

Abstract
The synthesis, characterization, and in vitro evaluation of a combination delivery of multiblock poly(N-2-hydroxypropyl)methacrylamide (HPMA), gemcitabine (GEM) and paclitaxel (PTX) conjugates is described in this study. Multiblock copolymer conjugates of a large molecular weight (Mw > 200 kDa) were studied and compared to traditional, small molecular weight (Mw < 45 kDa) conjugates. Stability of the conjugates in different pH was assessed, and their cytotoxicity in combination toward A2780 human ovarian cancer cells was evaluated by combination index analysis. Treatment duration (4 and 72 h) and sequence of addition were explored. In addition, an HPMA copolymer conjugate with both GEM and PTX in the side chains was evaluated in a similar manner and compared to a physical mixture of individual conjugates. Conjugates with narrow molecular weight distribution (Mw/Mn < 1.1) were obtained via RAFT polymerization, and drug loadings of between 5.5 and 9.2 wt% were achieved. Conjugates demonstrated moderate stability with less than 65% release over 24 h at pH 7.4, and near complete drug release in the presence of the lysosomal enzyme cathepsin B in 3 h. In combination, the cytotoxic effects of a mixture of the conjugates were primarily additive. Synergistic effects were observed when A2780 human ovarian cancer cells were treated simultaneously for 4 h with multiblock conjugates (CI < 0.7). When both GEM and PTX were conjugated to the same copolymer backbone, moderate antagonism (CI 1.3–1.6) was observed. These results demonstrate that multiblock HPMA copolymer–GEM and –PTX conjugates, when delivered as a mixture of individual agents, are promising for the treatment of ovarian cancer.
Keywords: HPMA copolymers, Biodegradable polymers, Combination therapy, Ovarian cancer, Gemcitabine, Paclitaxel
1. Introduction
Ovarian cancer was responsible for approximately 16,000 deaths in the United States during 2012 and an estimated 22,000 new cases were diagnosed during the same period. It is the most deadly cancer of the female reproductive system. When the disease is found at an early stage and is localized, greater than 90% of patients live longer than 5 years. This, however, is only the case in about 15% of patients. The majority of cases are diagnosed during stage III, where the 5 year survival rate is only 34% (Mould, 2012). Standard treatments for such patients often involve a combination of surgery and chemotherapy. A major goal of surgery is to remove as much tumor tissue as possible in a process called debulking (Wakabayashi et al., 2008). Chemotherapy often yields positive results in terms of tumor size reduction and even apparent disappearance. However, the vast majority of patients relapse. For these patients, tumor resistance is often encountered, with subsequent chemotherapy treatments yielding fewer and fewer positive results (Kim et al., 2012; Herzog and Pothuri, 2006). There is therefore a critical need for new treatment options for patients with drug resistant late stage ovarian cancer.
The standard chemotherapy approach involves treatment with a platinate (i.e., carboplatin) and a taxane (i.e., paclitaxel). A number of other chemotherapeutic agents are also helpful in combatting ovarian cancer. These include (in no particular order): topote-can, liposomal doxorubicin, GEM, cyclophosphamide, vinorelbine, ifosfamide, etoposide, altretamine, capecitabine, irinotecan, melphalan, pemetrexed, and albumin bound PTX (Herzog, 2006). While the goal of chemotherapy treatment is to inhibit the growth and kill cancer cells, significant toxicity also occurs in normal cells. This unintended toxicity can result in an array of side effects including nausea, mouth sores, gastrointestinal complications, fatigue, neutropenia, risk of infection, and hair loss. Strategies that can possibly enhance the efficacy and reduce the toxicity of existing chemotherapeutics are therefore needed.
One approach that can potentially increase the clinical utility of anticancer drugs is conjugation of these agents to water-soluble polymers (Kopeček, 1977; Vicent et al., 2009; Larson and Ghandehari, 2012; Kopeček and Kopečková, 2012). This affords several advantages. First, a number of chemotherapeutic agents are poorly water soluble, resulting in challenges during formulation and administration. Via conjugation to water-soluble polymers, aqueous solubility can be substantially improved without the use of organic solvents or surfactants. Second, conjugation to a carrier allows for opportunities to modify the biodistribution and pharmacokinetics of the conjugated system. In particular, it has been previously demonstrated that nanoscale sized conjugates can preferentially accumulate in tumor tissues via the enhanced permeability and retention (EPR) effect, where a combination of increased vascular permeability and decreased lymphatic drainage in the tumor microenvironment can drive tumor delivery (Matsumura and Maeda, 1986; Maeda, 2012). This is especially advantageous in anticancer applications, where these improvements can translate into a higher therapeutic index for a chemotherapy drug, thereby giving a clinician the option for more aggressive treatment, in hopes of achieving a better clinical response.
Copolymers of N-(2-hydroxypropyl)methacrylamide (HPMA) have been widely investigated as carriers for chemotherapy drugs (Kopeček, 1990, 2013; Duncan and Vicent, 2010; Kopeček and Kopečková, 2010; Lammers and Ulbrich, 2010). They are advantageous due to their chemical simplicity and ability to incorporate drug molecules and other functionalities (i.e., imaging agents, targeting moieties, etc.) with relative ease (Liu et al., 2009; Hongrapipat et al., 2008; Lammers et al., 2008). However, a major limitation in the past has been their non-biodegradability (Duncan and Vicent, 2010). To take full advantage of increased systemic circulation time and increased tumor accumulation via the EPR effect, HPMA copolymers must be sufficiently large to evade renal filtration (i.e., greater than 45 kDa). However, an eventual route of elimination is also required due to safety concerns over cumulative polymer accumulation throughout the body. For example, PK-1 (Duncan et al., 1998; Thomson et al., 1999), an early HPMA copolymer-doxorubicin conjugate evaluated in clinical trials was synthesized with a size of 28 kDa to ensure eventual renal elimination. However, this conjugate demonstrated only marginal efficacy during phase II studies (Seymour et al., 2009). This lack of efficacy can partially be explained by the conjugate's rapid elimination and lack of ability to take full advantage of the EPR effect. Recently, we have described a new generation of backbone biodegradable HPMA copolymers synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization and subsequent polymer coupling (Pan et al., 2011a,b; Yang et al., 2011; Luo et al., 2011). These polymers are based on the same biocompatible HPMA chemistry, but contain enzymatically degradable sequences within the backbone to impart biodegradability. Such conjugates will be able to circulate systemically in the blood for long periods of time and take full advantage of the EPR effect while maintaining the ability to be eliminated via renal filtration following enzymatic degradation. Backbone degradable HPMA copolymer conjugates with doxorubicin (Pan et al., 2013) or PTX (Zhang et al., 2013) have demonstrated enhanced antitumor efficacy in human ovarian carcinoma xenografts when compared to first generation (low molecular weight) conjugates.
GEM (trade name Gemzar®) is a synthetic nucleoside analog of cytidine. Its triphosphate analog is incorporated into DNA, thereby halting cell division. GEM has demonstrated activity both in vitro and in vivo in ovarian cancer models (Touma et al., 2006; Gallo et al., 2006; Peters et al., 1996) and is currently approved by the US FDA in combination with carboplatin for patients with advanced ovarian cancer who have experienced relapse after completion of platinum-based therapy. It is also currently under clinical investigation in combination with a number of other anticancer agents (Garcia et al., 2012; Hendrickson et al., 2012). Following intravenous administration, GEM can be converted to an inactive uracil metabolite (2′-deoxy-2′-2′-difluorouridine (dFdU)) (Heinemann et al., 1992). It is anticipated that conjugation of GEM to HPMA copolymers can possibly act to prevent this metabolism, thereby allowing more of the active form to be delivered to the tumor environment, where it can subsequently be enzymatically cleaved and provide its intended effect.
PTX (trade name Taxol®), is a mitotic inhibitor which acts by stabilizing microtubules, thereby inhibiting their breakdown during cellular division (Dumontet and Sikic, 1999). It is currently indicated as first-line and subsequent therapy for the treatment of advanced stage ovarian cancer. When used as a first-line therapy, it is indicated in combination with cisplatin. Due to its poor water solubility, the current formulation of Taxol® utilizes Cremophor® EL, which has been shown to cause severe hypersensitivity reactions in some patients (Rowinsky et al., 1993; Weiss et al., 1990). It is, therefore, anticipated that conjugation of PTX to HPMA copolymers will allow administration without the need of such surfactants, thereby reducing these side effects.
As previously discussed, a primary challenge in ovarian cancer chemotherapy is the development of drug resistance (Naumann and Coleman, 2011; Bookman, 2010). A great amount of research has, therefore, focused on the development of combination strategies, wherein administration of anticancer agents with different mechanisms of action can be utilized as a multi-pronged approach to provide a more universal cytotoxic effect, thereby reducing the chance for cancer cells to adapt and develop resistance. In particular, a combination approach utilizing GEM and taxanes has shown clinical promise in this regard (Garcia et al., 2012; Friedlander et al., 2007). From a mechanistic standpoint, one study (Zupi et al., 2005) demonstrated, via cell cycle analysis, that GEM alone halts cells in the S-phase while PTX arrested cells in the G2-phase. This resulted in a more robust and complete disruption of the cell cycle, due to the action of two agents via independent mechanisms of action. The enhanced cytotoxic effect of the combination was attributed to this irreversible perturbation of the cell cycle, resulting in the induction of apoptosis in a synergistic manner. This particular drug combination, therefore, may be useful in the treatment of advanced ovarian cancer.
The current study describes the synthesis and characterization of biodegradable HPMA copolymer–GEM and –PTX conjugates and their evaluation in combination. Conjugates with GEM and PTX on separate, as well as on the same, backbone were prepared to determine if there was any observed advantage to having both agents present on the same backbone. Their hydrolytic stability and enzymatic drug release were evaluated in vitro. The ability of the conjugates to induce cytotoxicity in A2780 human ovarian cancer cells was evaluated when used as single agents and in combination. Combination index analysis was then performed to determine if their combined effects were antagonistic, additive, or synergistic.
2. Materials and methods
2.1. Materials
Common reagents were purchased from Sigma–Aldrich (St. Louis, MO) and used as received unless otherwise specified. N-α-Fmoc protected amino acids, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 2-Cl-trityl chloride resin (100–200 mesh, 1.27 mmol/g) were purchased from AAPPTec Biosciences (Louisville, KY). Paclitaxel (>99.5%) was purchased from LC Laboratories (Woburn, MA). Gemcitabine hydrochloride (≥99.0%) was purchased from NetQem LLC (Research Triangle Park, NC). Papain, cathepsin B (from bovine spleen) and cathepsin B substrate (Z-Arg-Arg-p-nitroanalide) were obtained from Sigma–Aldrich. HPMA (Kopeček and Bažilová, 1973), N-methacryloylglycylphenylalanylleucylglycyl GEM/PTX (MA-GFLG-GEM and MA-GFLG-PTX) (Yang et al., 2011; Zhang et al., 2013), 4-cyanopentanoic acid dithiobenzoate (Mitsukami et al., 2001), and peptide2CTA (Pan et al., 2011b) were synthesized as previously described. 4,4′-Azobis(N,N′-propargyl-4-cyanopentanamide) (dialkyne-V-501) and Nα,Nδ-(bis(azidobenzoylglycylphenylalanylleucylglycylalanyl)lysine (diazide-GFLGK) were prepared according to described procedures (Pan et al., 2011a).
2.2. Synthesis and characterization of HPMA copolymer drug conjugates
2.2.1. Synthesis of multiblock HPMA copolymer drug conjugates
Long circulating multiblock HPMA copolymer–GEM conjugate (mP-GEM) was prepared by RAFT copolymerization of HPMA with a polymerizable derivative of GEM followed by Cu (I) catalyzed alkyne-azide click reaction as previously reported (Yang et al., 2011) with modifications. A typical synthesis is briefly described below.
2.2.1.1. Synthesis of clickable telechelic HPMA copolymer-gemcitabine conjugate
An ampoule containing MA-GFLG-GEM (127 mg, 0.18 mmol) was attached to the Schlenk-line. After three vacuum-nitrogen cycles to remove oxygen, 1 mL degassed DMSO acidified with 5 μL acetic acid was added and a clear colorless solution was obtained. A degassed HPMA aqueous solution (260 mg, 1.82 mmol in 0.8 mL DI H2O) was added into the ampoule via syringe under vigorous stirring. Following addition of RAFT agent peptide2CTA and initiator V-501, the ampoule was sealed, and copolymerization was performed at 70°C for 16 h. The polymer was obtained by precipitation into acetone and purified by re-dissolving in methanol and precipitation in acetone two more times. The copolymer was isolated as a light pink powder and dried under vacuum (yield: 230 mg, 60%). The average molecular weight and the polydispersity of the conjugates were determined by size exclusion chromatography (SEC) on an AKTA FPLC system equipped with a UV detector (GE Healthcare), miniDAWN TREOS and OptilabrEX (refractive index, RI) detector (Wyatt Technology, Santa Barbara, CA) using a Superose 6 HR10/30 column with sodium acetate buffer containing 30% acetonitrile (pH 6.5) as mobile phase. HPMA homopolymer fractions were used as molecular weight standards.
The copolymer was post-polymerization modified to replace dithiobenzoate groups at chain termini by alkyne groups (Yang et al., 2011). Thus, the product (220 mg) was reacted with dialkyne-V-501 (28 mg, 0.08 mmol, over 40 times excess with respect to dithiobenzoate end groups) in 1 mL DMSO at 70°C for 3 h, puri-fied by precipitation into acetone twice, resulting in α,ω-dialkyne telechelic HPMA copolymer–GEM conjugate (tP-GEM).
2.2.1.2. Chain extension via Cu (I) assisted alkyne-azide click reaction and fractionation
Diazide-GFLGK (2.1 mg, 1.8 μmol) and tP-GEM (200 mg, 1.8 μmol) were weighed into an ampoule. The ampoule was evacuated and refilled with nitrogen three times before adding 1 mL of deoxygenated DMSO. Sodium ascorbate solution (3.6 mg, 10×) was put in a vial. Deoxygenated solution of CuSO4 (1.5 mg, 5×) was added and the click reaction was initiated. The solution was stirred at room temperature for 20 h. The polymer was precipitated into acetone and dried under vacuum, and further fractionated/purified by size exclusion chromatography using an XK50 column. After dialysis against water and freeze-drying, the conjugates (mP-GEM) with varied Mw were obtained. Fraction G2 (Mw 213 kDa, Mw/Mn 1.06) was used in this study. GEM content in the conjugate was estimated by UV in methanol (ε300 = 5710 Lmol−1 cm−1). Synthesis of biodegradable multiblock HPMA copolymer-paclitaxel conjugate (mP-PTX) was carried out as previously reported (Zhang et al., 2013).
2.2.2. Synthesis of traditional (first generation) HPMA copolymer drug conjugates
To enable comparison of the activity of biodegradable multiblock HPMA copolymer–drug conjugates (mP-GEM and mP-PTX) to that of traditional HPMA copolymer–drug conjugates (P-GEM and P-PTX with Mw < 50 kDa), the copolymerization of HPMA with MA-GFLG-GEM or MA-GFLG-PTX was conducted as described above but using 4-cyanopentanoic acid dithiobenzoate (CPA) as the chain transfer agent. In a typical copolymerization for preparing P-GEM, monomers (HPMA: 395 mg, 2.76 mmol and MA-GFLG-GEM: 169 mg, 0.24 mmol) were dissolved in DMSO/H2O under N2 atmosphere. CPA and V-501 at a molar ratio of 4:1 were added through syringe. The ampoule was sealed and the polymerization was carried out at 70°C for 16 h. The copolymer was precipitated in acetone, washed with acetone three times and dried under reduced pressure at room temperature. The dithiobenzoate end group was removed by radical-induced modification with excess 2,2′-azobis(2,4-dimethyl valeronitrile) (V-65). Yield was 330 mg white powder (60%) with Mw 32 kDa and Mw/Mn 1.05. P-PTX was synthesized as previously reported (Zhang et al., 2013).
2.2.3. Synthesis of HPMA copolymer conjugate containing gemcitabine and paclitaxel
HPMA copolymer conjugate containing both GEM and PTX (P-GEM-PTX) was synthesized by RAFT copolymerization of HPMA with MA-GFLG-GEM and MA-GFLG-PTX using CPA as the RAFT chain transfer agent and V-65 as the initiator. The polymerization was conducted in the mixture of DMSO and DI H2O at 50 °C for 24 h. The feed molar ratio was [HPMA]: [GEM]: [PTX] = 90:7:3. The copolymer was precipitated in acetone, washed with acetone three times and dried under reduced pressure at room temperature. The dithiobenzoate end group was removed by radical-induced modification with excess V-65.
2.3. Hydrolytic stability and enzymatic release of gemcitabine and paclitaxel from HPMA copolymers
Hydrolytic stability in terms of drug release from HPMA copolymer conjugates was evaluated in phosphate buffered saline (PBS) at pH 7.4. GEM and PTX equivalent concentrations were maintained at low concentrations to prevent saturation. In particular, 3 mg/mL solutions of P-GEM, P-PTX and mP-GEM were prepared and divided into 100 μL aliquots. Samples were incubated in a shaking water bath at 37 °C. At predetermined time points, one aliquot sample was taken. For GEM conjugates, the samples were directly filtered, and 10 μL of filtrate loaded onto a HPLC system for analysis. For PTX conjugates, 200 μL of methanol/H2O (8:1, v/v) was added before filtration. Then, 20 μL of solution was loaded via autosampler for analysis.
2.3.1. HPLC analysis
An HPLC apparatus (Agilent 1100 LC System, Agilent Technologies, Santa Clara, CA) equipped with C18 column (Zorbax 300SB, 5 μm, 4.6 mm × 250 mm) and a diode array detector was used for determination of free GEM and PTX. For GEM, a gradient method of 2–90% buffer B in 30 min with flow rate 1.0 mL/min was used (buffer A: H2O + 0.1% TFA; buffer B: acetonitrile + 0.1% TFA). The signal at 268 nm was monitored and the peak with elution time at 6.1 min was attributed to free GEM. For PTX, a gradient method of 30–90% buffer B in 30 min with flow rate 1.0 mL/min was used, and the signal at 250 nm was monitored. A series of standard solutions of GEM/PTX were prepared, and calibration curves were made for quantification of GEM and PTX respectively.
For drug release studies, HPMA copolymer conjugates were incubated with a 2.0 × 10−7 M cathepsin B buffer solution at pH 6.0 containing 0.1 M citrate phosphate, 2 mM EDTA, and 10 mM glu-tathione. The enzymatic activity of cathepsin B was verified using a standard cathepsin B substrate (Z-Arg-Arg-p-nitroanilide) (data not shown). Samples were incubated at 37 °C with periodic agitation and 100 μL aliquots sampled at each time point. Methanol (400 μL) was added to each sample to prevent further enzymatic release, followed by analysis by HPLC. Conjugates dissolved in mobile phase alone were analyzed by HPLC and used to determine concentrations of free drugs present at time zero.
2.4. Cell culture
The A2780 human ovarian cancer cell line was obtained from ATCC (Manassas, VA) and cultured at 37 °C in a humidified atmosphere of 5% CO2 in RPMI-1640 cell culture medium (ATCC) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific HyClone, Logan, UT). Cells were maintained in a logarithmic growth phase during all studies.
2.5. Single agent in vitro cytotoxicity against A2780 human ovarian cancer cells
The ability of the conjugates to inhibit the growth of A2780 human ovarian cancer cells was evaluated in vitro using a 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt (WST-8) cell viability assay (Dojindo Molecular Technologies, Inc., Rockville, MD). To ensure water solubility of the free drugs GEM and PTX, cell culture medium containing 0.5% (v/v) DMSO was used to prevent precipitation. A2780 cells (4000 cells per well) were plated in 96-well plates for 24 h followed by drug treatment. Both short (4 h) and long (72 h) treatment times were evaluated. For short treatment cases, cells were washed with PBS following treatment, followed by further incubation (68 h) in growth media prior to assessment of viability (pulse-chase style experiment). Medium was then removed and cell viability quantified by WST-8 assay (modified MTT assay) using a SpectraMax M2 microplate UV spectrophotometer (Molecular Devices, Sunnyvale, CA). Relative viability was calculated by normalization of UV absorbance against untreated cells in each plate. Relative viability as a function of log drug concentration was plotted and non-linear least-squares regression analysis and calculation of the concentrations necessary to result in 50% cell viability as compared to controls (IC50 values) was performed using GraphPad Prism. A minimum of 3 samples was evaluated in each experiment, and a minimum of 3 independent experiments was performed for each treatment case.
2.6. In vitro combination treatment and combination index analysis
The combination treatments of GEM + PTX (free drug combination), P-GEM + P-PTX (traditional conjugates combination), and mP-GEM + mP-PTX (multiblock, biodegradable conjugate combination) were evaluated for cytotoxicity against A2780 cells in vitro. The ratio of GEM to PTX was maintained at 1:1 for all experiments. The effect of treatment sequence and treatment duration was also investigated. Treatment duration was tested via simultaneous incubation with GEM and PTX treatments for either 4 or 72 h. When treatment occurred for only 4 h, experiments were performed in a “pulse-chase” style as previously described. Treatment sequence was investigated by incubation for 4 h with drug A (GEM or PTX), followed by 68 h incubation with drug B (PTX or GEM). Following treatment, cells were assessed for viability by WST-8 assay as previously described. Drug effect was defined as (1 – [% relative viability/100]). Combination index analysis was performed and combination index values at 50% relative viability (CI at EC50) were calculated using CalcuSyn combination index software (Biosoft, Inc.) based on the Chou–Talalay method (Chou and Talalay, 1984). A minimum of 3 samples was evaluated in each experiment, and a minimum of 3 independent experiments was performed for each combination treatment case.
2.7. Evaluation of gemcitabine/paclitaxel dual agent copolymer
The combination polymer bearing both GEM and PTX on the same backbone was analyzed in a similar manner for its cytotoxicity against A2780 cells. In this evaluation, only treatment duration (4 and 72 h) was varied. Copolymers containing GEM and PTX separately (P-GEM and P-PTX) served as controls, and were mixed at a ratio that corresponded to that obtained for the combination polymer (P-GEM-PTX). Following either 4 or 72 h incubation, cell viability, drug effects, and combination index values were calculated as previously described. A minimum of 3 samples was evaluated in each experiment, and a minimum of 3 independent experiments was performed for each combination treatment case.
2.8. Statistical analysis
Differences in in vitro growth inhibition (IC50) and combination index (CI) values were determined by one-way ANOVA. Where differences were detected, Tukey's post-test was used to test for significance between groups. The default significance level was set at α = 0.05 for all statistical tests.
3. Results and discussion
3.1. Synthesis and characterization of HPMA copolymer conjugates
Both PTX and GEM have been conjugated to hydrophilic polymers for enhancing their bioavailability. For example, poly(l-glutamic acid)-PTX conjugates (Chipman et al., 2006) and HPMA copolymer-PTX conjugates (Meerum Terwogt et al., 2001) have been evaluated in clinical trials. In those conjugates PTX was covalently bound to polymer precursors through an ester bond at its 2′-OH position via variable linkers. GEM was also bound to poly(l-glutamic acid) via either 5′ ester bond in the presence of dicyclohexylcarbodiimide (DCC) and dimethylaminopyridine (DMAP) (Kiew et al., 2010), or via amide bond by the reaction of GEM with active ester on HPMA copolymer side chains (Lammers et al., 2009). The significant synthetic feature in this study is the utilization of a polymerizable drug derivative and controlled polymerization chemistry (Fig. 1). Copolymerization of HPMA with MA-GFLG-gemcitabine and/or paclitaxel was advantageous (reproducibility, yield) when compared to polymer analogous attachment. It provides a facile way to adjust drug content in a given conjugate by changing the feed ratio. The resulting HPMA copolymer conjugates possess molecular weights close to the theoretical value and very narrow molecular weight distribution as shown in Table 1. Moreover, a new architecture of HPMA copolymer–drug conjugates, biodegradable, long-circulating multiblock conjugates has been designed and prepared. Recent studies showed that this new generation of conjugates containing PTX (mP-PTX) or doxorubicin (mP-DOX) significantly enhanced therapeutic effect against human ovarian xenografts in nude mice when compared to low molecular weight conjugates.
Fig. 1.
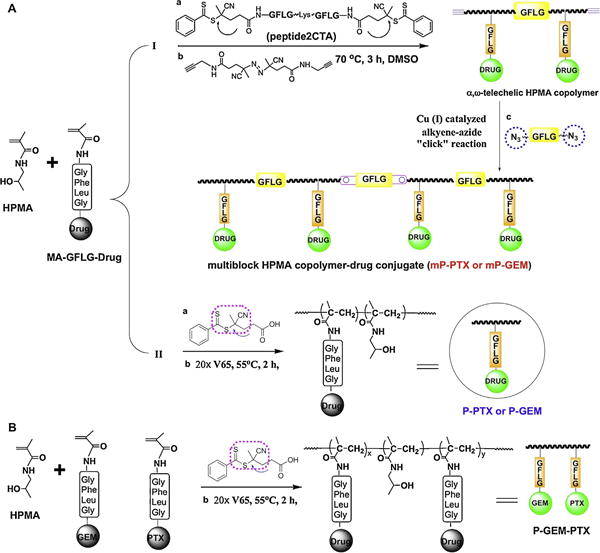
Schematic synthesis of HPMA copolymer-gemcitabine/paclitaxel conjugates. (A) (I): Synthesis of backbone degradable multiblock conjugates (mP-GEM and/or mP-PTX) via three steps: (1) RAFT copolymerization using peptide2CTA as RAFT chain transfer agent; (2) end-modification to obtain clickable telechelic diblock drug conjugates, and (3) chain extension by alkyne-azide click reaction (Zhang et al., 2013). (II): Synthesis of 1st generation conjugate (P-GEM and/or P-PTX). (B) Synthesis of HPMA copolymer conjugate containing dual-drugs per chain through RAFT copolymerization.
Table 1.
Characterization of HPMA copolymer–gemcitabine and HPMA copolymer–paclitaxel conjugates.
| Conjugates | Mw, kDa | Mw/Mn | Drug content (wt%) | |
|---|---|---|---|---|
| Gemcitabine conjugates | P-GEM | 32 | 1.07 | 7.7 |
| mP-GEM | 213 | 1.06 | 5.5 | |
| Paclitaxel conjugates | P-PTX | 40 | 1.06 | 9.2 |
| mP-PTX | 228 | 1.06 | 7.1 | |
| HPMA copolymer containing GEM and PTX on chain (P-GEM-PTX) | 55 | 1.13 | GEM 3.6 | |
| PTX 2.4 | ||||
3.2. Hydrolytic stability and enzymatic release of gemcitabine and paclitaxel from HPMA copolymers
Stability during systemic circulation and drug release following accumulation in tumor tissues are critical parameters for polymeric drug conjugates. If drug release occurs prematurely, the chemotherapeutic has the opportunity to elicit cytotoxicity in non-specific organs resulting in adverse effects. If drug release occurs too slowly following accumulation in tumor tissues, the concentration of the chemotherapeutic may never reach the levels necessary to effectively elicit its pharmacological effect, and efficacy may be compromised. Therefore, both stability and drug release were assessed.
Drug susceptibility to hydrolysis was assessed in PBS at pH 7.4. GEM and PTX conjugates demonstrated moderate stability. For PTX conjugates, 46% release occurs over 24 h at 37 ° C from the traditional (P-PTX) conjugates as compared to 65% release over the same period for the multiblock (mP-PTX) conjugates (Fig. 2b). This minor difference is most likely due to slightly higher drug loading for the traditional conjugate (9.1 wt%) compared to the multiblock conjugate (7.1 wt%). This can possibly be explained by intermolecular interactions between highly hydrophobic PTX molecules (c Log P = 3.06), resulting in a more compact copolymer conformation, which has less opportunity for drug hydrolysis. For GEM conjugates, approximately 50% release was observed over 24 h, and no differences were observed in release from traditional (P-GEM) or multiblock (mP-GEM) conjugates (Fig. 2a), even though traditional (P-GEM) conjugates also have slightly higher drug loading (7.7 wt%) as compared to multiblock (mP-GEM) conjugates (5.5 wt%). GEM is relatively hydrophilic (c Log P = −1.39), therefore, polymer conformation is less likely to be as dependent on drug load for GEM conjugates as compared to PTX conjugates.
Fig. 2.
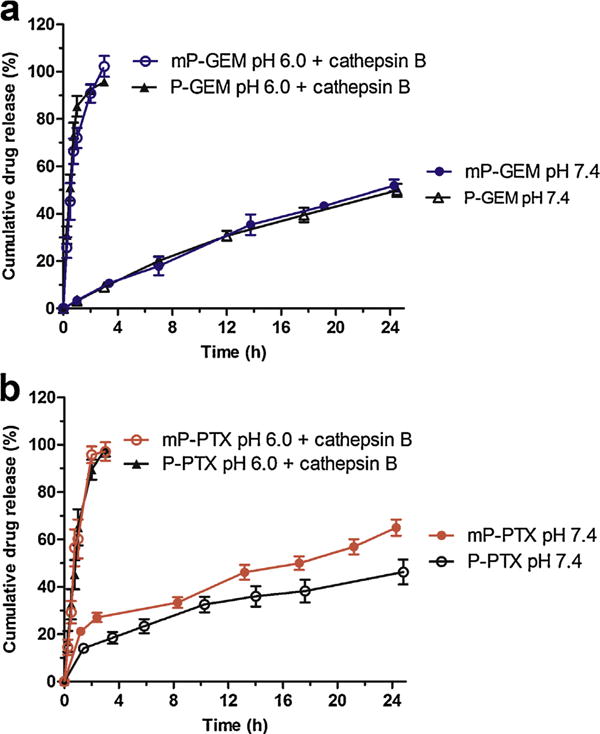
Stability of and drug release from HPMA copolymer-gemcitabine/paclitaxel conjugates. (a) Release of GEM from traditional (P-GEM) and multiblock (mP-GEM) conjugates at pH 7.4 and pH 6.0 in the presence of the lysosomal enzyme cathepsin B. (b) Release of PTX from traditional (P-PTX) and multiblock (mP-PTX) conjugates at pH 7.4 and pH 6.0 in the presence of the lysosomal enzyme cathepsin B. In the presence of cathepsin B, near complete drug release was achieved in 3 h. Conjugates also demonstrated moderate stability, with approximately 40–60% drug release over 24 h at pH 7.4. All experiments were performed at 37 °C. Data expressed as mean ± SD of three independent experiments.
Enzymatic release of GEM and PTX was evaluated in the presence of the enzyme cathepsin B at pH 6.0, in an attempt to mimic lysosomal conditions following cellular uptake. Under these conditions, rapid drug release occurred. For GEM conjugates, approximately 50% drug release occurred in 0.5 h (Fig. 2a). Drug release was slightly slower for PTX conjugates, with 50% drug release achieved in approximately 0.75 h (Fig. 2b). Again, this is possible due to interactions between hydrophobic PTX molecules, resulting in less accessibility of GFLG spacer and reduced enzymatic cleavage. However, these results, combined with the results from the stability studies, demonstrate that these conjugates are moderately stable at systemic pH (7.4), and demonstrate favorable drug release in simulated lysosomal conditions.
Overall, these data demonstrate that drug release is mostly independent of architecture (multiblock or traditional). Intuitively, it might be expected that a higher molecular weight conjugate would exhibit slower drug release, due to reduced enzymatic access to cleavage sites via a decreased surface area to volume ratio. The situation is slightly more complicated, however, as the larger multiblock conjugates contain additional backbone degradable sequences, which may facilitate faster degradation and act to compensate for their large size. However, further detailed studies are required to investigate these phenomena.
3.3. Single agent in vitro cytotoxicity against A2780 human ovarian cancer cells
The ability of the conjugates to inhibit the growth of A2780 human ovarian cancer cells was evaluated in vitro using a WST-8 cell viability assay. Multiblock, biodegradable HPMA copolymer conjugates, traditional HPMA copolymers conjugates, and free drugs were evaluated for each agent (GEM and PTX). The effect of treatment duration was also evaluated by incubation for either 4 or 72 h (Fig. 3). For polymer–drug conjugates, assessment of cell viability after a short period of incubation, followed by a longer growth phase (i.e., pulse-chase style cytotoxicity) is important in that it allows parameters such as the kinetics of cellular uptake and drug release to make a larger impact of cell viability. These assays are therefore generally better at discriminating these differences.
Fig. 3.

Single agent cytotoxicity. The ability of the conjugates to inhibit the growth of A2780 human ovarian cancer cells was evaluated in vitro using a WST-8 cell viability assay. The effect of treatment duration was assessed by varying incubation time. (A) Cells treated continuously for 72 h. All treatment groups (free drugs, traditional conjugates, and multiblock conjugates) exhibited potent activity in the low nanomolar range. (B) Cells treated for 4 h, followed by incubation in growth media for 68 h. Traditional and multiblock conjugates demonstrate less toxicity, most likely due to the kinetics of cellular uptake and drug release. Data expressed as mean ± SD of three independent experiments.
When treatments were applied continuously for 72 h, mP-GEM and mP-PTX conjugates were very potent with growth inhibition IC50 values of 10 and 7 nM respectively (Fig. 3A). These values were similar to those for the traditional conjugates (P-GEM and P-PTX), which were 9 and 3 nM respectively. Free GEM and PTX exhibited toxicity in a similar range with IC50 values of 4 and 5 nM respectively. In these 72 h treatment studies, the cytotoxicity values for each agent (conjugates or free drug controls) were similar, as the kinetics of drug release occurs quite quickly in comparison to the long incubation time. However, these results do demonstrate that the potency of GEM and PTX is maintained following conjugation to either multiblock or traditional HPMA copolymers.
When treatments were applied for only 4 h, some additional observations were made. First, as anticipated, significantly higher concentrations were required to induce cytotoxicity due to the short incubation time. Free GEM and PTX exhibited IC50 values of 33 and 47 nM respectively (Fig. 3B), while all of the conjugates exhibited IC50 value above 100 nM. During incubation, the conjugates must first enter the cell via endocytosis, and a fixed amount of time is also required for drug release to occur in lysosomal compartments. The free drugs, on the other hand, can freely diffuse through the cell membrane and immediately act on their intended target. For both GEM and PTX multiblock conjugates, activity was somewhat reduced as compared to the traditional conjugates. It is possible that differences in size and molecular weight may play a role in the kinetics of cellular uptake. However, further detailed investigation is necessary to fully elucidate which parameters are most significant.
3.4. In vitro combination treatment and combination index analysis
The administration of multiple chemotherapeutics in combination strategies has proven very successful in an array of various cancers, including ovarian cancer. First-line treatment for ovarian cancer currently consists of platinum and taxane combination therapy. When multiple drugs are used in combination, their overall effects can be antagonistic, additive, or synergistic. Synergistic effects most often occur when chemotherapeutic agents act via different mechanisms of action, or when the effects of one agent sensitizes cells to the effects of another. A common approach used to determine if a combination treatment is antagonistic, additive, or synergistic is that described by Chou and Talalay (1984). Following evaluation of drug effect alone and in combination, a combination index (CI) parameter is calculated. When CI is greater than 1, the combination treatment is antagonistic. When CI equals 1, the combination treatment is additive, and when CI is less than 1, the treatment is synergistic. Throughout this study, combination index values were calculated and compared at 50% effect (EC50), or where the combination resulted in 50% cell viability.
In this study, three different combination treatments were evaluated. They were: GEM + PTX (free drug combination), P-GEM + P-PTX (traditional conjugates combination), and mP-GEM + mP-PTX (multiblock conjugates combination). In each combination treatment, the molar ratio of GEM to PTX was maintained at 1:1, based on previous preliminary data with free GEM and PTX (data not shown), and due to the observation during single agent cytotoxicity studies wherein the activities of the conjugates (GEM vs. PTX) were similar. Where cells were exposed to both treatments simultaneously, both long (72 h) and short (4 h) incubation times were evaluated to help discriminate conjugates which may vary in cellular uptake and drug release kinetics.
During long incubation time (72 h) experiments, all combination treatments demonstrated slight antagonistic to additive combined effects, with CI values of 1.3, 1.1, and 1.0 for free drugs, traditional conjugates, and multiblock conjugates respectively (Fig. 4). Due to the long incubation time, the concentration of the treatment groups in various compartments of the biological system (i.e., outside the cellular space, within the lysosome, within the cytosol, etc.) have an opportunity to reach a state of equilibrium, causing the combined effects to approach additive effects. During the short incubation time (4 h) experiments, the combination treatment demonstrated additive to synergistic combined effects, with CI values of 1.0, 0.9, and 0.7 for free drugs, traditional conjugates, and multiblock conjugates, respectively (Fig. 4). At this stage, the exact mechanism or underlying phenomenon which causes this level of synergism for the multiblock conjugates is unclear. However, based on the fact that this level of synergism was only observed in the short incubation experiments, kinetic phenomena are most likely involved. A major difference between the multiblock conjugates and their traditional counterparts is their size and molecular weight, which may impact their cellular uptake. Also, as previously discussed, additional degradable segments present in the backbone of the multiblock conjugates may increase their hydrophobicity, thereby enhancing the ability to interact with lipid membranes or altering their rate of internalization and/or sub-cellular distribution. However, additional studies are required to justify these hypothetical explanations. The results do, nonetheless, demonstrate an additional advantage at the pharmacological level for the use of these multiblock, biodegradable systems as compared to traditional HPMA copolymer conjugates.
Fig. 4.

In vitro combination index analysis. A2780 cells were treated with a 1:1 molar ratio combination of GEM and PTX. A combination of free drugs, traditional conjugates, and multiblock conjugates were each evaluated. Cells were treated with both drugs simultaneously (for either 72 or 4 h), or sequentially. In the majority of cases, additive combined effects were observed, with combination index (CI) values close to 1.0. However, during simultaneous incubation for 4 h, where kinetic phenomena can be discriminated, a combination of the multiblock conjugates demonstrated synergism with a CI value of 0.7 ± 0.1. Data expressed as mean ± SD of three independent experiments.
Additional combination experiments were performed where A2780 cells were first exposed to drug A (GEM or PTX) for 4 h, followed by a prolonged treatment with drug B (PTX or GEM) for the remaining 68 h. The primary purpose of these studies was to determine if a discrete sequence of treatment was advantageous. Theoretically, it is possible that exposure to drug A may sensitize or “prime” the cells for subsequent treatment with drug B. In this study, no major changes in the combination index were observed during these studies, as the combined effects were primarily additive, with CI values ranging from approximately 0.9 to 1.1. However, minor synergism was observed for the multiblock conjugates when cells were exposed to PTX, followed by exposure to GEM (CI = 0.88, Fig. 4). This observation is consistent with other reports, which have suggested that this sequence of treatment is synergistic, due to a block of cells in the G0/G1 cell cycle phase and an increase in the number of apoptotic cells (Zupi et al., 2005; Oliveras-Ferraros et al., 2008). Another report has suggested that the combined effects PTX and GEM are highly sequence dependent and demonstrated antagonism for the reverse sequence (GEM → PTX) in breast cancer cells (Sui et al., 2006).
3.5. Evaluation of gemcitabine/paclitaxel dual agent copolymer
An HPMA copolymer bearing both GEM and PTX on the same backbone (P-GEM-PTX, dual agent conjugate) was synthesized and evaluated for its cytotoxic effect against A2780 cells in comparison with a physical mixture of individual HPMA copolymer conjugates (P-GEM + P-PTX). The molar ratio of GEM to PTX in the dual agent conjugate was 4.87:1, due to some loss of PTX during purification. The physical mixture, was, therefore, mixed in the same molar ratio (4.87:1, GEM:PTX) for direct comparison. Treatment duration was evaluated by incubation for 72 or 4 h as previously described. During long incubation time (72 h) experiments, no significant differences were observed in the activity on the conjugates, with IC50 values ranging from 4 to 10 nM (Fig. 5A). However, during short incubation time (4 h) experiments (Fig. 5B), a significant decrease in potency for the dual agent conjugate was observed as compared to the equivalent physical mixture. The IC50 value of the dual agent conjugate was 348 nM as compared to 211 nM for the physical mixture. When this data was analyzed by combination index analysis, the combined effects of each drug for the dual agent conjugate were antagonistic with a CI value of 1.6 as compared to 0.9 for the physical mixture (Fig. 5C). This could possibly be due to intramolecular interactions between drug molecules in the side chains of the dual agent conjugate, thereby altering parameters such as stability and drug release. The conclusions from this study, however, should not be overstated. This evaluation was conducted in vitro at the cellular level, where pharmacokinetics and biodistribution are not relevant. The possibility remains that a conjugate bearing two agents on the same backbone may benefit in vivo by sharing the same pharmacokinetics or biodistribution, as compared to a physical mixture, where each agent will exhibit its own pattern of distribution. This may be especially significant in conditions where pharmacological synergism for the two agents exists. However, the results do indicate that at the cellular level, conjugation of GEM and PTX to the same HPMA copolymer backbone resulted in an overall decrease in potency against A2780 cells.
Fig. 5.
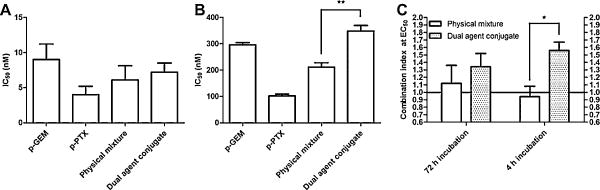
Evaluation of GEM/PTX dual agent copolymer. The cytotoxic effect of a dual agent copolymer bearing GEM and PTX on the same backbone was compared with a physical mixture of conjugates bearing GEM and PTX individually in A2780 human ovarian cancer cells. (A) Cells were exposed to treatments continuously for 72 h. All treatments were similar in activity with IC50 values between 4 and 10 nM. (B) Cells were exposed to treatments for 4 h, followed by incubation with growth media for 68 h. A significant decrease in the potency for the dual agent was observed as compared to a physical mixture of conjugates bearing GEM and PTX individually. (C) Combination index analysis of the dual agent conjugate as compared to the equivalent physical mixture. The dual agent conjugate demonstrated antagonistic combined effects as compared to the physical mixture, which showed nearly additive effects. Data expressed as mean ± SD of three independent experiments.
Other clinical considerations should be made when developing such combination polymers, where the ratio of two or more agents is fixed in the final drug product. A major disadvantage of such systems is the loss of flexibility in adjusting each drug dose independently. A particular patient may not tolerate one of the drugs in the combination product, thereby reducing the amount of the combination product that can be administered. This inflexibility can be of particular concern in anticancer applications, where the goal of the clinician is often to administer doses close to the threshold of tolerance in an attempt to achieve maximal efficacy.
4. Conclusion
Multiblock, backbone degradable HPMA copolymer-GEM and HPMA copolymer-PTX conjugates were synthesized and characterized. Conjugates with narrow polydispersity (Mw/Mn < 1.1) were obtained and drug loadings between 5.5 and 9.2 wt% were achieved. Conjugates demonstrated moderate stability with drug release less than 65% observed over 24 h at pH 7.4 and favorable drug release in the presence of the lysosomal enzyme cathepsin B. In combination, the cytotoxic effects of a mixture of the conjugates (traditional and multiblock) and free drug controls were generally additive. However, synergistic combined effects were observed when A2780 cells were treated simultaneously for 4 h with multiblock conjugates (CI < 0.7). When both GEM and PTX were conjugated to the same copolymer backbone, however, moderate antagonism (CI 1.3–1.6) was observed. These results demonstrate that multiblock HPMA copolymer-GEM and HPMA copolymer-PTX conjugates, when delivered as a mixture of individual agents, are promising in combination for the treatment of ovarian cancer.
Acknowledgments
The research was supported in part by the NIH grant R41 CA156933 from the National Cancer Institute.
References
- Bookman MA. The addition of new drugs to standard therapy in the first-line treatment of ovarian cancer. Ann Oncol. 2010;21(Suppl 7):vii211–vii217. doi: 10.1093/annonc/mdq368. [DOI] [PubMed] [Google Scholar]
- Chipman SD, Oldham FB, Pezzoni G, Singer JW. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugate. Int J Nanomed. 2006;1:375–383. doi: 10.2147/nano.2006.1.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol. 1999;17:1061–1070. doi: 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]
- Duncan R, Coatsworth JK, Burtles S. Preclinical toxicology of a novel polymeric antitumour agent HPMA copolymer-doxorubicin (PK1) Hum Exp Toxicol. 1998;17:93–104. doi: 10.1177/096032719801700204. [DOI] [PubMed] [Google Scholar]
- Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv Drug Deliv Rev. 2010;62:272–282. doi: 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Friedlander M, Buck M, Wyld D, Findlay M, Fitzharris B, De Souza P, Davies T, Kalimi G, Allan S, Perez D, Harnett P. Phase II study of carboplatin followed by sequential gemcitabine and paclitaxel as first-line treatment for advanced ovarian cancer. Int J Gynecol Cancer. 2007;17:350–358. doi: 10.1111/j.1525-1438.2007.00795.x. [DOI] [PubMed] [Google Scholar]
- Gallo D, Fruscella E, Ferlini C, Apollonio P, Mancuso S, Scambia G. Preclinical in vivo activity of a combination gemcitabine/liposomal doxorubicin against cisplatin-resistant human ovarian cancer (A2780/CDDP) Int J Gynecol Cancer. 2006;16:222–230. doi: 10.1111/j.1525-1438.2006.00304.x. [DOI] [PubMed] [Google Scholar]
- Garcia AA, Yessaian A, Pham H, Facio G, Muderspach L, Roman L. Phase II study of gemcitabine and docetaxel in recurrent platinum resistant ovarian cancer. Cancer Invest. 2012;30:295–299. doi: 10.3109/07357907.2012.657812. [DOI] [PubMed] [Google Scholar]
- Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533–539. [PubMed] [Google Scholar]
- Hendrickson AE, Oberg AL, Glaser G, Camoriano JK, Peethambaram PP, Colon-Otero G, Erlichman C, Ivy SP, Kaufmann SH, Karnitz LM, Haluska P. A phase II study of gemcitabine in combination with tanespimycin in advanced epithelial ovarian and primary peritoneal carcinoma. Gynecol Oncol. 2012;124:210–215. doi: 10.1016/j.ygyno.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog TJ. The current treatment of recurrent ovarian cancer. Curr Oncol Rep. 2006;8:448–454. doi: 10.1007/s11912-006-0074-9. [DOI] [PubMed] [Google Scholar]
- Herzog TJ, Pothuri B. Ovarian cancer: a focus on management of recurrent disease. Nat Clin Pract Oncol. 2006;3:604–611. doi: 10.1038/ncponc0637. [DOI] [PubMed] [Google Scholar]
- Hongrapipat J, Kopečková P, Liu J, Prakongpan S, Kopeček J. Combination chemotherapy and photodynamic therapy with Fab′ fragment targeted HPMA copolymer conjugates in human ovarian carcinoma cells. Mol Pharm. 2008;5:696–709. doi: 10.1021/mp800006e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiew LV, Cheong SK, Sidik K, Chung LY. Improved plasma stability and sustained release profile of gemcitabine via polypeptide conjugation. Int J Pharm. 2010;391:212–220. doi: 10.1016/j.ijpharm.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Kim A, Ueda Y, Naka T, Enomoto T. Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res. 2012;31:14. doi: 10.1186/1756-9966-31-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeček J. Soluble biomedical polymers. Polim Med. 1977;7:191–221. [PubMed] [Google Scholar]
- Kopeček J. The potential of water-soluble polymeric carriers in targeted and site-specific drug delivery. J Control Release. 1990;61:145–157. [Google Scholar]
- Kopeček J. Polymer-drug conjugates: origins, progress to date and future directions. Adv Drug Deliv Rev. 2013;65:49–59. doi: 10.1016/j.addr.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeček J, Bazilová H. Poly[N-(2-hydroxypropyl)methacrylamide]. 1. Radical polymerization and copolymerization. Eur Polym J. 1973;9:7–14. [Google Scholar]
- Kopeček J, Kopečková P. HPMA copolymers: origins early developments, present, and future. Adv Drug Deliv Rev. 2010;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeček J, Kopečková P. Design of polymer-drug conjugates. In: Kratz F, Senter P, Steinhagen H, editors. Drug Delivery in Oncology. Vol. 2. Wiley-VCH; Weinheim, Germany: 2012. pp. 485–512. Chapter 17. [Google Scholar]
- Lammers T, Šubr V, Peschke P, Kuhnlein R, Hennink WE, Ulbrich K, Kiessling F, Heilmann M, Debus J, Huber PE, Storm G. Image-guided and passively tumour-targeted polymeric nanomedicines for radiochemotherapy. Br J Cancer. 2008;99:900–910. doi: 10.1038/sj.bjc.6604561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers T, Šubr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, Storm G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30:3466–3475. doi: 10.1016/j.biomaterials.2009.02.040. [DOI] [PubMed] [Google Scholar]
- Lammers T, Ulbrich K. HPMA copolymers: 30 years of advances. Adv Drug Deliv Rev. 2010;62:119–121. doi: 10.1016/j.addr.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Larson N, Ghandehari H. Polymeric conjugates for drug delivery. Chem Mater. 2012;24:840–853. doi: 10.1021/cm2031569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kopečková P, Bühler P, Wolf P, Pan H, Bauer H, Elsässer-Beile U, Kopeček J. Biorecognition and subcellular trafficking of HPMA copolymer-anti-PSMA antibody conjugates by prostate cancer cells. Mol Pharm. 2009;6:959–970. doi: 10.1021/mp8002682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Yang J, Kopečková P, Kopeček J. Biodegradable multiblock poly[N-(2-hydroxypropyl)methacrylamide] via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- Meerum Terwogt JM, Ten Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, Rocchetti M, Rosing H, Koopman FJ, Beijnen JH. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs. 2001;12:315–323. doi: 10.1097/00001813-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34:2248–2256. [Google Scholar]
- Mould T. An overview of current diagnosis and treatment in ovarian cancer. Int J Gynecol Cancer. 2012;22(Suppl 1):S2–S4. doi: 10.1097/IGC.0b013e318251c8e3. [DOI] [PubMed] [Google Scholar]
- Naumann RW, Coleman RL. Management strategies for recurrent platinum-resistant ovarian cancer. Drugs. 2011;71:1397–1412. doi: 10.2165/11591720-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Oliveras-Ferraros C, Vazquez-Martin A, Colomer R, De Llorens R, Brunet J, Menendez JA. Sequence-dependent synergism and antagonism between paclitaxel and gemcitabine in breast cancer cells: the importance of scheduling. Int J Oncol. 2008;32:113–120. [PubMed] [Google Scholar]
- Pan H, Yang J, Kopečková P, Luo K, Kopeček J. Polymeric drug delivery conjugates and methods of making and using thereof. WO Patent WO/2011/112,482 2011a
- Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition-fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011b;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Sima M, Yang J, Kopeček J. Synthesis of long-circulating backbone degradable HPMA copolymer-doxorubicin conjugates and evaluation of molecular weight dependent antitumor efficacy. Macromol Biosci. 2013;13:155–160. doi: 10.1002/mabi.201200353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GJ, Ruiz Van Haperen VW, Bergman AM, Veerman G, Smitskamp-Wilms E, Van Moorsel CJ, Kuiper CM, Braakhuis BJ. Preclinical combination therapy with gemcitabine and mechanisms of resistance. Semin Oncol. 1996;23:16–24. [PubMed] [Google Scholar]
- Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC. Clinical toxicities encountered with paclitaxel (Taxol) Semin Oncol. 1993;20:1–15. [PubMed] [Google Scholar]
- Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, Boivin C, Hesslewood S, Twelves C, Blackie R, Schatzlein A, Jodrell D, Bissett D, Calvert H, Lind M, Robbins A, Burtles S, Duncan R, Cassidy J. Phase II studies of polymer-doxorubicin (PK1 FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34:1629–1636. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- Sui M, Xiong X, Kraft AS, Fan W. Combination of gemcitabine antagonizes antitumor activity of paclitaxel through prevention of mitotic arrest and apoptosis. Cancer Biol Ther. 2006;5:1015–1021. doi: 10.4161/cbt.5.8.2909. [DOI] [PubMed] [Google Scholar]
- Thomson AH, Vasey PA, Murray LS, Cassidy J, Fraier D, Frigerio E, Twelves C. Population pharmacokinetics in phase I drug development: a phase I study of PK1 in patients with solid tumours. Br J Cancer. 1999;81:99–107. doi: 10.1038/sj.bjc.6690657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma R, Kartarius S, Harlozinska A, Gotz C, Montenarh M. Growth inhibition and apoptosis induction in ovarian cancer cells. Int J Oncol. 2006;29:481–488. [PubMed] [Google Scholar]
- Vicent MJ, Ringsdorf H, Duncan R. Polymer therapeutics: clinical applications and challenges for development. Adv Drug Deliv Rev. 2009;61:1117–1120. doi: 10.1016/j.addr.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Wakabayashi MT, Lin PS, Hakim AA. The role of cytoreductive/debulking surgery in ovarian cancer. J Natl Compr Canc Netw. 2008;6:803–810. doi: 10.6004/jnccn.2008.0060. [DOI] [PubMed] [Google Scholar]
- Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, Baker JR, Jr, Van Echo DA, Von Hoff DD, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. React Funct Polym. 2011;71:294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Luo K, Yang J, Sima M, Sun Y, Janat-Amsbury M, Kopeček J. Synthesis and evaluation of a backbone degradable multiblock HPMA copolymer nanocarriers for delivery of paclitaxel. J Control Release. 2013;166:66–74. doi: 10.1016/j.jconrel.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupi G, Scarsella M, D'angelo C, Biroccio A, Paoletti G, Lopez M, Leonetti C. Potentiation of the antitumoral activity of gemcitabine and paclitaxel in combination on human breast cancer cells. Cancer Biol Ther. 2005;4:866–871. doi: 10.4161/cbt.4.8.1895. [DOI] [PubMed] [Google Scholar]