

Abstract
Objectives
There is an enrichment of immune response genes that are subject to copy number variations (CNVs). However, there is limited understanding of their impact on susceptibility to human diseases. CC chemokine ligand 3 like-1 (CCL3L1) is a potent ligand for the HIV coreceptor, CC chemokine receptor 5 (CCR5), and we have demonstrated previously an association between CCL3L1- gene containing segmental duplications and polymorphisms in CCR5 and HIV/AIDS susceptibility. Here, we determined the association between these genetic variations and risk of developing systemic lupus erythaematosus (SLE), differential recruitment of CD3+ and CD68+ leukocytes to the kidney, clinical severity of SLE reflected by autoantibody titres and the risk of renal complications in SLE.
Methods
We genotyped 1084 subjects (469 cases of SLE and 615 matched controls with no autoimmune disease) from three geographically distinct cohorts for variations in CCL3L1 and CCR5.
Results
Deviation from the average copy number of CCL3L1 found in European populations increased the risk of SLE and modified the SLE-influencing effects of CCR5 haplotypes. The CCR5 human haplogroup (HH)E and CCR5-Δ32-bearing HHG*2 haplotypes were associated with an increased risk of developing SLE. An individual’s CCL3L1–CCR5 genotype strongly predicted the overall risk of SLE, high autoantibody titres, and lupus nephritis as well as the differential recruitment of leukocytes in subjects with lupus nephritis. The CCR5 HHE/HHG*2 genotype was associated with the maximal risk of developing SLE.
Conclusion
CCR5 haplotypes HHE and HHG*2 strongly influence the risk of SLE. The copy number of CCL3L1 influences risk of SLE and modifies the SLE-influencing effects associated with CCR5 genotypes. These findings implicate a key role of the CCL3L1–CCR5 axis in the pathogenesis of SLE.
Introduction
Copy number variations (CNVs) are a common polymorphism, reflecting 12% or more of the human genome.1–3 Many of these structural variations are segmental duplications that contain immune response genes,4 and consequently such CNVs might underlie in part inter-individual differences in susceptibility to complex diseases with immunological underpinnings. Consistent with this, recent studies showed that susceptibility to systemic lupus erythaematosus (SLE) is associated with CNVs in FCGR3B5,6 and C47 genes and susceptibility to Crohn disease is associated with CNVs in DEFB4.8 We found that segmental duplications that contain the gene encoding CC chemokine ligand 3 like-1 (CCL3L1), the most potent agonist and HIV-suppressive ligand for the HIV-1 coreceptor CC chemokine receptor 5 (CCR5), is associated with variable chemokine expression, numbers of CD4+CCR5+ T cells, risk of acquiring HIV and disease progression rates.9 We also found an association between copy number of CCL3L1 and risk of Kawasaki disease, a childhood vasculitis;10 a recent study reported the association between CCL3L1 CNV and rheumatoid arthritis.11 Based on the extensive data implicating members of the chemokine system (including CCR5 and its ligands) in autoimmunity including SLE pathogenesis, 12–14 here, we tested the hypothesis that CCL3L1-containing segmental duplications affect the risk of developing SLE. We tested this hypothesis within the context of investigating the overall impact of the CCL3L1–CCR5 axis on SLE susceptibility. To date, all the genetic epidemiological studies that have examined the role of this axis in SLE are limited to analysis of the CCR5-Δ32 mutation, which is associated with loss of CCR5 surface expression.15 However, we have previously demonstrated that CCR5 is highly polymorphic and these variations can be organized into haplotype groups designated as human haplogroups (HH) A through G*2.16 Thus, to gain greater insights into the role of this axis on SLE development, we first determined the individual impact of the copy number of CCL3L1 and polymorphisms in CCR5, and then their conjoint effects. Finally, we also investigated whether the genetic background conferred by a low, average or high CCL3L1 gene dose modifies the phenotypic effects of CCR5 haplotypes on risk of developing SLE.
METHODS
Study cohorts
The San Antonio SLE cohort
This cohort consisted of prospectively collected data from patients enrolled into the San Antonio Lupus Study of Neuropsychiatric Disease (SALUD), and was as described previously.17 Individuals were enrolled into SALUD if they met four or more of the American College of Rheumatology (ACR) revised criteria for SLE18,19 and controls were healthy, non-blood-related family members and friends of the enrolled patients with lupus. This cohort comprised of 134 patients with SLE and 60 healthy controls; 73 (54.48%) cases and 33 (55%) controls were Hispanic whereas 45 (33.58%) cases and 26 (43.33%) controls were Caucasian. For the cases, the mean (SD) age was 43.04 (11.95) years, and 122 (91.04%) were females. For the controls, the mean (SD) age was 39.31 (15.45) years and 38 (63.33%) were females.
The Colombian SLE cohort
This cohort consisted of 143 patients with SLE and 421 healthy controls. All patients fulfilled four or more of the ACR criteria for SLE18, 19 and were recruited at the Rheumatology Unit at the Clinica Universitaria Bolivariana in Medellin, Colombia. The subjects were predominantly of European (ie, Spanish) descent. 20 The mean (SD) age of the patients was 32.77 (11.15) years and the cohort included 140 (97.90%) females. The controls were selected from the geographic neighborhood of the cases and matched for age, sex, ethnicity and socioeconomic status. Additional characteristics of this cohort and results of genetic epidemiological studies using this cohort have been described previously.21–25
The Ohio SLE cohort
This cohort consisted of 340 patients and 287 controls. Information on race was available for 268 (78.82%) cases and 258 (89.90%) controls. Of these 192 (71.64%) cases and 134 (51.94%) controls were European Americans and only these subjects were included in the present analyses. The Ohio SLE cohort used the 1997 ACR revised criteria for diagnosis of SLE.18 The controls were healthy volunteers from central Ohio who had no personal or family history of autoimmune diseases.
Genotyping
Methods used for genotyping the copy number of CCL3L19 and CCR5 polymorphisms26 were as described previously. The CCR5 haplotype classification system was also as described previously. 9, 16 CCR5 haplotypes (linked polymorphisms) are designated as human haplogroups (HH) A through G*2; HHF*2 and HHG*2 denote the haplotypes that contain the CCR2-64I and CCR5-Δ32 polymorphisms, respectively.16
Antibody assays
Antinuclear antibodies were determined by indirect immunofluorescence in HEp-2 cells and were considered positive at a titre of .1:80 dilution. Anti-double stranded DNA (antiDNA), anti-Ro, anti-La, anti-RNP and anti-Sm were measured by ELISA using commercial kits (INOVA, San Francisco, California, USA).
Assessment of renal involvement
Renal involvement was considered present when a renal biopsy demonstrated World Health Organization (WHO) class II–V histopathology, active urinary sediment, proteinuria .500 mg/ 24 h or nephrotic syndrome. Nephrotic syndrome was defined as .3.5 g/day of proteinuria, hypoalbuminaemia (<2.8 g/dl), hyperlipidaemia and oedema. Renal biopsies of 22 patients with lupus nephritis from the Ohio cohort were quantified for infiltrating CD68+ (macrophages) and CD3+ (T cells) cells as described previously.27
Statistical analyses
Allele and haplotype frequencies were estimated and Hardy– Weinberg equilibrium was determined using the PowerMarker (version 3.23) software.28 We conducted the statistical analysis for the SLE cohorts in two steps. First, we conducted the analysis for all patients with SLE pooled together vs. all the controls. Second, to exclude the possibility of confounding by ethnicity or geographic origin of the cohorts, we conducted the analysis separately for each cohort and compared the cases against their respective set of controls (fig 1A). As another measure to reduce the confounding by ethnicity, and because of small numbers of subjects of African descent in the three cohorts, we limited our analyses to subjects of Hispanic or European descent.
Figure 1.

Study cohorts and distribution of CCL3L1 copy number and CCR5 haplotypes. A. Systemic lupus erythaematosus (SLE) cohorts. Cohorts were from three different geographic regions, namely San Antonio, Texas, USA, Columbus, Ohio, USA and Medellin, Colombia. n, number of study subjects. The up/down arrow indicates a set of cases being compared against the corresponding set of controls as well as between all cases and all controls. The colors corresponding to each study subset are used in the remaining panels. Overall, all subjects. B, C. Distribution of CCL3L1 copy number in each study cohort (B) and all cohorts combined (C). D, E. Distribution of CCR5 haplotypes in each study cohort (D) and all cohorts combined (E). The overall difference of distribution between cases and controls was tested for significance using the χ2 test. The asterisks indicate significance values for differences in the frequency of the indicated CCL3L1 copy number and CCR5 haplotypes between cases and controls. p Values for possessing one and four copies of CCL3L1 (C) were 0.012 and 0.020, whereas for possessing CCR5 human haplogroup (HH)E, HHF*2 and HHG*2 haplotypes (E) they were p=0.034, 0.002 and <0.001, respectively.
We used unconditional multiple logistic regression analysis to assess the association between copy number of CCL3L1 or CCR5 haplotypes or genotypes (haplotype pairs) and risk of developing SLE. Crude odds ratios (OR) were used as an estimate of the relative risk and were determined along with 95% confidence interval (CI). By this multivariate approach the independent effect of each variable was assessed while simultaneously adjusting for the effects of the covariates in a single model, thus minimizing multiple comparisons. We also assessed the association between variations in CCL3L1–CCR5 with the risk of lupus nephritis and autoantibody titres. For this, we used clustered multinomial logistic regression analyses, where the study-cohort membership was the clustering variable. We used multivariate analysis of variance (MANOVA) to determine the association between CCL3L1–CCR5 genotypes and the number of inflammatory cells in different renal compartments in patients with lupus nephritis. All statistical analyses were conducted using the Stata 8.0 (Stata Corp, College Station, Texas, USA) statistical software.
RESULTS
CCL3L1 copy number and risk of developing SLE Consistent with our previous findings in subjects of European descent,9 the median copy number of CCL3L1 in the SLE cases and controls we studied here was also two (fig 1B–C). However, the overall frequency distribution of CCL3L1 copy number was significantly different between all cases and controls (p=0.032), and the frequency of one and four copies of CCL3L1 was higher in cases than controls (fig 1C). This suggested that deviation from the average copy number found in these study populations (two) might be a risk factor for developing SLE. Consistent with this, a copy number lower than or greater than two was associated with an increased risk of developing SLE (fig 2A). Evaluation of the SLE cohorts separately indicated that the maximal impact of the CCL3L1 CNV on SLE risk was in the cohorts from Colombia and Ohio (fig 2)
Figure 2.

Copy number of CCL3L1 and CCR5 haplotypes influence risk of developing systemic lupus erythaematosus (SLE). Association between CCR5 haplotype or CCL3L1 copy number and the risk of developing SLE was determined by logistic regression analyses in all cohorts combined (A) and in each SLE cohort separately (B–D). The error bars indicate 95% CIs around the point estimates (diamonds). In (A), the results are from the final model of a stepwise logistic regression using a probability criterion of p<0.1 and red color indicates statistical significance whereas pink color indicates statistical non-significance.
CCR5 haplotypes and SLE risk
The overall distribution of CCR5 haplotypes in the cohorts studied were similar to those observed previously in subjects of European descent.26 CCR5 HHC was the most common haplotype, and this was followed by HHE and HHF*2 (fig 1D–E). All the haplotypes were in Hardy–Weinberg equilibrium (data not shown). A highly statistically significant difference was detected in the distribution of CCR5 haplotypes between all cases and controls (p<0.001; fig 1E). To determine which CCR5 haplotype conveyed this SLE-influencing effect, we used logistic regression analysis. We found that in the pooled analysis of all cohorts, CCR5 HHE and the CCR5-Δ32-containing HHG*2 haplotype were each associated with an increased risk of developing SLE whereas CCR5 HHF*1 was associated with a trend towards protection (fig 2A). Concordantly, when examined in the individual cohorts, possession of HHE was associated with an increased risk of developing SLE, and a similar trend was observed for HHG*2 (fig 2B–D); an effect for HHF*1 was not observed in the individual cohorts, possibly because of its low frequency (figs 1D and 2B–D).
Impact of CCR5 genotypes on SLE development
To determine the influence of CCR5 haplotype pairs (genotypes) on SLE susceptibility, we selected the 12 most common CCR5 genotypes found in all cohorts. Six of these contained CCR5 HHE (fig 3A). Among the genotypes containing HHE, an increased risk of SLE was conferred mainly by CCR5 HHA/HHE and HHE/HHG*2 genotypes, and a trend was observed for HHE/HHF*2 (fig 3A).
Figure 3.
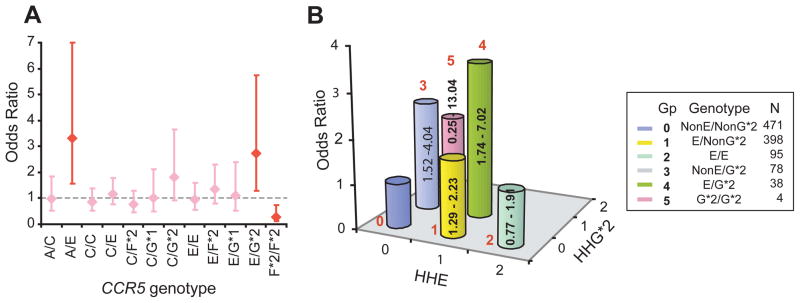
CCR5 genotypes and risk of systemic lupus erythaematosus (SLE). A. Association between the 12 most common CCR5 genotypes and risk of developing SLE. The diamonds and error bars represent point and 95% confidence intervals of odds ratio, respectively. Red indicates statistical significance while pink indicates statistical non-significance. B. Interactive effects of CCR5 human haplogroup (HH)E and HHG*2 on the risk of developing SLE. The numbers in red indicate the six color-coded genotypic groups; n, number of subjects in each group (Gp). For example, those who possess HHE but not HHG*2 are designated as E/NonG*2. For this analysis, group 0 (those lacking HHE and HHG*2) was considered as the reference category. The height of the bar indicates the odds ratio of developing SLE and the numbers in the bar indicate 95% CI.
Among the 12 most common CCR5 genotypes, 2 contained the CCR5-Δ32-containing HHG*2 haplotype. We found that in addition to possession of the HHE/HHG*2 genotype, the HHC/ HHG*2 genotype was also associated with a trend for an increased risk of developing SLE (fig 3A). Among the HHF*2- containing genotypes, homozygosity for this haplotype was associated with a marked reduction in the risk of SLE and HHC/ HHF*2 was associated with a trend towards protection (fig 3A). As an effect of the CCR2-64I-bearing HHF*2 haplotype on developing SLE was not detected in the individual cohorts (fig 2B–D), this indicated that the protective effects of HHF*2 are present mainly when it is in the homozygous state (fig 3A). Collectively, these findings identified CCR5 HHE-, HHG*2- and HHF*2-containing genotypes as determinants of SLE susceptibility, with dominant effects for the HHE- and HHG*2- containing genotypes.
Additive and independent impact of CCR5 HHE and HHG*2 on SLE
When examined at the level of haplotypes, HHE and HHG*2 were each associated with an increased risk of SLE (fig 2). Concordantly, HHE/HHG*2 genotype was associated with an increased risk of developing SLE (fig 3A). However, it was unclear whether the effects of HHG*2 were direct, or merely due its association with HHE. To address this, based on possession of these two haplotypes, we divided subjects into six CCR5 genotypic groups (fig 3B). Subjects who did not possess either HHE or HHG*2 were the reference category for this analysis. We found that CCR5 genotypic group 1 (possession of HHE but lacking HHG*2) and group 3 (possession of HHG*2 but lacking HHE) were each independently associated with an increased risk of developing SLE (fig 3B), indicating that HHE and HHG*2 had independent detrimental effects. Consequently, pairing of HHE and HHG*2 (CCR5 genotypic group 4 in fig 3B) had additive effects (OR=3.50, 95% CI 1.74 to 7.02; p<0.001).
CCL3L1–CCR5 genotype and histopathological phenotype
We next determined whether genotype–phenotype associations are also evident at the cellular level, i.e., whether recruitment of infiltrating leukocytes differed by CCL3L1–CCR5 genotype. CCR5 genotypes were categorized as shown in fig 3B and concordant with the genotype–phenotype associations presented above, we found that recruitment of CD68+ and CD3+ cells to the interstitium was greatest in subjects possessing the HHE/HHG*2 genotype (fig 4A). Other HHE- and HHG*2- containing genotypes along with CCL3L1 CNV also influenced cellular recruitment patterns (fig 4A). Statistical analyses revealed that the impact of CCL3L1 CNV and CCR5 genotypes on recruitment of CD3+ and CD68+ cells was restricted mainly to interstitium (fig 4B) and explained 72% of the variability in the number of cells per high power field. In this compartment, possession of HHE-containing genotypes was associated with a significant increase in recruitment of CD3+ and CD68+ cells (fig 4A, B). Possession of a copy number of CCL3L1 other than the population average (two) was also associated with a trend for a reduced recruitment of macrophages into the interstitial compartment (fig 4B).
Figure 4.

Association of CCL3L1–CCR5 genotypes with number of inflammatory cells in the indicated renal compartment in patients with lupus nephritis. A. Average number of inflammatory cells per high power field (hpf) in the indicated renal compartments stratified by CCL3L1– CCR5 genotype. The bars show mean number of cells/hpf. Macrophages (orange bars) and lymphocytes (brown bars) were identified by the presence of CD68 and CD3 markers, respectively. B. Results of multivariate analysis of variance (MANOVA) for association between CCL3L1–CCR5 genotypes with number of inflammatory cells in the indicated renal compartments. p Indicates the significance value estimated from the Snedecor F statistic and R2 indicates the variability explained by the full model including the CCL3L1–CCR5 genotypes. In this analysis non-HHE/non-HHG*2 was considered as the reference genotype for CCR5 and two copies was the reference category for CCL3L1.
Combined effects of CCL3L1 and CCR5 on SLE development
The aforementioned findings suggested that deviation from the average copy number of CCL3L1 (two) in conjunction with possession of CCR5 HHE and HHG*2 haplotypes might have the greatest impact on risk of developing SLE. To examine their combined effects, we stratified the SLE cohorts into three mutually exclusive CCL3L1–CCR5 genotypic risk groups (fig 5A). As a general rule, in each of the cohorts, relative to individuals in CCL3L1–CCR5 genotypic risk group 1 (two copies of CCL3L1 and lacking HHE and HHG*2), those with <2 or >2 CCL3L1 copies and HHE or HHG*2 (group 3) had a nearly twofold greater risk of developing SLE, whereas all others (group 2) had an intermediate risk (fig 5B).
Figure 5.

Conjoint effects of CCR5 and CCL3L1 on systemic lupus erythaematosus (SLE) susceptibility, development of renal lupus and autoantibody titres. A. CCL3L1–CCR5 genotypic groups. Group 1 comprised of subjects who did not possess CCR5 human haplogroup (HH)E or HHG*2 (Non-HHE and Non-HHG*2) and had two copies of CCL3L1. Group 2 comprised of subjects who either (a) did not possess CCR5 HHE or HHG*2 (Non-HHE and Non- HHG*2) and had either <2 or >2 copies of CCL3L1, or (b) those who possessed HHE or HHG*2 and had two copies of CCL3L1. Group 3 comprised of subjects who possessed HHE or HHG*2 CCR5 haplotypes and had <2 or >2 copies of CCL3L1. The color codes for the indicated genotypic risk groups are used in the rest of the figure. B. Association of the genotypic risk groups with risk of SLE. For these analyses, group 1 in (A) was considered as the reference category. For each cohort the odds ratios were estimated in a single model by multivariate logistic regression analysis. Letters (a–h) indicate significance values: a, 0.015; b, 0.041; c, 0.187; d, 0.020; e, 0.283; f, 0.009; g, 0.002; h, <0.001. C. Association of CCL3L1–CCR5 genotypic risk groups with risk of lupus nephritis in patients with SLE from the Colombia and Ohio cohorts. Letters (i) and (j) indicate significance values: i, <0.001; j, 0.071. In (B) and (C) the diamonds and error bars represent point and 95% confidence intervals of odds ratio, respectively. D. Association of the CCL3L1–CCR5 genotypic risk groups and autoantibody titres. Letters (k–n) indicate significance values: k, 0.037; l, 0.006; m, <0.001; n, <0.001. The anti-Ro and anti-La data was not available for the San Antonio cohort. Statistical significance for results shown in (C) and (D) was assessed using clustered multinomial logistic regression analyses.
CCL3L1–CCR5 genotypic risk groups and renal involvement and autoantibodies
The cohorts from Ohio and Colombia had sufficient number of subjects with lupus nephritis to determine whether the CCL3L1–CCR5 genotypic groups affected risk of renal involvement. We found that compared to individuals in CCL3L1–CCR5 genotypic risk group 1, those in groups 2 and 3 had a significantly higher risk of renal SLE (fig 5C).
We next determined the association between the CCL3L1– CCR5 genotypic risk groups and autoantibody titres. Compared to individuals in CCL3L1–CCR5 genotypic risk group 1, titres of anti-DNA were significantly higher in those in group 2 (OR=1.49, 95% CI 1.02 to 2.16) and group 3 (OR=1.39, 95% CI 1.10 to 1.75) (fig 5D). Additionally, relative to CCL3L1– CCR5 genotypic risk group 1, titres of anti-Ro (OR=1.6, 95% CI 1.58 to 1.62) and anti-La (OR=3.07, 95% CI 2.08 to 4.55) were significantly higher in group 3 (fig 5D). A similar but statistically non-significant association was detected for the CCL3L1–CCR5 genotypic risk groups and ANA levels (data not shown).
Modifier effects of CCL3L1 in SLE
To determine whether CCL3L1 copy number modified the SLEinfluencing phenotypic effects associated with CCR5 haplotypes, we stratified the subjects based on copy number of CCL3L1 (i.e., <2 2 or >2copies, table 1). The increased risk of SLE associated with CCR5 HHE was maximal, moderate and absent in subjects with 2, <2 and >2 CCL3L1 copies, respectively. Whereas the increased risk of developing SLE associated with CCR5 HHG*2 was not evident in those possessing two CCL3L1 copies, and instead was most evident in those with a copy number greater or lower than two (models 2 and 4 in table 1). This analysis also revealed that although a SLE susceptibility-reducing effect associated with the HHF*2 haplotype was not detected in the pooled analyses (fig 2A), such an effect is evident in the context of subjects who possessed >2 CCL3L1 copies (model 4 in table 1).
Table 1.
Overall and CCL3L1 gene dose–dependent effects of CCR5 haplotypes on risk of developing SLE.
| Model, CCR5 haplotype, CCL3L1 gene dose | OR (95% CI) | P |
|---|---|---|
| Model 1 (N=1059) | ||
| HHE | 1.61 (1.25 – 2.07) | <0.001 |
| HHG*2 | 2.57 (1.71 – 3.86) | <0.001 |
| HHF*1 | 0.24 (0.05 – 1.10) | 0.067 |
|
| ||
| Model 2 (<2 CCL3L1 copies, N=235) | ||
| HHG*2 | 3.03 (1.35 – 6.79) | 0.007 |
| HHE | 1.83 (1.06 – 3.15) | 0.030 |
| HHD | 4.24 (0.82 – 21.99) | 0.086 |
|
| ||
| Model 3 (2 CCL3L1 copies, N=428) | ||
| HHE | 1.94 (1.31 – 2.88) | 0.001 |
|
| ||
| Model 4 (>2 CCL3L1 copies, N=396) | ||
| HHG*2 | 4.12 (1.79 – 9.41) | 0.001 |
| HHF*2 | 0.64 (0.42 – 0.98) | 0.042 |
Model 1, stepwise unconditional logistic regression analysis for association between possession of CCR5 haplotypes and SLE disease susceptibility before accounting for CCL3L1 gene dose; Models 2 to 4, indicate stepwise unconditional logistic regression for association of the CCR5 haplotypes in the context of different CCL3L1 gene dose strata (i.e., <2, 2 and >2 CCL3L1 copies). All stepwise regression models used a probability criterion of P < 0.1. CI, confidence interval; OR, odds ratio. N, number of subjects. The analysis is for the combined three cohorts of SLE.
HH, human haplogroup
DISCUSSION
This study has four major findings. First, the genotype– phenotype associations suggest that the CCR5-CCR5 ligand axis plays an important role in SLE pathogenesis. This conclusion is supported by consistent genotype–phenotype associations detected at multiple levels: SLE risk, renal involvement, antibody titres and recruitment of leukocytes to the kidney. Previously, we9 and Townson et al29 found that there is a correlation between CCL3L1 copy number and chemokine production, with a plateau in chemokine production at higher copy numbers. Here, we found that relative to the average gene dose in the study population, a lower and higher copy number of CCL3L1 was associated with an enhanced risk of developing SLE, suggesting that development of SLE might be highly sensitive to chemokine production, such that an optimal level might be protective whereas low and high chemokine expression might be detrimental. With respect to variations in CCR5, the CCR5 HHE haplotype (which is associated with higher CCR5 transcriptional activity16 and protein expression30) and the CCR5 D32-containing HHG*2 haplotype (which is associated with reduced CCR5 surface expression31) were associated with an increased risk of SLE. Consistent with the latter finding, Aguilar et al32 found that the CCR5 D32 mutation was associated with an increased risk of having anti-dsDNA antibodies, and Gomez-Reino et al33 found that the frequency of the CCR5Δ32 homozygosity was higher in subjects with SLE (0.027) than in controls (0.009). Thus, from these associations we infer that deviation from a homeostatic balance of CCL3L1 and CCR5 expression levels might be detrimental with respect to SLE susceptibility.
Second, the results might also provide a genetic correlate into the proposed role by which CCR5 and its ligands mediate inflammatory processes in SLE pathogenesis. Inflammatory processes in SLE are initiated by lymphocytic infiltration into tissue spaces,34–37 and the histological distribution of lymphocytes, for example in lupus nephritis, appears to be determined by the expression of chemokine receptors, including CCR5.38–40 CCR5+ cells are invariably distributed in the interstitial regions.39, 40 Once inflammation is established, these lymphocytes release chemokines such as CCL3 and undergo apoptosis. 41–43 This locally accentuated chemokine gradient is thought to mediate two processes that might help promote the resolution phase: recruitment of additional CCR5+ lymphocytes and macrophages to facilitate termination of inflammation and clearance of apoptotic cells.44 Within this context, it is conceivable that higher expression levels of CCR5 (eg, with CCR5 HHE-containing genotypes) aggravates the inflammatory process by increasing initial lymphocytic infiltration. Similarly, since the resolution phase will depend on the chemokine gradient produced by apoptotic lymphocytes it is possible that the CCL3L1 copy number will independently influence the risk of SLE. The third major finding pertains to the observation that there is similarity in the CCL3L1 and CCR5 genetic determinants that influence SLE and HIV/AIDS susceptibility (see supplementary material). For example, several studies have demonstrated that CCR5 HHE-containing genotypes are associated with an increased risk of HIV/AIDS susceptibility,26, 45–47 and here we find that this haplotype is associated with an increased risk of SLE. We suggest that these findings might provide support for the common variants/multiple disease (CV/MD) hypothesis, which has been invoked to underlie the pathogenesis of several complex disease states.48, 49 The CV/MD hypothesis has also been invoked as a basis for susceptibility to clinically distinct autoimmune phenotypes.11, 50, 51 Our results suggest that the CV/MD hypothesis might be applicable when two diseases share some clinical autoimmune phenotypes, but when one entity such as SLE is a prototypical autoimmune disease and the other entity, such as infection with HIV-1 that can also be associated with autoimmunity,52 is not (supplementary material). Consistent with this, SLE and HIV share some common clinical, pathogenic and laboratory features (supplementary material). One possible implication of these results is that CCR5 blockade during HIV infection might increase risk of autoimmune phenotypes in subjects with specific CCR5 and CCL3L1 genotypes.
The fourth major finding pertains to gene modifiers, which is a principal challenge for the future of understanding the genetics of non-Mendelian diseases. Phenotype modification occurs when expression of one gene alters the phenotype normally conferred by another gene. Notably, the modifier can cause subtle or profound changes in the expression of the phenotype caused by mutation at another gene locus.53, 54 Modifier genes usually affect the phenotypic outcome of a given gene by interacting in the same or a parallel biological pathway as a disease gene.53, 54 In this light, given the ligand-receptor relationship of CCL3L1 and CCR5 and the finding that CCR5 haplotypes must reside in a permissive genetic background dictated by CCL3L1 gene copy numbers to manifest their full phenotypic effects in Kawasaki disease,10 SLE (table 1) and HIV/AIDS (data not shown) provide, to our knowledge, the first examples for genetic modification by copy number variations. The consistency of the gene modifier effects across three distinct disease states suggests that CCL3L1 copy number dependent phenotype modification might be a common phenomenon in disease states where the CCL3L1–CCR5 axis affects disease pathogenesis. Thus, failure to account for intercohort variations in the underlying distribution of such copy number variations might explain, in part, the many association studies that cannot be replicated, especially because CNV is a very common polymorphism in the human genome.1–3
Supplementary Material
Acknowledgments
Funding: This work was supported by the Veterans Administration Center on AIDS and HIV infection of the South Texas Veterans Health Care System and a MERIT (R37046326) and other awards (AI043279 and MH069270) from the NIH and other organisations (Elizabeth Glaser Scientist Award and the Burroughs Wellcome Clinical Scientist Award in Translational Research) to SKA, and the DK55546 award to BHR and the Fernando Chalem Rheumatology Award to J-MA. This work was also supported by NIH grants NS35477 (RB) and M01 RR001346, a shared resources grant to the University of Texas Health Science Center.
We thank G Crawford, S S Ahuja and R A Clark for invaluable programmatic support at UTHSCSA, and A S Ahuja for forbearance.
Footnotes
Competing interests: None.
References
- 1.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–46. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 2.Kehrer-Sawatzki H. What a difference copy number variation makes. Bioessays. 2007;29:311–3. doi: 10.1002/bies.20554. [DOI] [PubMed] [Google Scholar]
- 3.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 5.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 6.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007 doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene Copy-Number Variation and Associated Polymorphisms of Complement Component C4 in Human Systemic Lupus Erythematosus (SLE): Low Copy Number Is a Risk Factor for and High Copy Number Is a Protective Factor against SLE Susceptibility in European Americans. Am J Hum Genet. 2007;80:1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–48. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 10.Burns JC, Shimizu C, Gonzalez E, Kulkarni H, Patel S, Shike H, et al. Genetic variations in the receptor-ligand pair CCR5 and CCL3L1 are important determinants of susceptibility to Kawasaki disease. J Infect Dis. 2005;192:344–9. doi: 10.1086/430953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, Highton J, et al. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 12.Dong VM, McDermott DH, Abdi R. Chemokines and diseases. Eur J Dermatol. 2003;13:224–30. [PubMed] [Google Scholar]
- 13.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–15. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 14.Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, et al. Elevated Serum Levels of Interferon-Regulated Chemokines Are Biomarkers for Active Human Systemic Lupus Erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 16.Mummidi S, Bamshad M, Ahuja SS, Gonzalez E, Feuillet PM, Begum K, et al. Evolution of human and non-human primate CC chemokine receptor 5 gene and mRNA. Potential roles for haplotype and mRNA diversity, differential haplotype-specific transcriptional activity, and altered transcription factor binding to polymorphic nucleotides in the pathogenesis of HIV-1 and simian immunodeficiency virus. J Biol Chem. 2000;275:18946–61. doi: 10.1074/jbc.M000169200. [DOI] [PubMed] [Google Scholar]
- 17.McLaurin EY, Holliday SL, Williams P, Brey RL. Predictors of cognitive dysfunction in patients with systemic lupus erythematosus. Neurology. 2005;64:297–303. doi: 10.1212/01.WNL.0000149640.78684.EA. [DOI] [PubMed] [Google Scholar]
- 18.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 19.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 20.Correa PA, Whitworth WC, Kuffner T, McNicholl J, Anaya JM. HLA-DR and DQB1 gene polymorphism in the North-western Colombian population. Tissue Antigens. 2002;59:436–9. doi: 10.1034/j.1399-0039.2002.590515.x. [DOI] [PubMed] [Google Scholar]
- 21.Camargo JF, Tobon GJ, Fonseca N, Diaz JL, Uribe M, Molina F, et al. Autoimmune rheumatic diseases in the intensive care unit: experience from a tertiary referral hospital and review of the literature. Lupus. 2005;14:315–20. doi: 10.1191/0961203305lu2082oa. [DOI] [PubMed] [Google Scholar]
- 22.Correa PA, Gomez LM, Cadena J, Anaya JM. Autoimmunity and tuberculosis. Opposite association with TNF polymorphism. J Rheumatol. 2005;32:219–24. [PubMed] [Google Scholar]
- 23.Gomez LM, Anaya JM, Gonzalez CI, Pineda-Tamayo R, Otero W, Arango A, et al. PTPN22 C1858T polymorphism in Colombian patients with autoimmune diseases. Genes Immun. 2005;6:628–31. doi: 10.1038/sj.gene.6364261. [DOI] [PubMed] [Google Scholar]
- 24.Serrano NC, Paez C, Correa PA, Anaya JM. Endothelial nitric oxide synthase gene polymorphism is associated with systemic lupus erythematosus. J Rheumatol. 2004;31:2163–8. [PubMed] [Google Scholar]
- 25.Tobon GJ, Correa PA, Gomez LM, Anaya JM. Lack of association between TNF-308 polymorphism and the clinical and immunological characteristics of systemic lupus erythematosus and primary Sjogren’s syndrome. Clin Exp Rheumatol. 2005;23:339–44. [PubMed] [Google Scholar]
- 26.Gonzalez E, Bamshad M, Sato N, Mummidi S, Dhanda R, Catano G, et al. Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc Natl Acad Sci U S A. 1999;96:12004–9. doi: 10.1073/pnas.96.21.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, et al. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99:13795–800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21:2128–9. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 29.Townson JR, Barcellos LF, Nibbs RJ. Gene copy number regulates the production of the human chemokine CCL3-L1. Eur J Immunol. 2002;32:3016–26. doi: 10.1002/1521-4141(2002010)32:10<3016::AID-IMMU3016>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 30.Salkowitz JR, Bruse SE, Meyerson H, Valdez H, Mosier DE, Harding CV, et al. CCR5 promoter polymorphism determines macrophage CCR5 density and magnitude of HIV-1 propagation in vitro. Clin Immunol. 2003;108:234–40. doi: 10.1016/s1521-6616(03)00147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort. ALIVE Study Science. 1996;273:1856–62. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 32.Aguilar F, Nunez-Roldan A, Torres B, Wichmann I, Sanchez-Roman J, Gonzalez-Escribano MF. Chemokine receptor CCR2/CCR5 polymorphism in Spanish patients with systemic lupus erythematosus. J Rheumatol. 2003;30:1770–4. [PubMed] [Google Scholar]
- 33.Gomez-Reino JJ, Pablos JL, Carreira PE, Santiago B, Serrano L, Vicario JL, et al. Association of rheumatoid arthritis with a functional chemokine receptor, CCR5. Arthritis Rheum. 1999;42:989–92. doi: 10.1002/1529-0131(199905)42:5<989::AID-ANR18>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 34.Hoffman RW. T cells in the pathogenesis of systemic lupus erythematosus. Front Biosci. 2001;6:D1369–78. doi: 10.2741/hoffman. [DOI] [PubMed] [Google Scholar]
- 35.Josefsson E, Carlsten H, Tarkowski A. Neutrophil mediated inflammatory response in murine lupus. Autoimmunity. 1993;14:251–7. doi: 10.3109/08916939309077373. [DOI] [PubMed] [Google Scholar]
- 36.Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. 2001;12:1369–82. doi: 10.1681/ASN.V1271369. [DOI] [PubMed] [Google Scholar]
- 37.Yamada M, Yagita H, Inoue H, Takanashi T, Matsuda H, Munechika E, et al. Selective accumulation of CCR4+ T lymphocytes into renal tissue of patients with lupus nephritis. Arthritis Rheum. 2002;46:735–40. doi: 10.1002/art.10112. [DOI] [PubMed] [Google Scholar]
- 38.Furuichi K, Wada T, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Distinct expression of CCR1 and CCR5 in glomerular and interstitial lesions of human glomerular diseases. Am J Nephrol. 2000;20:291–9. doi: 10.1159/000013603. [DOI] [PubMed] [Google Scholar]
- 39.Segerer S, Mac KM, Regele H, Kerjaschki D, Schlondorff D. Expression of the C-C chemokine receptor 5 in human kidney diseases. Kidney Int. 1999;56:52–64. doi: 10.1046/j.1523-1755.1999.00544.x. [DOI] [PubMed] [Google Scholar]
- 40.Stasikowska O, Danilewicz M, Wagrowska-Danilewicz M. The significant role of RANTES and CCR5 in progressive tubulointerstitial lesions in lupus nephropathy. Pol J Pathol. 2007;58:35–40. [PubMed] [Google Scholar]
- 41.Anders HJ, Belemezova E, Eis V, Segerer S, Vielhauer V, Perez de Lema G, et al. Late onset of treatment with a chemokine receptor CCR1 antagonist prevents progression of lupus nephritis in MRL-Fas(lpr) mice. J Am Soc Nephrol. 2004;15:1504–13. doi: 10.1097/01.asn.0000130082.67775.60. [DOI] [PubMed] [Google Scholar]
- 42.Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. Faseb J. 2004;18:534–6. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- 43.Caproni M, Torchia D, Cardinali C, Volpi W, Del Bianco E, D’Agata A, et al. Infiltrating cells, related cytokines and chemokine receptors in lesional skin of patients with dermatomyositis. Br J Dermatol. 2004;151:784–91. doi: 10.1111/j.1365-2133.2004.06144.x. [DOI] [PubMed] [Google Scholar]
- 44.Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE, Luster AD, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–16. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaslow RA, Dorak T, Tang JJ. Influence of host genetic variation on susceptibility to HIV type 1 infection. J Infect Dis. 2005;191 (Suppl 1):S68–77. doi: 10.1086/425269. [DOI] [PubMed] [Google Scholar]
- 46.Mangano A, Gonzalez E, Dhanda R, Catano G, Bamshad M, Bock A, et al. Concordance between the CC chemokine receptor 5 genetic determinants that alter risks of transmission and disease progression in children exposed perinatally to human immunodeficiency virus. J Infect Dis. 2001;183:1574–85. doi: 10.1086/320705. [DOI] [PubMed] [Google Scholar]
- 47.Martin MP, Dean M, Smith MW, Winkler C, Gerrard B, Michael NL, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science. 1998;282:1907–11. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- 48.Becker KG. The common variants/multiple disease hypothesis of common complex genetic disorders. Med Hypotheses. 2004;62:309–17. doi: 10.1016/S0306-9877(03)00332-3. [DOI] [PubMed] [Google Scholar]
- 49.Yang Q, Khoury MJ, Friedman J, Little J, Flanders WD. How many genes underlie the occurrence of common complex diseases in the population? Int J Epidemiol. 2005;34:1129–37. doi: 10.1093/ije/dyi130. [DOI] [PubMed] [Google Scholar]
- 50.Anaya JM, Gomez L, Castiblanco J. Is there a common genetic basis for autoimmune diseases? Clin Dev Immunol. 2006;13:185–95. doi: 10.1080/17402520600876762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet. 2005;76:561–71. doi: 10.1086/429096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329–37. doi: 10.1016/s1568-9972(02)00086-1. [DOI] [PubMed] [Google Scholar]
- 53.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–74. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 54.Nadeau JH. Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev. 2003;13:290–5. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.