

Abstract
PURPOSE
Eleven patients diagnosed with various hematologic malignancies receiving an HLA-haploidentical hematopoietic cell transplant (HCT) participated in an ancillary biomarker trial. The goal of the trial was to evaluate potential pharmacologic biomarkers pertinent to the conditioning regimen (fludarabine monophosphate (fludarabine) and cyclophosphamide (CY)) or postgrafting immunosuppression (CY and mycophenolate mofetil (MMF)) in these patients.
METHODS
We characterized the interpatient variability of nine pharmacologic biomarkers. The biomarkers evaluated were relevant to fludarabine (i.e., area under the curve (AUC) of 2-fluoro-ara-A or F-ara-A); CY (i.e., AUCs of CY and four of its metabolites); and MMF (i.e., total mycophenolic acid (MPA) AUC, unbound MPA AUC, and inosine monophosphate dehydrogenase (IMPDH) activity).
RESULTS
Interpatient variability in the pharmacologic biomarkers was high. Among those related to HCT conditioning, the interpatient variability ranged from 1.5-fold (CY AUC) to 4.0-fold (AUC of carboxyethlphosphoramide mustard, a metabolite of CY). Among biomarkers evaluated as part of postgrafting immunosuppression, the interpatient variability ranged from 1.7-fold (CY AUC) to 4.9-fold (IMPDH area under the effect curve). There was a moderate correlation (R2=0.441) of within-patient 4-hydroxycyclophosphamide formation clearance.
CONCLUSIONS
Considerable interpatient variability exists in the pharmacokinetic and drug-specific biomarkers potentially relevant to clinical outcomes in HLA-haploidentical HCT recipients. Pharmacodynamic studies are warranted to optimize the conditioning regimen and postgrafting immunosuppression administered to HLA-haploidentical HCT recipients.
Keywords: fludarabine, cyclophosphamide, mycophenolate mofetil, IMPDH, hematopoietic cell transplant, biomarkers, pharmacokinetics
INTRODUCTION
While hematopoietic cell transplantation (HCT) offers the possibility of a lasting remission to patients with hematologic malignancies and disorders, its curative potential has traditionally been limited to those patients with a closely matched donor. As many as a third of Caucasian patients in need of HCT are unable to find a suitably matched donor; patients from racial or ethnic minorities face even greater difficulties in finding an appropriate donor[1]. Additionally, unrelated donor searches are costly and time-consuming, with the risk that disease progression will outpace the search process[1]. Since an overwhelming majority of patients of all ethnic backgrounds have at least one readily available HLA-haploidentical donor, broadening the scope of HCT to include this type of donor grafts is clearly desirable. Unfortunately, initial attempts to expand HCT to patients with HLA-mismatched donors had historically been unsuccessful, characterized by increased risk of graft failure, severe graft-versus-host disease (GVHD), and significant non-relapse mortality[1].
The addition of post-transplant cyclophosphamide (CY) to postgrafting immunosuppression, used in conjunction with a calcineurin inhibitor and mycophenolate mofetil (MMF), appears to have overcome these barriers. The combination of reduced-intensity conditioning, an HLA-haploidentical donor graft, and postgrafting immunosuppression that includes CY has led to acceptable rates of GVHD and improved survival[2]. It is desirable, however, to further lower rates of graft rejection, grades II-IV GVHD (currently 2% and 32%, respectively) and relapse [3]. We hypothesize the interpatient variability in the outcomes of an HLA-haploidentical HCT is associated with pharmacokinetic and drug-specific pharmacodynamic variability. Thus, we sought to characterize the interpatient variability in the pharmacokinetics and drug-specific pharmacodynamics of the various medications administered as part of the conditioning regimen (CY and fludarabine) and postgrafting immunosuppression (CY and MMF), as shown in Figure 1. Since the pharmacodynamics of these drugs differ between HCT conditioning regimens[4] and graft sources[5, 6], we sought to characterize the interpatient variability of nine biomarkers previously associated with clinical outcomes in HCT patients to determine the feasibility of pharmacodynamic studies in larger patient populations.
Figure 1.
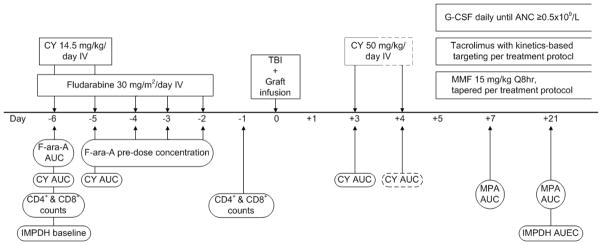
Schema of pharmacologically-based biomarkers. Abbreviations: AUC: area under the curve; AUEC: area under the effect curve; CY: cyclophosphamide, includes CY and four of its metabolites; fludarabine: fludarabine monophosphate; IMPDH: inosine monophosphate dehydrogenase; MMF: mycophenolate mofetil; MPA: mycophenolic acid, includes both total and unbound MPA; TBI: total body irradiation
METHODS
Patient Selection
Eleven patients were enrolled onto this ancillary biomarker study between June 2010 and November 2011; participation did not influence the HCT procedure. Participants were eligible for this study if they were 18 years of age or older and receiving conditioning that included fludarabine, an HLA-haploidentical graft, and postgrafting immunosuppression that included post-transplant CY and MMF. Patients were ineligible if their hemoglobin was less than 8g/dL, if they could not speak or read English, if they had been diagnosed with an immunodeficiency disorder (including HIV), if they were pregnant or breastfeeding, if their life expectancy was severely limited by diseases other than malignancy, or if their conditioning regimen or post-graft immunosuppression included a radiolabeled monoclonal antibody. Among the 22 patients receiving an HLA-haploidentical donor HCT, 16 were eligible to participate in this ancillary biomarker study. Among eligible patients, eleven (69%) agreed to participate and eight participants (73%) complied with all the blood sampling (Figure 1). This ancillary study was registered on clinicaltrials.gov with identifier NCT01141959; patients’ treatment protocols were registered with identifiers NCT01028716, NCT01008462, and NCT00789776. This research was conducted with the approval of the Fred Hutchinson Cancer Research Center (FHCRC) Institutional Review Board, and written consent was obtained from all participants prior to study conduct.
All participants were adults receiving HCT from HLA-haploidentical donors to treat high-risk hematological malignancies; the majority of participants were enrolled on NCT01028716. The stem cell source was bone marrow for three participants (27%) and peripheral blood stem cells (PBSC) for the remaining eight participants. One participant was enrolled on a phase I/II protocol and received an additional infusion of donor natural killer cells one week after the initial stem cell infusion in a trial to boost the graft versus tumor effect and to enhance early immune reconstitution after HCT[7].
Method of HCT
All participants received the same reduced-intensity conditioning regimen prior to infusion of their HLA-haploidentical donor graft. Conditioning consisted of the following: 1) CY, 14.5mg/kg per day, administered on days −6 and −5 (29 mg/kg total); 2) fludarabine monophosphate (fludarabine), 30mg/m2 per day, administered on days −6 through −2 (150 mg/m2 total); 3) one 200cGy fraction of total body irradiation (TBI) administered on day −1. Intravenous (IV) CY was dosed according to adjusted ideal body weight (AIBW), where AIBW = ideal weight + 0.25(actual weight - ideal weight). Ideal weight was calculated as follows: for men, ideal weight = 50kg + (2.3kg/inch of height over 5ft); for women, ideal weight = 45.5kg + (2.3kg/inch of height over 5ft). Fludarabine was dosed by body surface area (BSA), which was calculated using actual weight. CY was administered over 60 minutes (min); fludarabine was administered over 30 or 60 min, according to treatment protocol. Likewise, the order of administration of CY and fludarabine varied depending on the treatment protocol.
Post-grafting immunosuppression for all patients consisted of CY, MMF and tacrolimus. All patients were administered CY 50 mg/kg per day as a 60 min infusion. Three patients received one dose of post-transplant CY on day +3 (50 mg/kg total) and the remaining eight patients received two doses of post-transplant CY on days +3 and +4 (100 mg/kg total). The addition of the second dose of post-transplant CY has been shown to reduce chronic, but not acute, GVHD.[2] MMF and tacrolimus were initiated the day after the final post-transplant CY dose. MMF was given orally every 8 hour (hr) at a dose of 15 mg/kg based on AIBW and rounded to the nearest 250mg increment. In patients who could not tolerate oral MMF, the IV equivalent was given instead at a one-to-one substitution. All patients began tacrolimus therapy at a dose of 1 mg IV daily or 1 mg PO every 12 hr. Tacrolimus doses were adjusted to reach target trough concentrations of 5–15 ng/mL, based on patients’ treatment protocols. All patients also received daily G-CSF, beginning one day after the last dose of post-transplant CY, until the third consecutive day with an absolute neutrophil count greater than 0.5 × 109/L. One patient received an infusion of donor natural killer cells on day +7, as per treatment protocol (NCT00789776).
Supportive care, including antiemetics, antifungals, and antibiotics, was similar among all patients and followed FHCRC standard practice guidelines; three patients received corticosteroids during conditioning. On the days that pretransplant CY was administered, nine of eleven patients received 2-mercaptoethane sulfonate (mesna) for uroepithelial prophylaxis at a dose equivalent to that of CY. With post-transplant CY, all patients received an equivalent dose of mesna. All patients received antimicrobial prophylaxis against Pneumocystis jiroveci, Candida albicans, and herpes viruses.
Patients were monitored at least weekly for cytomegalovirus reactivation and treated pre-emptively with ganciclovir or foscarnet as needed. Patients were considered to have attained neutrophil engraftment on the first of three consecutive days with an absolute neutrophil count ≥0.5 × 109/L. Platelet engraftment was defined as the first of seven days, in the absence of transfusion, with a platelet count ≥20 × 106/L. GVHD was diagnosed according to established criteria[5, 8]. Relapse was assessed according to treatment protocols or as clinically necessary.
HCT conditioning biomarkers
2-fluoro-ara-A (F-ara-A) pharmacokinetics were evaluated as follows: on day −6, three blood samples were collected to evaluate the area under the concentration-time curve (AUC) of F-ara-A. On days −5 through −2, one blood sample was collected immediately before the start of each fludarabine infusion. On day −1, one blood sample was collected 24hr after the start of the final dose of fludarabine. The samples used to determine the AUC of F-ara-A were collected immediately at the end of the infusion, five minutes after the end of the infusion, 90 min after the start of the infusion, and 24 hr after the start of the infusion[9]. Our earlier experience from a similar ancillary biomarker study (clinicaltrials.gov #NCT00764829) was that this shorter sample schedule improved patient participation due to its shorter duration than the optimal schedule (i.e., 0.583, 1.5, 6.5, and 24 hr). The shorter sample schedule had an acceptable scaled mean squared error of 13.5 compared to 12.3 for the optimal schedule.[9] F-ara-A concentrations were quantified as previously described[9], with the following modifications: 100μL plasma was combined with 50μL methanol, 20μL clofarabine (internal standard), and 100μL acetonitrile. The mixture was vortexed and centrifuged. The supernatant was analyzed using high-performance liquid chromatography/tandem mass spectroscopy (LC/MS/MS) running a gradient mobile phase of 0.1% formic acid and methanol through an Agilent Zorbax SB-C18 column (Agilent, Santa Clara, CA), monitoring m/z 304 (internal standard) and 286 (F-Ara-A). The assay’s dynamic range was 0.1 to 5.0μg/mL, and the interday precision was less than 10%. The AUCs were calculated using post-hoc Bayesian estimates from NONMEM version 7.2 (Icon Development Solutions, Ellicott City, MD). We did not assess the accumulation rate of the active metabolite fludarabine triphosphate (F-ara-ATP) in CD4+ and CD8+ T-lymphocytes due to concerns regarding participant safety[10]. Specifically, the assessed biomarkers required 165 to 230mL of blood drawn over four weeks. Considering that F-ara-ATP requires an additional 60 mL blood draw, we chose not to assess F-ara-ATP after extensive discussions with the IRB.
Cyclophosphamide pharmacokinetics were obtained with each CY dose. For patients treated in the outpatient setting, samples were collected at 1(end of infusion), 2, 4, 8, and 24hr from the start of the infusion. For patients who had been admitted for inpatient care, samples were collected at 1 (end of infusion), 2, 4, 8, 16, 20, and 24 hr from the start of the infusion[11]. When applicable, the 24hr samples were collected before the start of the following dose. Samples were placed at 4 C within five minutes of collection and were processed within 18 hr. Concentrations of CY,4-hydroxycyclophosphamide (4HCY), carboxyethylphosphoramide mustard (CEPM), deschloroethylcyclophosphamide (DCCY), and 4-oxocyclophosphamide (KetoCY) were measured as previously reported[12], except using LC/MS/MS. The dynamic range of the assays of CY and its metabolites were as follows: CY, 0.96 to 192μM; 4HCY, 0.125 to 25μM; CEPM 0.043 to 17μM; DCCY 0.063 to 25μM; KetoCY 0.022 to 9.1μM. All assays had an interday precision less than 10%. The AUCs were estimated using noncompartmental modeling with SAS (SAS, Version 9.2, Cary, NC)[13, 14].
Fludarabine and cyclophosphamide pharmacodynamics were assessed by two blood samples, collected before and after conditioning administration to evaluate circulating CD4+ and CD8+ T-cell counts. The first sample was collected on day −6 before administration of either CY or fludarabine. The second was collected on day −1, 24hr after the start of the final dose of fludarabine and before TBI. Samples were assayed by a single-platform method using BD Trucount tubes (BD Biosciences, San Jose, CA) and Beckman Coulter Cytostat Tetra One monoclonal antibody reagent with a Coulter FC-500 flow cytometer running Beckman Coulter CXP software (all Beckman Coulter, Brea, CA). CD45/SS gating was used to identify lymphocytes and distinguish CD4+ and CD8+ T-cell populations in peripheral blood.
Post-transplant immunosuppression biomarkers
Cyclophosphamide pharmacokinetics were evaluated with each post-transplant CY dose, using the methods described above in the conditioning regimen biomarker section.
Two MMF biomarkers were evaluated, specifically MPA pharmacokinetics and a MMF-specific pharmacodynamic marker of IMPDH activity. The pharmacokinetics of MPA were measured on days +7 and +21. On each day, five blood samples were collected for measurement of the total and free MPA AUCs. For patients on oral MMF, samples were collected immediately before the dose and 1.25, 2, 3, and 4 hr after the dose[15]. For patients on IV MMF, samples were collected at 2 (end of infusion), 2.5, 3, 4, and 6 hr from the start of the two-hour infusion[16]. Total plasma MPA was measured by LC/MS as previously described[5]. Free MPA was determined by equilibrium dialysis using the Pierce RED (Rapid Equilibrium Dialysis) Device (Thermo Fisher Scientific, Waltham, MA). AUCs were estimated using noncompartmental modeling with SAS (SAS, Version 9.2, Cary, NC).
IMPDH activity in peripheral blood mononuclear cells was assessed on two occasions. Pre-transplant baseline (i.e., before MMF administration) IMPDH activity was determined with a sample collected on day −6, concurrent with the first F-ara-A pharmacokinetic blood draw. Post-transplant IMPDH activity was measured on day +21; samples were collected at the same times as the MPA pharmacokinetic draws, above.
Blood samples were stored at 4 °C until processing. Peripheral blood mononuclear cells (PMNC) were isolated within 6 hr of collection. PMNC were isolated by diluting blood in PBS at a 1:1 v:v ratio, then layering the mixture on Ficoll. The height ratio of Ficoll to diluted whole blood sample was 3:4. This suspension was then centrifuged at 298 g for 30 min at 22 °C. PMNC were collected from the interface and diluted with PBS to a volume of 10 mL to wash, then centrifuged at 405 g for 15 min at 22 °C. To facilitate cell counting and limit the variability in cell concentration, all but 1.1 mL of the supernatant was removed. The PMNC pellet was resuspended in the remaining PBS, and 1.0 mL of the PBS-cell slurry was transferred to a 2 mL tube. White blood cell counts in this sample were quantitated using a Horiba Diagnostics ABX Micro 60 (requires <10 μL) (Irving, CA). Following cell counting, the sample was centrifuged at 325 g for 10 min at room temperature. From the microcentrifuge tube, 920 μL of the supernatant was removed; distilled water was added to adjust the cell concentration to 0.5 × 107 cells/mL lysate. The cells were subsequently stored at −80°C until incubation. After thawing, insoluble fragments of disrupted cells were removed by centrifugation at 16000 g for 2 min. The supernatant was used for IMPDH activity assay.
IMPDH activity in lysed lymphocytes was determined from the conversion of inosine-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) according to a procedure adapted from Glander et al.[17] and Daxecker et al[18]. For each incubation a fresh reaction mixture was prepared from stock solutions: 1.6 mL NaH2PO4 (120 mmol/L, stored at 4 °C), 1.6 mL KCl (300 mmol/L, stored at 4 °C), 0.8 mL IMP (6.0 mmol/L, stored at −20 °C), 0.8 mL NAD (4.5 mmol/L, made fresh each day); the pH was adjusted to 7.4 using 1M NaOH, and the total volume brought to 5.2 mL with deionized water. From this reaction mixture 130 μL was used. The incubation was carried out in a 37 °C shaking water bath. The reaction was started via the addition of 130 μL of reaction mixture to the 50 μL pre warmed (5 min) cell lysate (standardized concentration of 0.5×107 cells/mL). After 2.5 hr, the enzymatic reaction was terminated by the addition of 1250 μL methanol. Internal standard (20 μL of approximately 130 pmol/μL 8-Bromo adenosine 5′-monphosphate (BMP) in deionized water) was then added to each incubate via repeating syringe, followed by centrifugation at 16000 g for 10 min. The supernatant was then transferred to 12 × 75 disposable culture tubes and evaporated to dryness under a stream of air at 37°C. The residue was dissolved in 75μL of deionized water and 5μL were injected on the HPLC-MS.
The HPLC separation was performed on an Agilent HPLC/MS series 1100 system equipped with a thermostated autosampler (Agilent Technologies, Palo Alto, CA). Agilent ChemStation (version B.01.03) was used for instrument control. Separation was achieved using a Thermo Scientific Hypercarb column (2.0mm × 100mm × 5μ, part no. 35005-102130, Thermo Scientific, Bellefonte, PA). The mobile phase consisted of acetonitrile and 0.1 M ammonium acetate adjusted to pH 8.5 with ammonium hydroxide. A gradient system was used starting at 5% acetonitrile for 0.5 min, increasing to 30% at 4 min, held at 30% until 5 min, then returning to 5% at 5.1 min. The total run time was 10 min. The injector was maintained at 4 °C. The injection volume was 5 μL. The column thermostat was set to 30.0 °C and the solvent flow was maintained at 0.3 mL/min.
An Agilent G1946D MSD (Agilent Technologies, Palo Alto, CA) API-ES in positive ion mode was used. The temperature of the drying gas (N2) was maintained at 350 °C at a flow of 11 L/min. The nebulizing pressure was 35 psi; the capillary voltage was 2400 V, and the fragmentor voltage was 100 V. The MSD was run in the SIM mode. Monitored ions included m/z 365 for the (M+H+) ion of xanthosine monophosphate, m/z 348 for the (M+H+) ion of adenosine 5′-monophosphate, and m/z 426 for the (M+H+) ion of BMP, the internal standard. The MSD conditions for quantification were as described above; the fragmentor and capillary voltage were optimized under analytical conditions with Chemstation FIA software. During analysis, eluent before 3 min and after 7.5 min was diverted to waste. Typical retention times were 4.2 min for IMP, 5.5 min for XMP, and 6.9 min for BMP (Supplemental Figure 1). The area under the effect curves (AUECs) on day 21 were estimated using noncompartmental modeling with SAS (SAS, Version 9.2, Cary, NC).
RESULTS
Patient Population
Eleven patients were enrolled, of whom six were female and five were male. The median age of participants was 50.8 years (range 21.8 – 68.4). Five patients were treated for Non-Hodgkin lymphoma, three patients for Hodgkin lymphoma, and three patients for myelodysplastic syndrome (MDS) and/or acute myelogenous leukemia (AML). All patients had HLA-haploidentical related donors - six donors were patients’ children, three were parents, and two were siblings. The median number of graft-versus-host mismatches was five (range: 3–5); the median number of host-versus-graft mismatches was also five (range: 2–5). Patient characteristics are summarized in Table 1.
Table 1.
Patient characteristicsa
| Nb (%) | |
|---|---|
| Age (yr) | 50 (21 – 68) |
| Male sex | 5 (45%) |
| AIBW (kg) | 64.6 (51.1 – 82.8) |
| Donor relationship | |
| Mother | 2 (18%) |
| Father | 1 (9%) |
| Brother | 2 (18%) |
| Daughter | 3 (27%) |
| Son | 3 (27%) |
| Disease | |
| Non-Hodgkin lymphoma | 5 (45%) |
| Hodgkin lymphoma | 3 (27%) |
| Acute myeloid leukemia/myelodysplastic syndrome (MDS) | 3 (27%) |
| Cytomegalovirus serostatus | |
| Recipient +/Donor − | 5 (45%) |
| Recipient +/Donor + | 4 (36%) |
| Recipient −/Donor − | 1 (9%) |
| Recipient −/Donor + | 1 (9%) |
| Graft source | |
| Peripheral blood stem cells | 8 (73%) |
| Bone marrow | 3 (27%) |
| Post-transplant CYc | |
| One dose (Day +3) | 3 |
| Two doses (Days +3 and +4) | 8 |
All patients received the same conditioning regimen prior to HCT;
data are presented as median (range) or number (%);
all patients received tacrolimus and MMF. One patient’s second dose of post-transplant CY was delayed by 24hr because the patient was not clinically stable.
Clinical Outcomes
All patients attained neutrophil engraftment. The median (range) day of neutrophil engraftment, defined as the first of three consecutive days with an absolute neutrophil count 0.5 × 109/L, was day +15 (11 – 20). Nine participants attained platelet engraftment at a median (range) of day +15 (12 – 40) days post-transplant; platelet engraftment was defined as the first of seven consecutive days with a platelet count 20 × 106/L without transfusion support. Eight patients (73%) developed acute GVHD at a median (range) of 29 (15 – 97) days post-transplant. All patients had grade II GVHD (i.e., no grade 1, 3 or 4 GVHD was observed) and were treated with corticosteroids, as well as continuation of tacrolimus and MMF. Of the nine participants who were cytomegalovirus (CMV) positive prior to HCT, eight (89%) participants reactivated CMV. Six participants are currently alive, with a median follow-up of 19 months (range 14–29) post-transplant, one of whom relapsed on day + 534 and is in CR after undergoing additional treatment Two participants died of relapse (days +46 and +85). Three participants died of non-relapse mortality (Table 3): one of pneumonia (day +126), one of multi-organ failure with GVHD (day +259), and one of acute respiratory distress syndrome (day +265); all three were on immunosuppression at the time of death.
Table 3.
Post-transplant pharmacologic biomarkers and clinical outcomes
| Cyclophosphamide (μM×hr) | Total MPA AUC0-8hr (μg/mL×hr) | Free MPA AUC0-8hr (ng/mL×hr) | IMPDH AUEC (pmol × 106 cells) | Outcome (days) | Length of follow-up (months) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | CY | 4HCY | CEPM | DCCY | KetoCY | Day +7 | Day +21 | Day +7 | Day +21 | |||
| One CY dose (day +3); AUC0-24hr | ||||||||||||
| 3 | 2760 | 41.0 | 80.5 | 223 | 56.0 | 20.23 | 12.74 | 327 | 161 | 5676 | Deceased; relapse (85) | |
| 4 | 2663 | 65.2 | 133 | 215 | 69.8 | 35.35 | 25.18 | 469 | 305 | 4858 | Relapsed (534), now in CR | 25 |
| 10 | 1932 | 58.3 | 142 | 227 | 79.2 | 12.90 | 9.25 | 170 | 141 | 4039 | Deceased; relapse (46) | |
| Two CY doses (days +3 and +4); AUC0-48hr | ||||||||||||
| 1 | 3839 | 228 | 520 | 297 | 184 | 21.60 | 15.55 | 237 | 208 | 1532 | 29 | |
| 2 | 4796 | 174 | 489 | 508 | 158 | 9.83 | 8.51 | 158 | 145 | 5781 | Deceased; ARDSa, renal failure; on ISPb (265) | |
| 5 | 4295 | 116 | 321 | 211 | 89.0 | 23.18 | 15.25 | 432 | 235 | 1171 | Deceased; pneumonia; on ISPb (126) | |
| 6 | 3812 | 121 | 160 | 555 | 95.5 | 12.09 | --c | 154 | -- | -- | 23 | |
| 7 | 3685 | 134 | 228 | 258 | 122 | 9.46 | 11.46 | 192 | 179 | 4377 | Deceased; MOF with GVHD; on ISPb,d (259) | |
| 8 | 3123 | 120 | 269 | 635 | 111 | 12.20 | --c | 238 | -- | -- | 16 | |
| 9 | 3425 | 35.7e | 408 | 283 | 132 | 10.50 | 6.77 | 181 | 108 | 1930 | 15 | |
| 11 | 2754 | 150 | 161 | 423 | 169 | 10.44 | --c | 196 | -- | -- | 14 | |
| Median (range)f | 3748 (2754 – 4796) | 134 (116 – 228) | 295 (160 – 520) | 360 (211 – 635) | 127 (89.0 – 184) | 12.20 (9.46 – 35.35) | 12.10 (6.77 – 25.18) | 196 (154 – 469) | 170 (108 – 305) | 4208 (1171 – 5781) | ||
| Fold rangef | 1.7 | 2.0 | 3.2 | 3.0 | 2.1 | 3.7 | 3.7 | 3.0 | 2.8 | 4.9 | ||
Acute respiratory distress syndrome;
Immunosuppression; pt was on immunosuppressants at time of death;
Patients 6, 8, and 11 declined to participate in the second of the two MMF pharmacokinetic sampling days;
Multi-organ failure with graft versus host disease;
4HCY pharmacokinetics only collected with first dose of CY; due to delay in second dose, 4HCY derivatizing agent was no longer viable at time of sample collection;
Median and fold range values include only those patients receiving two doses of post-transplant CY.
Pharmacokinetic and pharmacodynamic biomarkers
Considerable interpatient variability was seen in both pre- and post-transplant biomarkers, as listed in Tables 2 and 3. The majority of participants had a substantive decline in their circulating CD4+ or CD8+ cells with 10 of the 11 participants having a greater than 90% decline (with 100% decline meaning no circulating CD4+ or CD8+ cells found). The percent decline varied 3.3-fold and 1.1-fold, respectively. The majority of patients experienced an increase in the ratio of CD4+ to CD8+ cells after conditioning: the pre-transplant median (range) CD4+/CD8+ ratio was 0.6 (0.12 – 2.5, N=11), while the post-transplant median (range) was 1.5 (0 – 6.2, N=10). Fludarabine AUC0-24hr varied by a factor of 2.1 (maximum/minimum). Among F-ara-A trough (pre-dose) concentrations, the median within-patient coefficient of variation (the standard deviation divided by the average) was 16% (range 9–36%) and the within-patient variability was 1.5-fold (range 1.2- to 2.6-fold).
Table 2.
Pre-transplant pharmacologic biomarkers
| Cyclophosphamide AUC0-48hr (μM×hr) | F-ara-A AUC (μM×hr) | % Decline in Circulating T-cells | Recipient IMPDH (pmol×106 cells/hr) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient # | CY | 4HCY | CEPM | DCCY | KetoCY | CD4+ cells | CD8+ cells | ||
| 1 | 1303 | 84.8 | 155 | 57.8 | 83.5 | 17.7 | 97 | 100 | 649 |
| 2 | 1623 | 34.0 | 140 | 134 | 53.1 | 17.7 | --a | --a | 588 |
| 3 | 1590 | 28.0 | 59.1 | 123 | 31.6 | 13.2 | 97 | 96 | 660 |
| 4 | 1541 | 51.6 | 65.2 | 83.4 | 33.5 | 15.5 | 94 | 100 | 858 |
| 5 | 1594 | 27.1 | 98.7 | 52.0 | 35.9 | 16.4 | 93 | 98 | 1118 |
| 6 | 1190 | 43.0 | 51.2 | 149 | 44.1 | 11.8 | 100 | 100 | 983 |
| 7 | 1447 | 40.0 | 86.7 | 51.0 | 45.1 | 12.8 | 96 | 95 | 1260 |
| 8 | 1125 | 64.7 | 116 | 199 | 58.8 | 18.6 | 96 | 100 | 1214 |
| 9 | 1451 | 34.8 | 124 | 115 | 53.1 | 21.5 | 100 | 100 | 424 |
| 10 | 1637 | 35.0 | 122 | 167 | 57.3 | 18.8 | 100 | 100 | 1140 |
| 11 | 1055 | 31.2 | 38.5 | 156 | --b | 10.4 | 30 | 88 | 820 |
| Median (range) | 1451 (1055 – 1637) | 35.0 (27.1 – 84.8) | 98.7 (38.5 – 155) | 123 (51.0 – 199) | 49.1 (31.6 – 83.5) | 16.4 (10.4 – 21.5) | 96 (30 – 100) | 100 (88 – 100) | 858 (424 – 1260 |
| Fold range | 1.6 | 3.1 | 4.0 | 3.9 | 2.6 | 2.1 | 3.3 | 1.1 | 3.0 |
Post-transplant circulating T-cell counts not collected for this patient;
KetoCY AUC not evaluable in this patient.
The AUCs of CY and its metabolites varied widely both before and after transplant. During conditioning, CY AUC0-48hr varied 1.6-fold, while the AUCs of CY’s metabolites were more widely distributed with a 3.1-fold range for 4HCY AUCs, a 4.0-fold range for CEPM AUCs, a 3.9-fold range for DCCY AUCs, and a 2.6-fold range for KetoCY AUCs. Similar variation was seen in post-transplant CY. Among patients receiving two doses of post-transplant CY, CY AUC0-48hr had a 1.7-fold variation. In this setting, the metabolites of CY varied as follows: 2.0-fold for 4HCY, 3.2-fold for CEPM, 3.0-fold for DCCY, and 2.1-fold for KetoCY. Both pre- and post-transplant formation clearances of 4HCY and DCCY (the AUC of the metabolite divided by the AUC of the parent compound) showed great variation. The pre-transplant formation clearance for 4HCY varied 3.8-fold, while the DCCY formation clearance varied 5.4-fold. Post-transplant, there was 5.3-fold variability in the formation clearance of 4HCY and 4.1-fold variability for DCCY. Notably, the formation clearance of 4HCY was similar in six of the 11 participants after the lower (14.5 mg/kg), pre-transplant dose to that after the higher (50 mg/kg), post-transplant CY dose (Figure 2). There was a moderate correlation (R2=0.441) of within-patient 4HCY formation clearances. As expected, the maximum plasma concentrations (Cmax) increased from the first dose of pre-transplant CY (14.5 mg/kg) to the first dose of post-transplant CY (50 mg/kg). The median (range) Cmax for pre-transplant CY and 4HCY, respectively, were 91μM (75–113) and 1.4μM (1.0 – 6.2). Post-transplant, the median (range) Cmax for CY and 4HCY were 304μM (262 – 375) and 9.7μM (3.6 – 18.8), respectively. Three of the 11 participants declined the day +21 blood draws, which were used to estimate the MPA AUC and IMPDH activity. The AUC0-8hr of total MPA varied widely but consistently; there was a 3.7-fold difference on both days +7 and +21. Free MPA AUC0-8hr was similarly variable, showing 3.0-fold variation on day +7 and 2.8-fold variation on day +21. The variability in pre-and post-transplant IMPDH activity was, however, much greater. Before transplant, there was a 3.0-fold difference in the recipients’ baseline (i.e., before MMF) IMPDH activity (Table 2). After transplant, there was 4.9-fold variation in the IMPDH AUEC (Table 3).
Figure 2.

Comparison of 4HCY formation clearances (4HCY AUC/CY AUC) from first doses of CY before and after transplant.
DISCUSSION
The key findings from this pilot study are: 1) ancillary biomarker studies seeking to characterize the pharmacokinetics and drug-specific pharmacodynamics of several medications used for HLA-haploidentical HCT are feasible; 2) there is substantial interpatient variability in these potential biomarkers with conventional body weight or BSA-based dosing. The majority (69%) of eligible patients agreed to participate, and most participants (79%) completed all the blood sampling. Despite these encouraging participation rates, the study population is insufficient to evaluate whether potential drug-specific biomarkers are associated with clinical outcomes. As shown in Table 4, an adequately powered study would need 25–50 participants, which could be easily achieved through multi-center studies, to evaluate the pharmacodynamics in HLA-haploidentical HCT recipients. Such prospective studies are needed to determine whether personalized dosing of the conditioning regimen or post-grafting immunosuppression can improve survival. A complete biomarker analysis from 50 participants, would require 100 patients, assuming rates of participation (69%) and blood sampling compliance (Figure 1, 73%) similar to what we observed. Acute GVHD occurred in most (72%) of the patients, including the three participants who died of non-relapse mortality by day 265 post-HCT. Each of these three participants was taking immunosuppressants at the time of death. The wide interpatient variability in the potential biomarkers, combined with the prevalence of acute GVHD and non-relapse mortality raises the question of whether further biomarker studies could identify ways to personalize the HCT procedure to improve patient outcomes. Biomarkers, once validated, hold great promise to guide treatment selection (e.g., gene expression profiling for breast cancer)[19] or personalization of chemotherapy dosing (e.g., kinetics-based busulfan dosing[4] or genetics-based dosing of mercaptopurine)[19, 20]. However, the potential need for large sample sizes to draw definitive conclusions about infrequent toxicities[21] is a challenge for biomarker studies in patients with cancer. We sought to characterize multiple biomarkers for two reasons. First, often a single biomarker does not sufficiently explain the clinical outcomes in a patient population (e.g., estrogen receptor, HER-2 are used in breast cancer treatment[19, 21]). Second, quantitative and systems pharmacology (QSP), an evolving discipline which seeks to combine experimental research with computational modeling, presents a possible avenue to more quickly identify the optimal conditioning regimen and/or postgrafting immunosuppression in HLA-haploidentical HCT recipients. QSP studies are designed to advance the discovery, development, and clinical use of therapeutic drugs. As the use of alternative graft sources continues to increase, multi-center studies should take a QSP approach to evaluate various biomarkers and their clinical relevance. In this setting, QSP studies are necessary: their novel biochemical, pathway-oriented, and physiological approaches complement substantive investments in genomics, with the goal of understanding drug-target interactions and pathophysiology[22].
Table 4.
Power calculations showing feasibility of adequate participant accrual to conduct a meaningful pharmacodynamic analysis in HLA-haploidentical recipients
| Potential primary endpoint | Historical rate[3] | Expected event rate | Power | ||||
|---|---|---|---|---|---|---|---|
| Entire group | Lowest AUC groupa | Highest AUC groupa | Effect size | N=25 | N=50 | ||
| 2-year progression free survival | 35%b | 35% | 50% | 20% | 30% | 0.33 | 0.61 |
| 35% | 60% | 10% | 50% | 0.98 | 0.99 | ||
| 1-year progression free survival | 48% | 48% | 70% | 25% | 45% | 0.61 | 0.92 |
| 48% | 80% | 15% | 65% | 0.95 | 0.99 | ||
| 1-year overall survival | 62% | 62% | 90% | 34% | 56% | 0.86 | 0.99 |
| 1 year cumulative incidence of relapse | 45% | 45% | 10% | 80% | 70% | 0.90 | 0.99 |
| 45% | 20% | 70% | 50% | 0.72 | 0.97 | ||
| Non-relapse mortality | 7% | 7% | 1% | 15% | 14% | 0.24 | 0.44 |
Hypothesized event rate between lowest AUC group (i.e., < median) and highest AUC group (i.e., ≥ median). Effect size is the event rate in lowest AUC group minus the event rate in the highest AUC group;
event rate per BMT CTN version 1.0
Multiple biomarkers related to fludarabine and CY in the HCT conditioning regimen were evaluated. Because of its ability to decrease lymphocyte counts, fludarabine has become an essential part of many reduced-intensity HCT conditioning regimens. After administration, nucleotidases rapidly dephosphorylate fludarabine to F-ara-A[23] and subsequently form F-ara-ATP intracellularly. The F-ara-A AUCs from this study are consistent with those from patients conditioned with fludarabine (30–40 mg/m2) and 50 mg/kg cyclophosphamide (median AUC 5.0μg×h/mL, range 2.0 to 11.0) [24]. Intracellular accumulation of F-ara-ATP is similar between CD4+ and CD8+ isolated from HCT recipients[10]. Notably, we observed similar decreases in circulating CD4+ and CD8+ cells (Table 2). Fludarabine administration leads to a marked prolonged reduction in circulating CD4+ and CD8+ cells, but the immediate effects of its administration upon these cells has yet to be described[25]. In vitro data regarding the effects of fludarabine upon the proportion of CD4+ vs. CD8+ cells undergoing apoptosis are contradictory and suggest that CD4+ cells are similarly[26] or less susceptible[27] to fludarabine than CD8+ cells. The immediate effects of fludarabine upon recipients’ circulating CD4+ and CD8+ cell counts may influence the ratio of CD4+ to CD8+ cells which affects their sensitivity to TBI in vitro[28]. In turn, recipient T-cell suppression may be a critical determinant of engraftment[29] and long-term immune function[30]. As shown in Table 2, there was considerable variability in the decline in circulating CD4+ cell counts but less variability in CD8+ cell counts.
With fludarabine and TBI, CY is also an integral component of this reduced-intensity conditioning regimen. To our knowledge, we are the first to evaluate the pharmacokinetics of post-transplant CY. CY is a prodrug with multiple metabolites, of which 4HCY formation is critical because it is transported intracellularly and subsequently broken down to phosphoramide mustard, which covalently cross-links DNA. The growth of bone marrow progenitor[31, 32], CFU-GM[33], natural killer[34], and T-[35, 34] cells are suppressed ex-vivo by 4HCY in a concentration-dependent manner. The current method of dosing CY by body weight leads to considerable interpatient variability in the AUCs of CY, 4HCY, and additional metabolites (Tables 2 and 3), which is consistent with our data in patients receiving CY 120 mg/kg administered with TBI (CY/TBI) or targeted busulfan (TBU/CY)[4]. Variability in the exposure to CY and/or its metabolites may account for interpatient differences in the efficacy and toxicity of post-transplant CY. The AUCs of CY and/or its metabolites are biomarkers for the efficacy and toxicity of the CY/TBI conditioning regimen[11]. However, the concentration-effect relationships of CY vary between different conditioning regimens, as the AUCs of CY and its metabolites are not associated with clinical outcomes in patients conditioned with the TBU/CY regimen[4]. Similarly, pharmacodynamics of MPA differ based on graft source when MMF is a component of post-grafting immunosuppression[5, 6]. The pharmacokinetics and pharmacodynamics of CY have yet to be characterized in patients receiving HLA-haploidentical HCT. The use of post-transplant CY has lead to acceptable outcomes after receipt of an HLA-haploidentical donor graft[2, 3]. Thus, a pharmacokinetic study in a homogenous patient group receiving post transplant CY is necessary to determine the clinical relevance of the pharmacokinetic variability of CY and its metabolites. Unfortunately, the number of haploidentical HCT performed at our single center, despite our encouraging accrual rate (69%), did not provide a large enough sample to conduct such a pharmacodynamic analysis. To our knowledge, this study is novel as it is the first to characterize the pharmacokinetic variability of post-transplant CY.
Because CY was administered on two separate occasions, we could characterize the within-patient variability of 4HCY formation clearance, which is the 4HCY AUC divided by the CY AUC. As shown in Figure 2, six of the 11 participants had a consistent 4HCY formation clearance, which suggests that a test dose of CY may be possible to determine individual-specific 4HCY AUC. One participant, however, had a lower 4HCY formation clearance after the pre-transplant CY dose. Furthermore, four participants had a greater 4HCY formation clearance after the lower (14.5 mg/kg) pre-transplant CY dose compared to the higher (50 mg/kg) post-transplant CY dose. The latter group qualitatively agrees with finding in 16 breast cancer patients that the extent of bioactivation is greater with lower dose CY (500 mg/m2, equivalent to 12.5 mg/kg) compared to myeloablative dose CY (100 mg/kg)[36, 37]. No drug interactions were identified that could have affected the 4HCY formation clearance in this latter group (Supplemental Table 1). Of note, although CY autoinduces 4HCY formation[38], this is not expected to influence the between-dose comparison for two reasons. First, eight days elapsed between the last pre-transplant CY dose and the first post-transplant CY dose. Since the usual half-life of CY is 3–4 hr, there should be no residual CY at the time of post-transplant CY administration. Second, if autoinduction was a factor, 4HCY formation clearance would be expected to be higher with post-transplant CY which only occurred in one of the 11 participants (Figure 2). Notably, although concurrent medications did change, few CY-drug interactions were possible (Supplemental Table 1).
MMF is another component of postgrafting immunosuppression. MPA, MMF’s active metabolite, is a selective, reversible, and noncompetitive inhibitor of IMPDH, which is involved in the de novo pathway of purine synthesis in T- and B-lymphocytes[39, 40]. Other investigators have found associations between mycophenolic acid AUC or predose concentrations and clinical outcomes in patients receiving fludarabine/CY conditioning regimens and umbilical cord blood grafts[6]. To our knowledge, no one has evaluated MPA pharmacodynamics in HLA-haploidentical HCT recipients. We sought to characterize IMPDH activity, based on recent data from renal transplant patients reporting that high recipient IMPDH activity is associated with a higher risk of graft rejection[41]. Additional data from renal transplant patients shows considerable variability in IMPDH inhibition at a fixed MPA concentration. Of note, IMPDH had the greatest interpatient variability in this pilot study and would be an obvious candidate biomarker for future studies.
In summary, this pilot study demonstrates the feasibility of evaluating multiple biomarkers in HCT recipients treated in the ambulatory clinic. Adequately powered (Table 4) multi-center studies should be conducted to evaluate the clinical relevance of these biomarkers in HLA-haploidentical HCT recipients.
Supplementary Material
Acknowledgments
The authors would like to thank the participants and their caregivers for their time and cooperation, as well as the patient care staff for their support of this project. We would also like to acknowledge Ms. Megan Kelton-Rehkopf for her contributions to study coordination and Ms. Linda Risler and Mr. Brian Phillips for their analytical expertise. This study was supported by grants from the National Institutes of Health (HL91744S1, CA78902).
Footnotes
Conflict of interest statement: Disclosures; none.
References
- 1.Bayraktar UD, Champlin RE, Ciurea SO. Progress in Haploidentical Stem Cell Transplantation. Biol Blood Marrow Transplant. 2012;18:372–380. doi: 10.1016/j.bbmt.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luznik L, O’Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, Gooley TA, Piantadosi S, Kaup M, Ambinder RF, Huff CA, Matsui W, Bolaños-Meade J, Borrello I, Powell JD, Harrington E, Warnock S, Flowers M, Brodsky RA, Sandmaier BM, Storb RF, Jones RJ, Fuchs EJ. HLA-Haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14:641–650. doi: 10.1016/j.bbmt.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunstein CG, Fuchs EJ, Carter SL, Karanes C, Costa LJ, Wu J, Devine SM, Wingard JR, Aljitawi OS, Cutler CS, Jagasia MH, Ballen KK, Eapen M, O’Donnell PV on behalf of the Blood and Marrow Transplant Clinical Trials Network. Alternative donor transplantation after reduced intensity conditioning: results of parallel phase 2 trials using partially HLA-mismatched related bone marrow or unrelated double umbilical cord blood grafts. Blood. 2011;118:282–288. doi: 10.1182/blood-2011-03-344853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCune JS, Batchelder AL, Deeg HJ, Gooley TA, Cole S, Phillips BR, Schoch HG, McDonald GB. Cyclophosphamide following targeted oral busulfan as conditioning for hematopoietic cell transplantation: pharmacokinetics, liver toxicity, and mortality. Biol Blood Marrow Transplant. 2007;13:853–862. doi: 10.1016/j.bbmt.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Giaccone L, McCune JS, Maris MB, Gooley TA, Sandmaier BM, Slattery JT, Cole S, Nash RA, Storb RF, Georges GE. Pharmacodynamics of mycophenolate mofetil after nonmyeloablative conditioning and unrelated donor hematopoietic cell transplantation. Blood. 2005;106:4381–4388. doi: 10.1182/blood-2005-06-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson P, Rogosheske J, Barker JN, Green K, Ng J, Weisdorf D, Tan Y, Long J, Remmel R, Sawchuk R, McGlave P. Relationship of mycophenolic acid exposure to clinical outcome after hematopoietic cell transplantation. Clin Pharmacol Ther. 2005;78:486–500. doi: 10.1016/j.clpt.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Baron F, Petersdorf EW, Gooley TA, Sandmaier BM, Malkki M, Chauncey TR, Maloney DG, Storb RF. What Is the Role for Donor Natural Killer Cells after Nonmyeloablative Conditioning? Biol Blood Marrow Transplant. 2009;15:580–588. doi: 10.1016/j.bbmt.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SJ, Vogelsang G, Flowers MED. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215–233. doi: 10.1053/bbmt.2003.50026. [DOI] [PubMed] [Google Scholar]
- 9.Salinger DH, Blough DK, Vicini P, Anasetti C, O’Donnell PV, Sandmaier BM, McCune JS. A limited sampling schedule to estimate individual pharmacokinetic parameters of fludarabine in hematopoietic cell transplant patients. Clin Cancer Res. 2009;15:5280–5287. doi: 10.1158/1078-0432.CCR-09-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodahl EL, Wang J, Heimfeld S, Sandmaier BM, O’Donnell PV, Phillips B, Risler L, Blough DK, McCune JS. A novel phenotypic method to determine fludarabine triphosphate accumulation in T-lymphocytes from hematopoietic cell transplantation patients. Cancer Chemother Pharmacol. 2008;63:391–401. doi: 10.1007/s00280-008-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald GB, McCune JS, Batchelder AL, Cole S, Phillips BR, Ren AG, Vicini P, Witherspoon R, Kalhorn TF, Slattery JT. Metabolism-based cyclophosphamide dosing for hematopoietic cell transplant. Clin Pharmacol Ther. 2005;78:298–308. doi: 10.1016/j.clpt.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Kalhorn TF, Howald WN, Cole S, Phillips BR, Wang J, Slattery JT, McCune JS. Rapid quantitation of cyclophosphamide metabolites in plasma by liquid chromatography mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;835:105–113. doi: 10.1016/j.jchromb.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Yeh ST. Using trapezoidal rule for the area under a curve calculation. [Accessed 28 Jun 2012];Proceedings of the 27th Annual SAS User Group International. 2002 http://www2.sas.com/proceedings/sugi27/p229-27.pdf?iframe=true&width=100%&height=100%.
- 14.Soto Matos-Pita A, de Miguel Lillo B. [Accessed 28 Jun 2012];Noncompartmental pharmacokinetics and bioequivalence analysis. 2005 http://www.lexjansen.com/pharmasug/2005/statisticspharmacokinetics/sp07.pdf.
- 15.Li H, Mager DE, Sandmaier BM, Maloney DG, Bemer MJ, McCune JS. Population pharmacokinetics and dose optimization of mycophenolic acid in HCT recipients receiving oral mycophenolate mofetil. J Clin Pharmacol. 2013;53:393–402. doi: 10.1002/jcph.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Mager DE, Bemer MJ, Salinger DH, Vicini P, Sandmaier BM, Nash R, McCune JS. A limited sampling schedule to estimate mycophenolic acid area under the concentration-time curve in hematopoietic cell transplantation recipients. J Clin Pharmacol. 2012;52:1654–64. doi: 10.1177/0091270011429567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glander P, Sombogaard F, Budde K, Gelder T, Hambach P, Liefeldt L, Lorkowski C, Mai M, Neumayer HH, Vulto AG, et al. Improved assay for the nonradioactive determination of inosine 5′-monophosphate dehydrogenase activity in peripheral blood mononuclear cells. Ther Drug Monit. 2009;31:351–359. doi: 10.1097/FTD.0b013e31819c3f3d. [DOI] [PubMed] [Google Scholar]
- 18.Daxecker H, Raab M, Muller MM. Influence of mycophenolic acid on inosine 5′-monophosphate dehydrogenase activity in human peripheral blood mononuclear cells. Clin Chim Acta. 2002;318:71–77. doi: 10.1016/s0009-8981(01)00801-4. [DOI] [PubMed] [Google Scholar]
- 19.Duffy MJ, Crown J. A personalized approach to cancer treatment: How biomarkers can help. Clin Chem. 2008;54:1770–1779. doi: 10.1373/clinchem.2008.110056. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson L, Devita VT. Focus issue on biomarkers. Nat Rev Clin Oncol. 2010;7:295–295. doi: 10.1038/nrclinonc.2010.70. [DOI] [PubMed] [Google Scholar]
- 21.McLeod HL, Isaacs KL. Preemptive pharmacogenetic testing: insufficient data equal unsatisfactory guidance. Ann Intern Med. 2011;154:842–844. doi: 10.7326/0003-4819-154-12-201106210-00016. [DOI] [PubMed] [Google Scholar]
- 22.Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, Califano A, D’Argenio DZ, Iyengar R, Jusko WJ, Lalonde R. Quantitative and systems pharmacology in the post-genomic era: New approaches to discovering drugs and understanding therapeutic mechanisms. [Accessed 30 Aug 2012]; http://www.nigms.nih.gov/NR/rdonlyres/8ECB1F7C-BE3B-431F-89E6-A43411811AB1/0/SystemsPharmaWPSorger2011.pdf.
- 23.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41:93–103. doi: 10.2165/00003088-200241020-00002. [DOI] [PubMed] [Google Scholar]
- 24.Long-Boyle JR, Green KG, Brunstein CG, Cao Q, Rogosheske J, Weisdorf DJ, Miller JS, Wagner JE, McGlave PB, Jacobson PA. High fludarabine exposure and relationship with treatment-related mortality after nonmyeloablative hematopoietic cell transplantation. Bone Marrow Transplant. 2010;46:20–26. doi: 10.1038/bmt.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keating MJ, O’brien S, Lerner S, Koller C, Beran M, Robertson LE, Freireich EJ, Estey E, Kantarjian H. Long-term follow-up of patients with chronic lymphocytic leukemia (CLL) receiving fludarabine regimens as initial therapy. Blood. 1998;92:1165–1171. [PubMed] [Google Scholar]
- 26.Consoli U, El-Tounsi I, Sandoval A, Snell V, Kleine HD, Brown W, Robinson JR, DiRaimondo F, Plunkett W, Andreeff M. Differential induction of apoptosis by fludarabine monophosphate in leukemic B and normal T cells in chronic lymphocytic leukemia. Blood. 1998;91:1742–1748. [PubMed] [Google Scholar]
- 27.Gamberale R, Galmarini CM, Fernández-Calotti P, Jordheim L, Sánchez-Avalos J, Dumontet C, Geffner J, Giordano M. In vitro susceptibility of CD4+ and CD8+ T cell subsets to fludarabine. Biochem Pharmacol. 2003;66:2185–2191. doi: 10.1016/j.bcp.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Wilkins RC, Kutzner BC, Truong M, McLean JRN. The effect of the ratio of CD4+ to CD8+ T-cells on radiation-induced apoptosis in human lymphocyte subpopulations. Int J Radiat Biol. 2002;78:681–688. doi: 10.1080/09553000210144475. [DOI] [PubMed] [Google Scholar]
- 29.Mariotti J, Taylor J, Massey PR, Ryan K, Foley J, Buxhoeveden N, Felizardo TC, Amarnath S, Mossoba ME, Fowler DH. The Pentostatin Plus Cyclophosphamide Nonmyeloablative Regimen Induces Durable Host T Cell Functional Deficits and Prevents Murine Marrow Allograft Rejection. Biol Blood Marrow Transplant. 2011;17:620–631. doi: 10.1016/j.bbmt.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castermans E, Hannon M, Dutrieux J, Humblet-Baron S, Seidel L, Cheynier R, Willems E, Gothot A, Vanbellinghen J-F, Geenen V, Sandmaier BM, Storb R, Beguin Y, Baron F. Thymic recovery after allogeneic hematopoietic cell transplantation with non-myeloablative conditioning is limited to patients younger than 60 years of age. Haematologica. 2010;96:298–306. doi: 10.3324/haematol.2010.029702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhalla K, Bullock G, Lutzky J, Holladay C, Ibrado AM, Jasiok M, Singh S. Effect of combined treatment with interleukin-3 and interleukin-6 on 4-hydroperoxycyclophosphamide-mediated reduction of glutathione levels and cytotoxicity in normal and leukemic bone marrow progenitor cells. Leukemia. 1992;6:814–819. [PubMed] [Google Scholar]
- 32.Watters JW, Kloss EF, Link DC, Graubert TA, McLeod HL. A mouse-based strategy for cyclophosphamide pharmacogenomic discovery. J Appl Physiol. 2003;95:1352–1360. doi: 10.1152/japplphysiol.00214.2003. [DOI] [PubMed] [Google Scholar]
- 33.Jones RJ, Zuehlsdorf M, Rowley SD, Hilton J, Santos GW, Sensenbrenner LL, Colvin OM. Variability in 4-hydroperoxycyclophosphamide activity during clinical purging for autologous bone marrow transplantation. Blood. 1987;70:1490–1494. [PubMed] [Google Scholar]
- 34.Zhong RK, Donnenberg AD, Rubin J, Ball ED. Differential effect of 4-hydroperoxycyclophosphamide and antimyeloid monoclonal antibodies on T and natural killer cells during bone marrow purging. Blood. 1994;83:2345–2351. [PubMed] [Google Scholar]
- 35.Ozer H, Cowens JW, Colvin M, Nussbaum-Blumenson A, Sheedy D. In vitro effects of 4-hydroperoxycyclophosphamide on human immunoregulatory T subset function. I. Selective effects on lymphocyte function in TB cell collaboration. J Exp Med. 1982;155:276–290. doi: 10.1084/jem.155.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Busse D, Busch FW, Bohnenstengel F, Eichelbaum M, Fischer P, Opalinska J, Schumacher K, Schweizer E, Kroemer HK. Dose escalation of cyclophosphamide in patients with breast cancer: consequences for pharmacokinetics and metabolism. J Clin Oncol. 1997;15:1885–1896. doi: 10.1200/JCO.1997.15.5.1885. [DOI] [PubMed] [Google Scholar]
- 37.Busse D, Busch FW, Schweizer E, Bohnenstengel F, Eichelbaum M, Fischer P, Schumacher K, Aulitzky WE, Kroemer HK. Fractionated administration of high-dose cyclophosphamide: influence on dose-dependent changes in pharmacokinetics and metabolism. Cancer Chemother Pharmacol. 1999;43:263–268. doi: 10.1007/s002800050893. [DOI] [PubMed] [Google Scholar]
- 38.Salinger DH, McCune JS, Ren AG, Shen DD, Slattery JT, Phillips BR, McDonald GB, Vicini P. Real-time dose adjustment of cyclophosphamide in a preparative regimen for hematopoietic cell transplant: a Bayesian pharmacokinetic approach. Clin Cancer Res. 2006;12:4888–4898. doi: 10.1158/1078-0432.CCR-05-2079. [DOI] [PubMed] [Google Scholar]
- 39.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/s0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 40.Laverdière I, Caron P, Couture F, Guillemette C, Levesque E. Liquid Chromatography_Coupled Tandem Mass Spectrometry Based Assay to Evaluate Inosine-5′-monophosphate Dehydrogenase Activity in Peripheral Blood Mononuclear Cells from Stem Cell Transplant Recipients. Anal Chem. 2012;84:216–23. doi: 10.1021/ac202404y. [DOI] [PubMed] [Google Scholar]
- 41.Glander P, Hambach P, Braun K-P, Fritsche L, Giessing M, Mai I, Einecke G, Waiser J, Neumayer H-H, Budde K. Pre-transplant inosine monophosphate dehydrogenase activity is associated with clinical outcome after renal transplantation. Am J Transplant. 2004;4:2045–2051. doi: 10.1111/j.1600-6143.2004.00617.x. [DOI] [PubMed] [Google Scholar]
- 42.Upton A, McCune JS, Kirby KA, Leisenring W, McDonald GB, Batchelder AL, Marr KA. Fluconazole coadministration concurrent with cyclophosphamide conditioning may reduce regimen-related toxicity postmyeloablative hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13:760–764. doi: 10.1016/j.bbmt.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Jonge ME, Huitema ADR, Rodenhuis S, Beijnen JH. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 2005;44:1135–1164. doi: 10.2165/00003088-200544110-00003. [DOI] [PubMed] [Google Scholar]
- 44.Gilbert CJ, Petros WP, Vredenburgh J, Hussein A, Ross M, Rubin P, Fehdrau R, Cavanaugh C, Berry D, McKinstry C, et al. Pharmacokinetic interaction between ondansetron and cyclophosphamide during high-dose chemotherapy for breast cancer. Cancer Chemother Pharmacol. 1998;42:497–503. doi: 10.1007/s002800050851. [DOI] [PubMed] [Google Scholar]
- 45.Raccor BS, Claessens AJ, Dinh JC, Park JR, Hawkins DS, Thomas SS, Makar KW, McCune JS, Totah RA. Potential contribution of cytochrome P450 2B6 to hepatic 4-hydroxycyclophosphamide formation in vitro and in vivo. Drug Metab Dispos. 2012;40:54–63. doi: 10.1124/dmd.111.039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindley C, Hamilton G, McCune JS, Faucette S, Shord SS, Hawke RL, Wang H, Gilbert D, Jolley S, Yan B, Lecluyse EL. The effect of cyclophosphamide with and without dexamethasone on cytochrome P450 3A4 and 2B6 in human hepatocytes. Drug Metab Dispos. 2002;30:814–822. doi: 10.1124/dmd.30.7.814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.