

Abstract
Inflammation and autophagy are cellular defense mechanisms. When these processes are deregulated (deficient or overactivated) they produce pathologic effects, such as oxidative stress, metabolic impairments, and cell death. Unresolved inflammation and disrupted regulation of autophagy are common features of pancreatitis and pancreatic cancer. Furthermore, obesity, a risk factor for pancreatitis and pancreatic cancer, promotes inflammation and inhibits or deregulates autophagy, creating an environment that facilitates the induction and progression of pancreatic diseases. However, little is known about how inflammation, autophagy, and obesity interact to promote exocrine pancreatic disorders. We review the roles of inflammation and autophagy, and their deregulation by obesity, in pancreatic diseases. We discuss the connections among disordered pathways and important areas for future research.
Keywords: Pancreatic Acinar Cell, Cytokines, Neutrophil, Pancreatic Ductal Adenocarcinoma, K-Ras
Inflammation plays a key role in tissue repair by eliminating injured cells, through the action of neutrophils, macrophages, and other immune cells, controlled via secreted mediators such as cytokines and chemokines.1–3 One mechanism initiating this response is up-regulation of genes that encode cytokines, chemokines, and other inflammatory mediators, through activation of transcription factors such as nuclear factor-κB (NF-κB), activator protein-1, nuclear factor of activated T cells (NFAT), and signal transducer and activator of transcription-3 (STAT3).4,5 Another mechanism is activation of the inflammasome—a multiprotein complex that serves as a platform for caspase-1 activation and resulting proteolytic maturation and secretion of interleukin (IL)-1β and IL-18.6 Efficient development and resolution of the inflammatory response and restoration of tissue homeostasis depends on coordinated interactions among various types of immune cells.1–3
Inflammation is a key feature of exocrine pancreatic disorders. The severity of acute pancreatitis (AP) is largely determined by whether the inflammatory response resolves or amplifies into persistent systemic inflammatory response syndrome (SIRS). Persistent low-grade inflammation is a characteristic of chronic pancreatitis (CP) and an important factor in the development of pancreatic ductal adenocarcinoma (PDAC). Moreover, inflammation mediates the pathogenic effects of (defective) autophagy and obesity in patients with pancreatitis or pancreatic cancer. We discuss how strategies to alter the inflammatory response might be used to treat patients with pancreatic diseases.
Role of Inflammation in Pancreatitis
The initial injury that causes AP results in acinar cell necrosis and an inflammatory response, which are usually sterile.7 In most patients, the acute inflammatory response and pancreas damage ultimately resolve, but in severe cases unremitting SIRS leads to multiple organ (especially lung) failure, a major cause of mortality among patients with AP.8–11 Twenty years ago, patients with AP were reported to have increased levels of circulating inflammatory cytokines.12,13 Studies of models of AP (animals and the ex vivo model of hyperstimulated acinar cells) established that in response to the initial insult, acinar cells produce and release a variety of inflammatory mediators14–20 that initiate the inflammatory response (Figure 1). These mediators recruit neutrophils, and then macrophages, monocytes, and lymphocytes, to the pancreas. Recent studies21–24 have identified subtypes of macrophages and monocytes that infiltrate the pancreas in models of AP, as well as immune cell populations that reside in murine pancreas: macrophages and small numbers of CD4+ and CD8+ T cells. CD4+ cells are recruited during the development of cerulein-induced pancreatitis and mediate parenchymal damage.25
Figure 1.
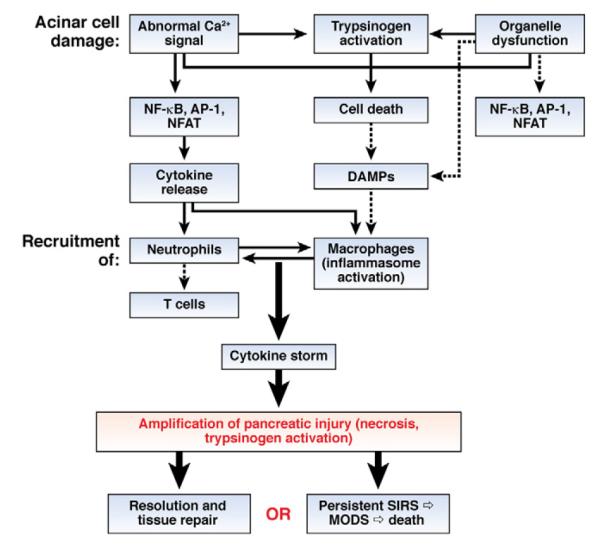
Inflammation in acute pancreatitis. Acinar cells respond to initial insult by activating transcription factors such as NF-κB, activator protein-1 (AP-1), and NFAT, leading to production of cytokines and other mediators that initiate the inflammatory response. Inflammation is also induced by release of DAMPs from damaged or dying cells, which activates inflammasomes. The inflammatory mediators recruit neutrophils, macrophages, and T cells into the pancreas, leading to cytokine storm and systemic inflammation. In most cases, pancreatic injury and inflammation resolves, but if perpetuated it can lead to potentially fatal SIRS and multiple organ dysfunction (MODS). Dashed lines indicate pathways that are likely but not proven in pancreatitis.
Development of AP in patients and experimental models is associated with strong up-regulation of inflammatory mediators such as tumor necrosis factor (TNF)α, IL-1β, IL-6, IL-8, platelet-activating factor, and chemokines of the CXC (such as CXCL2/MIP-2/KC) and CC families (such as CCL2/MCP-1) (for review, see articles by Pandol et al,10 Norman,13 Bhatia et al,19 Vonlaufen et al,26 Shanmugam and Bhatia,27 and Frossard et al28). IL-8, macrophage migration inhibitory factor, and especially IL-6 have been proposed to be early predictors of severe AP in human beings.29,30 It is not exactly clear how acinar cell injury leads to the induction of inflammatory mediators, but the process involves activation of specific transcription factors. NF-κB, in particular, is activated in experimental and human AP, and genetic and pharmacologic approaches have shown its important role in the cytokine storm.17,18,31–37 Activator protein-1 and NFAT also are activated in experimental pancreatitis.16,18,38 Acinar cell NF-κB and NFAT activation are Ca2+-dependent,38 – 40 and NF-κB activation also is mediated by novel isoforms isoforms of protein kinase C41,42 and by protein kinase D.43
Compared with the transcriptional activation of cytokines, much less is known about the mechanism and the role of inflammasome activation in pancreatitis. The inflammasome is activated in macrophages that infiltrate the pancreas; its blockade, by deletion of caspase-1 or components of inflammasome (such as the nucleotide binding domain leucine-rich repeat [NLR] family, pyrin domain containing-3 [NLRP3]), ameliorates cerulein pancreatitis in mice.7,24 The beneficial effect of deletion of Toll-like receptor-4 in AP models44,45 also can be mediated through inflammasome inhibition. Mechanisms of inflammasome activation in pancreatitis likely involve reactive oxygen species (ROS) (eg, resulting from mitochondrial damage) or damage/danger-associated molecular pattern molecules (DAMPs)–cellular components, such as high-mobility group box-1 protein, released from necrotic cells.6,7
The evidence that inflammation to a large extent determines the severity of pancreatitis comes from experimental studies, such as the seminal finding46 that mortality from severe AP was greatly reduced in mice with genetic deletions of receptors for TNF or IL-1β. These studies used various approaches to inhibit inflammation: inactivation or neutralization of cytokines, chemokines, adhesion molecules, and other mediators of inflammation13,14,34,47–53; deletion of Toll-like receptors to decrease the innate immune response44,45; blockade of neutrophil recruitment with neutralizing antibodies47,53–56 or genetic deletion of specific integrins48,57; inhibition of complement58; or multidrug strategies.59 Conversely, acinar cell-specific overexpression of IL-1β causes spontaneous pancreatitis.60 It is important, however, to remember that the pharmacologic or genetic manipulation of the inflammatory response in these models were mostly tested before induction (or early in the course) of AP.
A seemingly counterexample is the exacerbation of cerulein pancreatitis in mice with genetic deletion of the chemokine receptor CCR5,61 however, this deletion increases inflammatory cell infiltration, which can be prevented by simultaneous neutralization of several CC chemokines.61 One explanation for these results is that CCR5 depletes CC chemokines and thereby reduces further influx of neutrophils.3,62
The anti-inflammatory cytokines IL-10 and IL-22 protect against AP in animal models.63–66 However, although overexpression of IL-10 suppresses acute inflammation, it promotes the development of CP.63 Pancreatic and plasma levels of IL-6 increase during early stages of human and experimental AP and correlate with disease severity.12,19,28,29 However, this multifunctional cytokine is likely to have a complex role in the development of pancreatitis because it has proinflammatory and anti-inflammatory activities.67–69 The anti-inflammatory effects of IL-6 are mediated by a membrane-bound receptor, whereas its inflammatory activities are mediated by its binding to a soluble IL-6 receptor (sIL-6R).68,69 IL-6 neutralization ameliorates severe pancreatitis in mice,70 yet disruption of IL-6 increases levels of TNFα and IL-1β to increase the severity of cerulein-induced pancreatitis.71
Compared with AP, much less is known about the mechanisms of inflammation in CP.72 In a model in which CP is induced by administration of dibultyltin dichloride, early inflammation is mediated predominantly by macrophages, with increased levels of IL-1β and IL-10, whereas the chronic phase is associated with increases in IL-2, IL-4, and interferon-γ and T-cell infiltration.73,74 Infiltration by lymphocytes also is prominent in spontaneous CP in WBN/Kob rats.75 Disease-specific (in particular, regulatory) T cells recently were characterized in patients with CP76; increased numbers of cytotoxic T cells also were reported.77 Studies are needed to determine whether the persistent inflammation associated with CP results from an unresolved acute inflammatory response.
How does inflammation exacerbate pancreas damage? Inflammation shifts apoptosis-necrosis balance of acinar cell death toward necrosis,54 and greater amounts of parenchymal necrosis are associated with a worse prognosis for patients10 and more severe experimental pancreatitis.54,78–80 Pharmacologic or genetic inhibition of the inflammatory response invariably decreases necrosis in experimental models of AP. Furthermore, infiltrating inflammatory cells (both neutrophils and macrophages) mediate the pathologic, intra-acinar activation of trypsinogen, which is a key feature of pancreatitis.48,49,55,56 TNFα,14,46,48 ROS (generated by neutrophil reduced nicotinamide adenine dinucleotide phosphate oxidase),55 matrix metalloproteinase-9,49 and p5381 were proposed as mediators through which infiltrating inflammatory cells promote acinar cell death and trypsinogen activation. The persistent, low-grade inflammation also promotes pancreatic fibrosis by facilitating activation of stellate cells.72,82
To date, our knowledge of the inflammatory response in pancreatitis has not translated into effective therapies, and trials have been limited to patients with AP.19,28,83–85 Experimental studies strongly indicate that the acute inflammatory, especially neutrophilic, response in pancreatitis is overzealous or un(der)controlled, and that its down-regulation could have beneficial effects. Different approaches can be pursued to modulate inflammation in patients, such as dampening the cytokine storm by targeting specific inflammatory mediators. On the basis of experimental studies, promising strategies to explore include neutralizing TNFα,14,46,48 antagonizing sIL-6R,68,69 or, conversely, up-regulating IL-10 or IL-22.63–66
There are many challenges to this approach. The effects of inflammatory mediators overlap, and cytokines such as TNFα or IL-6 have diverse roles at different phases of the inflammatory response, and the therapeutic window for patients with AP is narrow.13 Another possible complication is an excessive or dysregulated compensatory anti-inflammatory response.85 These complexities could account for the lack of efficacy of lexipafant, a potent platelet-activating factor antagonist, in the only extensive trial to target a specific inflammatory mediator in patients with AP.28,83,84 A more promising strategy might be to target cytokine or chemokine receptors on distinct sub-populations of inflammatory cells.62 A caveat here is that subsets of immune cells and their repertoire of chemokines and receptors differ between human beings and rodents.3
An alternative strategy might be to block a master regulator of inflammation, such as NF-κB activation. However, this approach is likely to have too broad an effect. Moreover, NF-κB regulates not only inflammation but also cell survival and other processes in pancreatitis. Therefore, manipulating this pathway can have both beneficial and deleterious effects, as found in mouse models of AP.34–37,86,87
A promising approach (and research direction) that has not been explored in pancreatitis88 is to target mechanisms that could disrupt resolution of the inflammatory response (Table 1). Our general understanding of how inflammation resolves has increased greatly during the past few years.1–3,89 One cause of nonresolving inflammation in AP could be unremitting pancreatic injury, which continuously reinitiates inflammatory response—a vicious cycle of parenchymal necrosis and neutrophil infiltration. Another possibility involves defective interactions between neutrophils and macrophages that are required to resolve inflammation.3 For example, in the final phase of acute inflammation, macrophages switch from a proinflammatory to anti-inflammatory state in response to signals generated by neutrophils as they undergo apoptosis. Moreover, neutrophils are actively involved at this phase, producing pro-resolving lipid mediators (such as lipoxin A4) that inhibit further neutrophil recruitment.89 Lipoxins, along with other classes of inflammation-resolving lipid mediators and their receptors,1–3,89 therefore might be developed to treat patients with pancreatitis. It will be important to elucidate these mechanisms (Table 1) in experimental models and patients.
Table 1.
Mechanisms of Unresolving Inflammation to Explore in Pancreatitis
| Mechanisms that may cause unresolved inflammation in pancreatitis1–3 |
| Persistent/unresolved acinar cell injury |
| Excessive production of proinflammatory mediators |
| Weak production of anti-inflammatory mediators (IL-10, IL-22, TGF-β) |
| Weak production or defective function of pro-resolving lipid mediators |
| Defective scavenging of proinflammatory chemokines by neutrophils |
| Suppression of apoptosis and/or promotion of necrosis |
| Abnormal T-cell response (eg, deficiencies in T-regulatory cells) |
Inflammation in PDAC Development
PDAC is the most common type of pancreatic cancer and is notable for its aggressiveness, desmoplastic stromal response, immunosuppression, and resistance to therapies.90–93 Two types of inflammation contribute to PDAC initiation and progression. One is chronic inflammation caused by pancreatitis, obesity, or other genetic or environmental factors, and the other is tumor-associated inflammation. A typical feature of PDAC is its abundant desmoplasia, which comprises extracellular matrix components, fibroblasts, and vascular and immune cells.93,94 Immune cells make up around 50% of the tumor cell mass in PDAC94 and include innate immune cells (macrophages, myeloid-derived suppressor cells [MDSCs], neutrophils, and dendritic cells) and B and T cells.95 They are recruited in response to signals from tumor cells that express oncogenic Kras, and communicate with tumor cells and each other by producing cytokines and chemokines that control tumor growth.94,95 Oncogenic K-Ras promotes the inflammatory protumorigenic microenvironment and also the desmoplastic stromal response.94
It is not clear how inflammation promotes PDAC initiation and progression. Several mechanisms have been considered (Figure 2). First, inflammation facilitates cancer cell survival and proliferation. Oncogenic K-Ras promotes the inflammatory response through production of inflammatory mediators, such as cytokines. These mediators promote proliferation and survival of premalignant cells via activation of transcription factors such as STAT3 and NF-κB, which regulate transcription of genes that control cell survival, proliferation, and invasion, as well as cytokine and chemokine production.5,95 K-Ras activation causes pancreatic neoplastic epithelial (PanIN) cells to produce cytokines such as IL-6 and IL-11, which activate STAT3 in an autocrine manner96 and recruit immune (particularly myeloid) cells. The immune cells secrete IL-6 and sIL-6R to activate STAT3 in PanIN cells through IL-6 trans-signaling,97 creating a positive-feedback loop. In pancreatic epithelial cells, STAT3 controls transcription of multiple genes that regulate proliferation and apoptosis, including B-cell lymphoma-extra large, induced myeloid leukemia cell differentiation protein, cyclin D1, survivin, and c-Myc.98 STAT3 deficiency attenuates KrasG12D-induced formation of PanINs.96,97 NF-κB signaling also is activated by oncogenic K-Ras or by cytokines secreted from malignant or tumor-infiltrating immune cells in PDAC.99,100 NF-κB controls production of cytokines, including TNFα and IL-1α, which subsequently activate NF-κB in a positive-feedback loop.99 TNFα-mediated activation of NF-κB can activate Notch, and Notch signaling synergizes with K-Ras to accelerate PDAC development.100 In addition, activated Notch signaling suppresses the anti-inflammatory transcription factor peroxisome proliferator–activated receptor-γ, leading to constitutive production of inflammatory mediators by malignant cells in PDAC.100
Figure 2.

Inflammation in PDAC. Activation of Kras in pancreatic epithelial cells promotes production of inflammatory mediators (including IL-6 and IL-11, which activate STAT3 in an autocrine manner) or recruits myeloid cells that produce IL-6 and sIL-6R to activate STAT3 in a paracrine manner. NF-κB also can be activated in an autocrine or paracrine manner by TNFα and IL-1α, creating a positive-feedback loop to maintain the tumor’s inflammatory microenvironment. Inflammation-induced activation of STAT3 and NF-κB promotes cell survival, proliferation, and the EMT, which contribute to initiation and progression of PDAC. Pancreatic neoplastic cells that express oncogenic Kras also produce the inflammatory cytokine granulocyte-macrophage colony–stimulating factor (GM-CSF), leading to recruitment of myeloid progenitor cells and their subsequent differentiation into MDSCs, which suppress the immune surveillance function of CD8+ T cells. COX2, cyclooxygenase 2; MMP, matrix metalloproteinase.
In another mechanism, inflammation mediates suppression of immunosurveillance, which is a notable feature of PDAC.94 Pancreatic infiltration by immunosuppressive cells such as MDSCs and regulatory T cells dominates the early immune response to low-grade preinvasive lesions, and persists as PDAC becomes invasive, whereas anti–tumor effector T cells are scarce.101 A paracrine immune suppression circuit has been identified in PDAC: K-Ras activation causes malignant cells to secrete the inflammatory cytokine granulocyte-macrophage colony–stimulating factor, which promotes recruitment of Gr1−CD11b− myeloid progenitor cells and induces their differentiation into Gr1+CD11b+ MDSCs.102–104 In turn, MDSCs have immunosuppressive effects on CD8+ killer T cells. The imbalance in protumor and antitumor immune cells, the accumulation of tumorigenic and immunosuppressive MDSCs, and the absence of antitumor CD8+ killer T cells promote PDAC development.102
Inflammation also could inhibit oncogene-induced senescence (OIS). In mice with cerulein-induced pancreatitis, inflammation accelerates KrasG12D-induced formation of PanINs and their progression105; one mechanism is via inhibition of OIS in lower-grade PanINs.106 Treatment of patients with CP with anti-inflammatory drugs causes PanINs to undergo senescence, which could reduce the risk for PDAC development.106
Inflammation promotes metastasis by stimulating the epithelial-mesenchymal transition (EMT). PanIN cell dissemination into the bloodstream, through the EMT and subsequent acquisition of cancer stem cell features, precedes tumor formation.107 The EMT, as well as tumor cell invasiveness and dissemination, are stimulated by inflammation; anti-inflammatory therapy with dexamethasone blocks dissemination.107 Several cytokines, including transforming growth factor-β, IL-1, TNFα, and IL-6, promote the EMT, possibly via NF-κB or STAT3 activation, which induces expression of regulators of the EMT.95,108
Inflammatory stimuli also amplify and prolong Ras activity via an NF-κB–mediated, Ras–NF-κB–cyclooxygenase-2 positive-feedback loop.109 A low level of oncogenic K-Ras activity in adult pancreatic cells does not cause cancer until Ras activity is amplified and prolonged by inflammation.109 Inflammatory stimuli sustain Ras activity, which leads to production of more inflammatory mediators via NF-κB activation. This positive-feedback loop maintains high levels of Ras activity and promotes cancer development.109 Finally, inflammation promotes oncogenic mutagenesis. Inflammatory cells, via cytokines such as TNFα, stimulate ROS accumulation in epithelial cells, which causes DNA damage and genomic instability and thereby facilitates tumorigenesis.95,108
In summary, in pancreatic epithelial cells, activation of oncogenic Kras promotes production of inflammatory mediators, including IL-6, IL-11, TNFα, and IL-1α, which activate STAT3 and/or NF-κB in an autocrine or paracrine manner to promote cell survival and proliferation, and maintain an inflammatory tumor microenvironment. By producing granulocyte-macrophage colony–stimulating factor, oncogenic Kras-expressing neoplastic cells suppress immunosurveillance. Chronic inflammation inhibits OIS, stimulates the EMT, amplifies and prolongs Ras activity, and promotes oncogenic mutagenesis, all of which contribute to PDAC initiation, development, and metastasis (Figure 2).
The Process of Autophagy
Autophagy comprises several intracellular pathways of lysosome-mediated degradation and recycling of organelles, long-lived proteins, and lipids.110,111 Macroautophagy (hereafter referred to as autophagy) begins with the formation of an autophagosome, a unique double-membrane vacuole that sequesters material destined for degradation. Autophagosome formation is a complex process controlled by evolutionary conserved autophagy related (ATG) proteins.111,112 Autophagosomes fuse with lysosomes, generating single-membrane autolysosomes in which cargo is degraded by lysosomal hydrolases and the degradation products are recycled back to the cytoplasm. In the basal state, autophagy acts as a quality-control mechanism to eliminate protein aggregates and damaged organelles (eg, mitochondria). Equally important, autophagy is a protective mechanism that allows cells to adapt to stresses, such as nutrient deprivation (starvation is one of the strongest inducers of normal physiological autophagy). One important aspect of autophagy is its dynamic character110,111,113 Therefore, it is important to assess the flux through this pathway, that is, the rate of turnover of autophagic vacuoles. A number of diseases have been associated with impairments in autophagy.110,114,115 Dysfunction could occur at various steps of the autophagic pathway, including decreased formation of autophagosomes, their impaired fusion with lysosomes, or inefficient lysosomal proteolytic activity.110,115
Impaired Autophagy in Pancreatitis
The role of autophagy in pancreatitis recently was discussed in detail (see article by Gukovskaya and Gukovsky116). Evidence from animal models indicates that autophagy is impaired in pancreatitis, and that one mechanism involves defective functions of lysosomes. A hallmark of impaired autophagy is the accumulation of abnormally large vacuoles. Acinar cell vacuolation is a long-noted but poorly understood feature of experimental and human pancreatitis.117–122 Most of these vacuoles are autophagic–predominantly autolysosomes that contain poorly degraded cargo.123 Compared with starvation, AP induces many more, and strikingly larger, vacuoles in acinar cells. Vacuole accumulation is accompanied by increased pancreatic levels of the autophagy marker Atg8/LC3-II, observed in all models of AP.116,123–126 Pancreatitis decreases autophagic efficiency, as manifest by a reduced rate of degradation of long-lived proteins123 and an increased level of p62 (also called sequestosome 1), a multifunctional protein that mediates autophagic clearance of ubiquitinated protein aggregate. p62 is itself degraded by autophagy.127,128 Together, these data reveal that autophagic flux is impaired in AP.
The accumulation of large autolysosomes with partially degraded cargo further indicates that lysosomal hydrolytic activity is compromised. There is evidence that lysosomal function is impaired in pancreatitis through at least 2 mechanisms that reduce autophagic flux: deficient hydrolytic activity and alterations in lysosome-associated membrane proteins (LAMPs)116,123,125,129 (Mareninova et al, unpublished observations). Lysosomal and autophagic dysfunction have been observed in human beings with pancreatitis, all commonly used models of AP (such as cerulein-induced), and in alcohol- or coxsackievirus-induced pancreatitis.123–125,129–131 Importantly, genetic alterations that specifically disrupt lysosomal or autophagic functions induce pancreatitis-like injury. For example, defects in lysosomes and/or autophagy in LAMP-2 knockout mice cause spontaneous pancreatitis, characterized by vacuole accumulation, progressive pancreatic inflammation, and acinar cell necrosis116,129 (Mareninova et al, unpublished observations). Acinar cell injury also develops in mice with disruption of Gnptab, which encodes N-acetylglucosamine-1-phosphotransferase.116,132 This enzyme mediates the addition of mannose-6-phosphate onto acid hydrolases, a step required for their delivery to the lysosome.
Moreover, loss of lysosomal or autophagic function is involved in the induction of pancreatitis by genetic or molecular manipulations of other, seemingly unrelated, pathways.116 Disruption of Spink-3 (mouse ortholog of serine protease inhibitor Kazal-1, the main endogenous trypsin inhibitor in human beings) impairs autophagy and causes acinar cell vacuolation and, ultimately, pancreas degeneration.133,134 Overexpression or administration of IL-22 ameliorates cerulein-induced pancreatitis through effects on autophagy.64 Pancreas-specific deletion of the inhibitor of κB kinase (IKK)α, a component of the IKK kinase complex responsible for NF-κB activation, results in acinar cell damage that progresses from vacuole accumulation to severe chronic pancreatitis.135 Importantly, the role of IKKα in maintaining pancreatic homeostasis is independent of its kinase activity and unrelated to NF-κB. Instead, the IKKα deficiency impairs the completion of autophagy in acinar cells, with p62 accumulation as the key pathogenic mechanism.135
In addition to acinar cell vacuolation, impaired autophagy also mediates the pathologic, intra-acinar accumulation of trypsin.116,123,126 The intra-acinar conversion of trypsinogen to active trypsin is mediated by cathepsin B (CatB),20,116,123,136,137 and this process is counteracted by CatL, which degrades trypsin and trypsinogen.123,137 Lysosome dysfunction in pancreatitis appears to cause an imbalance between CatB and CatL, so that CatL activity is not sufficient to degrade trypsin, resulting in its accumulation.116,123 An important function of efficient autophagy is to limit inflammation (see later). The impaired autophagy in LAMP2- or Gnptab-deficient mice is associated with persistent inflammation and acinar cell necrosis116,129,132 (Mareninova et al, unpublished observations).
Defects in lysosome function and autophagy therefore appear to be a converging point of multiple disordered pathways that contribute to the development of pancreatitis.116 Studies of autophagy in experimental pancreatitis have been limited to models of AP. However, results from genetic models have indicated a similar role for impaired autophagy (in particular, reduced autophagic flux) in the pathogenesis of CP.
Elucidating the mechanisms that cause lysosomal and autophagic dysfunctions could lead to identification of therapeutic targets for pancreatitis. The challenge will be to normalize these pathways rather than simply stimulate or block autophagy in acinar cells. Because of defects in lysosomal proteolytic activity, stimulating autophagy in pancreatitis could even exacerbate acinar cell vacuolation. This might explain why inhibition of autophagy, via genetic or small interfering RNA–mediated knockdown of Atg5, reduces vacuole accumulation and trypsinogen activation in mice with cerulein-induced pancreatitis and ex vivo models of AP.123,126
Roles for Autophagy in PDAC Development
Autophagy is believed to have different roles during different stages of cancer development (Figure 3), suppressing tumorigenesis but promoting growth of established tumors.138–140 PDAC cells require autophagy to survive stressful conditions, such as hypoxia, nutrient deprivation, metabolic stress, and chemotherapy.138 They have higher basal levels of autophagy than other types of tumor cells, possibly owing to Kras activation.141 However, this makes PDAC cells more sensitive to inhibitors of autophagy such as with chloroquine, which blocks lysosome acidification and autophagosome degradation, or RNA interference to reduce levels of proteins required for autophagy.141 Inhibition of autophagy suppressed growth of pancreatic xenograft tumors in mice and tumor development in KrasG12D mice.141
Figure 3.
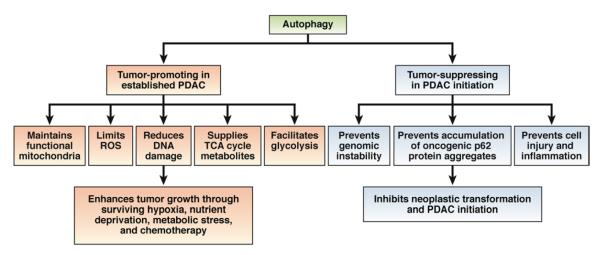
Dual role of autophagy in PDAC development. PDAC cells require efficient autophagy to survive hypoxia, nutrient deprivation, metabolic stress, and exposure to chemotherapeutic agents. Basal autophagy therefore promotes growth of PDACs by maintaining functional mitochondria, reducing ROS and DNA damage, supplying tricarboxylic acid (TCA) cycle metabolites, and facilitating glycolysis. In contrast, autophagy suppresses PDAC initiation: it limits ROS production, genomic damage, cell injury, and inflammation; and eliminates oncogenic aggregates of p62, to prevent transformation of epithelial cells.
Increased basal level of autophagy in PDAC cells prevents ROS accumulation and DNA damage, and maintains energy homeostasis, facilitating their survival and proliferation.142 It also provides PDAC cells with tricarboxylic acid cycle intermediates required for oxidative phosphorylation and maintenance of energy homeostasis.141 Consistent with these observations, Ras-induced autophagy supplies tricarboxylic acid cycle metabolites and maintains the functional mitochondria required for tumor growth.143 KrasG12D increases glucose uptake and glycolysis in fully established PDAC,143 and drives glycolysis flux into ribose biosynthesis and glycosylation in mitogen-activated protein kinase kinase 1– and Myc-dependent manners.144 It is not clear if autophagy has a role in KrasG12D-induced metabolic reprogramming in PDAC cells. It has been reported that autophagy facilitates glycolysis during Ras-induced, adhesion-independent transformation.145 In addition, oncogenic Kras-induced mitophagy (selective autophagic removal of damaged mitochondria) can supply nutrients and expedite glycolysis.146
The tumor microenvironment is hypoxic, and autophagy is induced by hypoxia-inducible factor-α signaling via BCL2/adenovirus E1B 19-kilodalton interacting protein-3, or through the adenosine monophosphate (AMP)-activated protein kinase–the mechanistic target of rapamycin (mTOR) pathway.147,148 Pancreatic cancer stem cells require autophagy to survive hypoxia and starvation and to undergo self-renewal.149 However, in apoptosis-resistant cancer cells, inhibition of autophagy leads to necrosis; the necrotic cells release tumorigenic cytokines and DAMPs, such as HMGB-1 and S100A9, to promote an inflammatory tumor microenvironment.150,151
In nontransformed epithelial cells, autophagy suppresses tumor formation by limiting genomic damage, cell death and inflammation, and by eliminating aggregates of oncogenic proteins. Impaired autophagy causes accumulation of protein aggregates and damaged organelles, including mitochondria, which is associated with increased production of ROS.110 ROS, in turn, can damage proteins, organelles, and the genome to promote tumorigenesis (Figure 3). Compared with high-grade PanINs, lower-grade PanINs and normal exocrine pancreas tissues have lower levels of autophagy, which could allow for more genomic instability and facilitate PDAC initiation.141 Genomic damage has been detected in low-grade PanINs in patients with CP.152
Long-term inhibition of autophagy induces cell death via apoptosis or necrosis, which promotes inflammation and tumorigenesis.151 For example, autophagy defects in liver result in tissue damage, inflammation, and formation of hepatic adenomas.153,154 As discussed earlier, impaired autophagy and lysosomal dysfunction in exocrine pancreas contribute to the pathogenesis of pancreatitis.116 Chronic pancreatic injury caused by defects in autophagy can activate compensatory proliferation of stem cells, leading to ductal metaplasia and a regenerative response that contributes to tumorigenesis. Multiple signaling pathways, including Notch, Hedgehog, and Wnt–β-catenin, which maintain stem cell homeostasis, are activated during the regenerative response in CP tissues.155 Dysregulation of such pathways has been linked to pancreatic tumorigenesis.
Normal, efficient autophagy maintains a low level of p62/sequestosome 1, whereas defects in autophagy result in accumulation of p62 and p62-positive aggregates of ubiquitinated proteins.156 p62 accumulation leads to stabilization and nuclear translocation of the transcription factor Nrf2, by scavenging the Nrf2 inhibitor Keap1. Nrf2 activation induces transcription of antioxidant defense genes to protect cells from oxidative stress.156,157 Persistent Nrf2 activation has been associated with pancreatic tumorigenesis.158 Persistent Nrf2 activation in mice with liver-specific disruption of Atg7 contributes to liver tumor development,153 which is blocked by p62 deletion.156 In addition to Nrf2 activation, p62 accumulation has been associated with other oncogenic pathways, such as those that involve NF-κB and mTOR.128
Because autophagy can suppress tumorigenesis yet maintain tumor growth (Figure 3), it would be interesting to disrupt autophagy in vivo in a time-dependent manner and to investigate its role in PDAC initiation, progression, and metastasis. It should be noted that the mechanisms that regulate autophagy are cell type- or context-specific. It is also important to remember that the cytotoxic response caused by defective autophagy can either eradicate cancer cells or lead to genomic instability, oxidative stress, and other alterations that promote tumorigenesis. The balance between cytotoxicity caused by defective autophagy and p62 accumulation-induced Nrf2 activation, which protects cells, could determine cell fate. Only when cells survive the cytotoxic effects of defective autophagy, ROS accumulation, and genomic instability can they initiate formation of PDAC.
Defects in Autophagy Promote Inflammation
Autophagy limits inflammation whereas defects in autophagy could mediate the pathogenesis of inflammatory diseases.110,159 Defective autophagy can promote inflammation through several mechanisms (Figure 4), in particular by activating specific transcriptional responses. For example, p62 accumulation in autophagy-deficient cells leads to TRAF6 oligomerization and NF-κB activation.99,128 Importantly, the combined activation of NF-κB by Ras and p62 promotes pancreatic tumorigenesis.99
Figure 4.
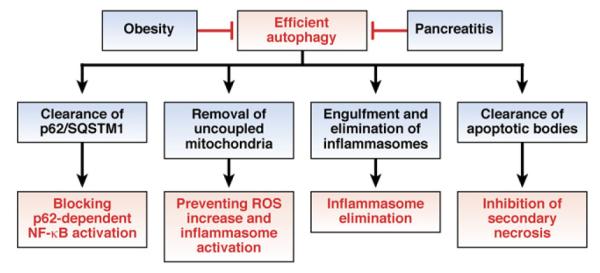
Autophagy limits inflammation. Pancreatitis and obesity each reduce the efficacy of, or lead to defects in, autophagy. Reduced autophagy promotes inflammation and tumorigenesis in patients with pancreatitis or obesity.
Leaky or depolarized mitochondria are removed continuously by mitophagy.160 Failure of defective autophagy to remove damaged mitochondria could increase levels of ROS, which is required for inflammasome (ie, NLRP3) activation.2,6,160 Inflammasomes recently were shown to be engulfed by autophagosomes and eliminated through autophagy. Furthermore, the induction of NLRP3-containing inflammasomes in macrophages induces formation of autophagosomes.161 Inflammasome activation therefore creates a negative-feedback loop by activating autophagy, which controls inflammation by eliminating active inflammasomes. This loop is disrupted if autophagy is impaired.
Efficient autophagy also limits tissue inflammation by clearing apoptotic material. The efficient clearance of apoptotic bodies prevents secondary necrosis and the release of DAMPs, which induce inflammation. Autophagy proteins, such as ATG5, are required for clearance of apoptotic cells.110
Obesity Predisposes to Pancreatic Disorders by Affecting Inflammation and Autophagy
The obesity epidemic is a growing threat to human health, worldwide.162,163 It is the most common cause of insulin resistance and greatly increases the risk for cardio-vascular disease, stroke, and some types of cancer. Obesity increases susceptibility to these diseases by causing low-grade chronic inflammation.162–165 The links between obesity and inflammation are not fully understood, but several mechanisms have been proposed. The expansion of fat mass, caused by adipocyte enlargement, fuels infiltration of adipose tissue by macrophages,162–165 which shift toward the inflammatory M1-type. Obesity reduces the number of T-regulatory cells and increases the number of CD8+ T cells, which promotes macrophage recruitment.165 The levels of TNFα, IL-1β, IL-6, and IL-18 increase within adipose tissue and systemically, such as through inflammasome activation in macrophages.166 The inflammatory mediators secreted by macrophages act not only locally, in a paracrine manner, but also contribute to general inflammation. Obesity increases levels of the proinflammatory hormone leptin and decreases its anti-inflammatory counterpart, adiponectin.
Normal, efficient autophagy limits inflammation (Figure 4). Obesity, or a high-fat diet (HFD), inhibits autophagy to promote inflammation. Obesity inhibits autophagy by activating anti-autophagic Akt and mTOR signaling pathways and down-regulating autophagic genes such as Ulk1/Atg1, Atg5, Atg7, and Atg6/Beclin1. Excess nutrients lead to accumulation of misfolded proteins in the endoplasmic reticulum (ER), causing ER stress.162,163 The association between ER stress and chronic inflammation has been increasingly recognized.167 Moreover, loss of autophagy greatly promotes ER stress induced by obesity and causes insulin resistance.168
Defects in autophagy can lead to accumulation of damaged mitochondria. The HFD causes mitochondrial damage, increases ROS production by mitochondria, shifts the cellular redox environment to a more oxidized state, and decreases redox-buffering capacity, as shown in rodent and human skeletal muscle.169 In turn, increases in ROS activate the inflammasome.6
Obesity is associated with increased risk and severity of pancreatitis.170,171 In mice, obesity caused by deletion of leptin (ob/ob) or leptin receptor (db/db), or by a HFD, increases the severity of pancreatitis.170–174 Studies have only begun to elucidate the mechanisms by which obesity increases the severity of pancreatitis, which likely include up-regulation of the inflammatory response. Pancreatic levels of IL-1β, IL-6, CCL2/MCP-1, and neutrophil infiltration in pancreas and lung are all greater in ob/ob and db/db mice with cerulein-induced pancreatitis, compared with their lean littermates.174 The combination of IL-18 and IL-12 induces severe AP in ob/ob but not wild-type mice; levels of these cytokines are increased in patients with pancreatitis, and levels of IL-18 are increased in obese patients.173 Increases in TNFα and reductions in IL-10 are associated with necrosis of adipose tissue in taurocholate-induced pancreatitis.175 Unsaturated fatty acids, generated from the breakdown of excess intrapancreatic fat, contribute to inflammation, parenchymal necrosis, multi-organ failure, and mortality in AP associated with obesity. Inhibition of lipolysis reduces these adverse outcomes.171
Based on epidemiologic and cohort studies, obesity and HFDs are risk factors for pancreatic cancer.176 Although the mechanisms of this association are not fully understood, they appear to involve inflammation and mitochondrial dysfunction. The stromal–vascular fraction of adipose tissue from obese individuals and the pancreatic tumor stroma each contain a large number of inflammatory cells, including macrophages and T cells, which produce many inflammatory mediators, including TNFα, IL-1β, and IL-6.163,177,178 Moreover, obese subjects and patients with pancreatic cancer have increased blood levels of TNFα and IL-6.163,179,180 In the p48Cre-K-rasLSL-G12D/+ (p48-Kras) mouse model of pancreatic cancer, HFD-induced obesity increases circulating levels of TNFα and IL-6 and infiltration of the pancreas with inflammatory cells, and also accelerates development of PanINs.181 Blocking TNF signaling by crossing p48-Kras and TNF-receptor-1–deficient mice significantly reduces the development of PanINs in mice fed HFDs, indicating a role for TNFα in pancreatic tumorigenesis.181
Interestingly, because of alterations in energy metabolism, p48-Kras mice fed HFDs remain insulin-sensitive, despite chronic inflammation. This indicates that TNFα could promote tumorigenesis independently of insulin resistance, which was thought to be a causal factor. In addition, p48-Kras mice on an HFD have increased mitochondrial β-oxidation of fatty acids, an important energy source for cancer cells. Increased β-oxidation of fatty acids compensates for the negative energy balance, a consequence of pancreatic exocrine insufficiency that results from chronic inflammation and fibrosis.
Mechanisms that link obesity and inflammation in other organs include activation of the IL-6–STAT3 pathway or NLRP3 inflammasome.136,182,183 Obesity-induced, IL-6–dependent activation of STAT3 promotes hepato-carcinogenesis in mice.184 STAT3 also has been shown to accelerate the development of PDAC precursors.96,97 STAT3 controls expression of matrix metalloproteinase-7, which is up-regulated in PanINs and human PDACs and could be involved in PDAC progression.185,186 Increased release of inflammatory mediators by fat tissue in obesity not only induces insulin resistance in adipocytes via paracrine effects but also decreases insulin sensitivity in other tissues, including liver and skeletal muscle.163 The resulting hyperinsulinemia induces the synthesis of insulin-like growth factor 1; both factors promote cell proliferation in vitro and tumor growth in animal models.187 This process is believed to involve activation of phosphatidylinositol 3-kinase–Akt–target of rapamycin complex 1 signaling, which controls proliferation and survival of PDAC cells.188,189
Levels of leptin increase, and those of adiponectin decrease, with increasing adiposity.190 Leptin is associated with tumor development.191 For example, leptin induces production of IL-6 by colon epithelial cells and is involved in early stages of tumorigenesis.192 It is correlated positively with cancer risk and can promote PDAC growth by activating phosphatidylinositol 3-kinase–Akt and JAK–STAT3 signaling. In contrast, plasma levels of adiponectin are associated inversely with pancreatic cancer progression.193 Adiponectin attenuates insulin–insulin-like growth factor 1 signaling, down-regulates TNF and IL-6, and activates AMP-activated protein kinase, a negative regulator of TORC.194,195
Epigenetic alterations also could contribute to PDAC development.196 In particular, the micro RNA (miR)-200 family members inhibit the EMT and could slow progression of PDAC.197,198 These miRs also have anti-inflammatory properties, suppressing signaling pathways that lead to NF-κB activation and production of TNFα and IL-6.199 Levels of other miRs, including miR-221, are increased in primary PDACs and pancreatic cancer cell lines.196,198 Interestingly, miR-200 family members are down-regulated, whereas miR-221 is up-regulated, in adipose tissue of obese mice.200
Histone and DNA methylation also have been implicated in pancreatic carcinogenesis.196 For example, these processes down-regulate cyclin-dependent kinase inhibitor/p16 to promote PDAC development. Regulatory links among obesity, inflammation, and the epigenetic mechanisms in pancreatic cancer pathogenesis remain to be elucidated.
Summary and Future Directions
Inflammation and defective autophagy are each involved in the pathogenesis of pancreatitis and pancreatic cancer, and mediate the effects of obesity in promoting exocrine pancreatic diseases. The severity of AP is largely determined by whether the inflammatory response resolves or amplifies into persistent SIRS and multiple organ failure. Chronic low-grade inflammation promotes fibrosis and cell death during progression of CP.
Although our knowledge of the mechanisms of inflammation in pancreatitis has increased significantly, we have much to learn about how inflammation is initiated (including pathways of inflammasome activation and the identity and involvement of DAMPs), the activities of cytokines (such as IL-6) in different stages of the inflammatory response, the role of anti-inflammatory factors (such as IL-22), the links between acute and chronic inflammation, and, importantly, the mechanisms that prevent its resolution (including the involvement of proresolving lipid mediators).
Chronic (in either CP or obesity) and tumor-associated inflammation each create an environment that promotes, via multiple mechanisms, PDAC development. Much is known about the roles of the IL-6–STAT3 and NF-κB pathways in cell survival and proliferation, whereas little is known about how inflammation stimulates the EMT and inhibits OIS. Elucidation of these mechanisms could lead to effective anti-inflammatory agents, which could block progression of premalignant lesions to PDAC.
Accumulating evidence indicates that defects in autophagy mediate many pathologic effects of AP, and findings from genetic models indicate that they also contribute to the development of CP. Research into the roles of autophagy in pancreatic diseases only recently has begun. Important areas of study include determining the mechanisms by which autophagy is impaired (such as via lysosomal dysfunction); identifying links among autophagy, inflammation, and parenchymal cell death; and determining the role of autophagy in the pathogenesis of CP.
Autophagy has a complex role in the development of PDAC, promoting growth of established tumors but suppressing early stages of tumorigenesis. The exact pathways that control the dual roles of autophagy in the pathogenesis of PDAC, and whether autophagy is efficient or defective, remain to be elucidated. Important mechanisms that could link aberrant autophagy to inflammation in pancreatitis and PDAC include accumulation of p62 and mitochondrial dysfunction, resulting in increased levels of ROS.
Obesity increases the severity of pancreatitis and facilitates progression of PDAC. Although the mechanisms of this process are not well understood, they likely involve chronic inflammation, mediated by molecules such as TNFα and IL-6, feedback loops associated with the obese state, and disruptions in autophagy that promote ER stress and mitochondrial dysfunction. Integrated studies of the mechanisms of inflammation, autophagy, and obesity could lead to new therapeutic targets and effective treatments for pancreatitis and pancreatic cancer.
Supplementary Material
Acknowledgments
The authors apologize to many authors whose studies could not be cited because of space limitations.
Funding Our research was supported by the Department of Veterans Affairs and National Institutes of Health grants DK59936, and, in part, P01 CA163200 (A.G.), AA19730 (I.G.), AI043477 (M.K.), CA163798 (M.K.), P50 AA011999 (A.G, I.G, Pilot Project to M.K.), and R03CA167120 (N.L.), by a Pancreatic Cancer Research Lustgarten Foundation grant RFP-B-007 (M.K.), and the Erwin Schrödinger Fellowship J 3233 of the Austrian Science Fund (J.T.).
Abbreviations used in this paper
- AMP
adenosine monophosphate
- AP
acute pancreatitis
- ATG
autophagy related
- Cat
cathepsin
- CP
chronic pancreatitis
- DAMP
damage/danger-associated molecular pattern molecule
- EMT
epithelial-mesenchymal transition
- ER
endoplasmic reticulum
- HFD
high-fat diet
- IKK
inhibitor of κB kinase
- IL
interleukin
- LAMP
lysosome-associated membrane protein
- MDSC
myeloid-derived suppressor cells
- miR
microRNA
- mTOR
mechanistic target of rapamycin
- NFAT
nuclear factor of activated T cells
- NF-κB
nuclear factor-κB
- NLR
nucleotide binding domain leucine-rich repeat
- OIS
oncogene-induced senescence
- PanIN
pancreatic neoplastic epithelial
- PDAC
pancreatic ductal adenocarcinoma
- ROS
reactive oxygen species
- sIL
soluble interleukin
- SIRS
systemic inflammatory response syndrome
- STAT
signal transducer and activator of transcription
- TNF
tumor necrosis factor
Biographies





Footnotes
Supplementary Material Note: The first 50 references associated with this article are available below in print. The remaining references accompanying this article are available online only with the electronic version of this article. To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://dx.doi.org/10.1053/j.gastro.2013.02.007.
References
- 1.Mantovani A, Cassatella MA, Costantini C, et al. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 4.Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumgart S, Ellenrieder V, Fernandez-Zapico ME. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut. 2013;62:310–316. doi: 10.1136/gutjnl-2011-301008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strowig T, Henao-Mejia J, Elinav E, et al. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 7.Hoque R, Malik AF, Gorelick F, et al. Sterile inflammatory response in acute pancreatitis. Pancreas. 2012;41:353–357. doi: 10.1097/MPA.0b013e3182321500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buter A, Imrie CW, Carter CR, et al. Dynamic nature of early organ dysfunction determines outcome in acute pancreatitis. Br J Surg. 2002;89:298–302. doi: 10.1046/j.0007-1323.2001.02025.x. [DOI] [PubMed] [Google Scholar]
- 9.Mofidi R, Duff MD, Wigmore SJ, et al. Association between early systemic inflammatory response, severity of multiorgan dysfunction and death in acute pancreatitis. Br J Surg. 2006;93:738–744. doi: 10.1002/bjs.5290. [DOI] [PubMed] [Google Scholar]
- 10.Pandol SJ, Saluja AK, Imrie CW, et al. Acute pancreatitis: bench to the bedside. [erratum appears in Gastroenterology 2007;133: 1056] Gastroenterology. 2007;132:1127–1151. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 11.Singh VK, Wu BU, Bollen TL, et al. Early systemic inflammatory response syndrome is associated with severe acute pancreatitis. Clin Gastroenterol Hepatol. 2009;7:1247–1251. doi: 10.1016/j.cgh.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Leser HG, Gross V, Scheibenbogen C, et al. Elevation of serum interleukin-6 concentration precedes acute-phase response and reflects severity in acute pancreatitis. Gastroenterology. 1991;101:782–785. doi: 10.1016/0016-5085(91)90539-w. [DOI] [PubMed] [Google Scholar]
- 13.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- 14.Gukovskaya AS, Gukovsky I, Zaninovic V, et al. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grady T, Liang P, Ernst SA, et al. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology. 1997;113:1966–1975. doi: 10.1016/s0016-5085(97)70017-9. [DOI] [PubMed] [Google Scholar]
- 16.Orlichenko LS, Behari J, Yeh TH, et al. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299:G867–G876. doi: 10.1152/ajpgi.00177.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 1998;275:G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 18.Vaquero E, Gukovsky I, Zaninovic V, et al. Localized pancreatic NF-kappaB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1197–G1208. doi: 10.1152/ajpgi.2001.280.6.G1197. [DOI] [PubMed] [Google Scholar]
- 19.Bhatia M, Brady M, Shokuhi S, et al. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;190:117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.Saluja AK, Lerch MM, Phillips PA, et al. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–269. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 21.Saeki K, Kanai T, Nakano M, et al. CCL2-induced migration and SOCS3-mediated activation of macrophages are involved in cerulein-induced pancreatitis in mice. Gastroenterology. 2012;142:1010–1020.e9. doi: 10.1053/j.gastro.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 22.Perides G, Weiss ER, Michael ES, et al. TNF-alpha-dependent regulation of acute pancreatitis severity by Ly-6C(hi) monocytes in mice. J Biol Chem. 2011;286:13327–13335. doi: 10.1074/jbc.M111.218388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bedrosian AS, Nguyen AH, Hackman M, et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology. 2011;141:1915–1926.e1-14. doi: 10.1053/j.gastro.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoque R, Sohail M, Malik A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358–369. doi: 10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demols A, Le Moine O, Desalle F, et al. CD4(+)T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. 2000;118:582–590. doi: 10.1016/s0016-5085(00)70265-4. [DOI] [PubMed] [Google Scholar]
- 26.Vonlaufen A, Apte MV, Imhof BA, et al. The role of inflammatory and parenchymal cells in acute pancreatitis. J Pathol. 2007;213:239–248. doi: 10.1002/path.2231. [DOI] [PubMed] [Google Scholar]
- 27.Shanmugam MK, Bhatia M. The role of pro-inflammatory molecules and pharmacological agents in acute pancreatitis and sepsis. Inflamm Allergy Drug Targets. 2010;9:20–31. doi: 10.2174/187152810791292881. [DOI] [PubMed] [Google Scholar]
- 28.Frossard JL, Morel P, Pastor CM. Why clinical trials might succeed in acute pancreatitis when they failed in septic shock. JOP. 2003;4:11–16. [PubMed] [Google Scholar]
- 29.Aoun E, Chen J, Reighard D, et al. Diagnostic accuracy of interleukin-6 and interleukin-8 in predicting severe acute pancreatitis: a meta-analysis. Pancreatology. 2009;9:777–785. doi: 10.1159/000214191. [DOI] [PubMed] [Google Scholar]
- 30.Dambrauskas Z, Giese N, Gulbinas A, et al. Different profiles of cytokine expression during mild and severe acute pancreatitis. World J Gastroenterol. 2010;16:1845–1853. doi: 10.3748/wjg.v16.i15.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinle AU, Weidenbach H, Wagner M, et al. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology. 1999;116:420–430. doi: 10.1016/s0016-5085(99)70140-x. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Ji B, Han B, et al. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122:448–457. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 33.Altavilla D, Famulari C, Passaniti M, et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab Invest. 2003;83:1723–1732. doi: 10.1097/01.lab.0000101734.82054.be. [DOI] [PubMed] [Google Scholar]
- 34.Rakonczay Z, Jr, Hegyi P, Takacs T, et al. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57:259–267. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 35.Neuhofer P, Liang S, Einwachter H, et al. Deletion of IkappaBalpha activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A. Gastroenterology. 2013;144:192–201. doi: 10.1053/j.gastro.2012.09.058. [DOI] [PubMed] [Google Scholar]
- 36.Huang H, Liu Y, Daniluk J, et al. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology. 2013;144:202–210. doi: 10.1053/j.gastro.2012.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gukovsky I, Gukovskaya A. Nuclear factor-kappaB in pancreatitis: jack-of-all-trades, but which one is more important? Gastroenterology. 2013;144:26–29. doi: 10.1053/j.gastro.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awla D, Zetterqvist AV, Abdulla A, et al. NFATc3 regulates trypsinogen activation, neutrophil recruitment, and tissue damage in acute pancreatitis in mice. Gastroenterology. 2012;143:1352–1360. doi: 10.1053/j.gastro.2012.07.098. [DOI] [PubMed] [Google Scholar]
- 39.Han B, Logsdon CD. CCK stimulates mob-1 expression and NF-kappaB activation via protein kinase C and intracellular Ca(2+) Am J Physiol Cell Physiol. 2000;278:C344–C351. doi: 10.1152/ajpcell.2000.278.2.C344. [DOI] [PubMed] [Google Scholar]
- 40.Tando Y, Algul H, Wagner M, et al. Caerulein-induced NF-kappaB/Rel activation requires both Ca2+ and protein kinase C as messengers. Am J Physiol Gastrointest Liver Physiol. 1999;277:G678–G686. doi: 10.1152/ajpgi.1999.277.3.G678. [DOI] [PubMed] [Google Scholar]
- 41.Satoh A, Gukovskaya AS, Nieto JM, et al. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 42.Satoh A, Gukovskaya AS, Reeve JR, Jr, et al. Ethanol sensitizes NF-kappaB activation in pancreatic acinar cells through effects on protein kinase C-epsilon. Am J Physiol Gastrointest Liver Physiol. 2006;291:G432–G438. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- 43.Thrower EC, Yuan J, Usmani A, et al. A novel protein kinase D inhibitor attenuates early events of experimental pancreatitis in isolated rat acini. Am J Physiol Gastrointest Liver Physiol. 2011;300:G120–G129. doi: 10.1152/ajpgi.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharif R, Dawra R, Wasiluk K, et al. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. 2009;58:813–819. doi: 10.1136/gut.2008.170423. [DOI] [PubMed] [Google Scholar]
- 45.Awla D, Abdulla A, Regner S, et al. TLR4 but not TLR2 regulates inflammation and tissue damage in acute pancreatitis induced by retrograde infusion of taurocholate. Inflamm Res. 2011;60:1093–1098. doi: 10.1007/s00011-011-0370-1. [DOI] [PubMed] [Google Scholar]
- 46.Denham W, Yang J, Fink G, et al. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology. 1997;113:1741–1746. doi: 10.1053/gast.1997.v113.pm9352880. [DOI] [PubMed] [Google Scholar]
- 47.Frossard JL, Saluja A, Bhagat L, et al. The role of intercellular adhesion molecule 1 and neutrophils in acute pancreatitis and pancreatitis-associated lung injury. Gastroenterology. 1999;116:694–701. doi: 10.1016/s0016-5085(99)70192-7. [DOI] [PubMed] [Google Scholar]
- 48.Sendler M, Dummer A, Weiss FU, et al. Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut. 2013;62:430–439. doi: 10.1136/gutjnl-2011-300771. [DOI] [PubMed] [Google Scholar]
- 49.Awla D, Abdulla A, Syk I, et al. Neutrophil-derived matrix metalloproteinase-9 is a potent activator of trypsinogen in acinar cells in acute pancreatitis. J Leukoc Biol. 2012;91:711–719. doi: 10.1189/jlb.0811443. [DOI] [PubMed] [Google Scholar]
- 50.Frossard JL, Lenglet S, Montecucco F, et al. Role of CCL-2, CCR-2 and CCR-4 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. J Clin Pathol. 2011;64:387–393. doi: 10.1136/jcp.2010.088500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.