staiger JF, Petersen CC. Membrane potential dynamics of GABAergic neurons in the barrel cortex of behaving mice. Neuron. 2010;65:422–435. doi: 10.1016/j.neuron.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Gentet LJ, et al. Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex. Nature neuroscience. 2012;15:607–612. doi: 10.1038/nn.3051. [DOI] [PubMed] [Google Scholar]
- 37.McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Progress in neurobiology. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- 38.Alitto HJ, Dan Y. Cell-type-specific modulation of neocortical activity by basal forebrain input. Frontiers in systems neuroscience. 2012;6:79. doi: 10.3389/fnsys.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Botly LC, De Rosa E. Impaired visual search in rats reveals cholinergic contributions to feature binding in visuospatial attention. Cereb Cortex. 2012;22:2441–2453. doi: 10.1093/cercor/bhr331. [DOI] [PubMed] [Google Scholar]
- 40.Soma S, Shimegi S, Osaki H, Sato H. Cholinergic modulation of response gain in the primary visual cortex of the macaque. Journal of neurophysiology. 2012;107:283–291. doi: 10.1152/jn.00330.2011. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nature neuroscience. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- 42.Langlois M, et al. Involvement of the thalamic parafascicular nucleus in mesial temporal lobe epilepsy. The Journal of neuroscience. 2010;30:16523–16535. doi: 10.1523/JNEUROSCI.1109-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchanan JA, Bilkey DK. Transfer between atropine-induced spiking in the perirhinal cortex and electrical kindling of the amygdala. Brain research. 1997;771:63–70. doi: 10.1016/s0006-8993(97)00778-6. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, McCormick DA. Control of firing mode of corticotectal and corticopontine layer V burst-generating neurons by norepinephrine, acetylcholine, and 1S, 3R-ACPD. The Journal of neuroscience. 1993;13:2199–2216. doi: 10.1523/JNEUROSCI.13-05-02199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormick DA, Wang Z, Huguenard J. Neurotransmitter control of neocortical neuronal activity and excitability. Cereb Cortex. 1993;3:387–398. doi: 10.1093/cercor/3.5.387. [DOI] [PubMed] [Google Scholar]
- 46.Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. The Journal of neuroscience. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. The Journal of neuroscience. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. The Journal of neuroscience. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annual review of neuroscience. 2005;28:403–450. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 50.Klinkenberg I, Sambeth A, Blokland A. Acetylcholine and attention. Behavioural brain research. 2011;221:430–442. doi: 10.1016/j.bbr.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 51.Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomedical engineering online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ringach DL, Shapley RM, Hawken MJ. Orientation selectivity in macaque V1: diversity and laminar dependence. The Journal of neuroscience. 2002;22:5639–5651. doi: 10.1523/JNEUROSCI.22-13-05639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.