

Abstract
Anergy is a key physiological mechanism for restraining self-reactive B cells. A marked portion of peripheral B cells are anergic B cells that largely depend on B cell-activating factor (BAFF) for survival. BAFF activates the canonical and noncanonical NF-κB pathways, both of which are required for B cell survival. Here we report that deficiency of the adaptor protein B cell lymphoma 10 (Bcl10) impaired the ability of BAFF to support B cell survival in vitro, and specifically increased apoptosis in anergic B cells in vivo, dramatically reducing anergic B cells in mice. Bcl10-dependent survival of self-reactive anergic B cells was confirmed in the IgHELsHEL double-transgenic mouse model of B cell anergy. Further, we found that BAFF stimulation induced Bcl10 association with IKKβ, a key component of the canonical NF-κB pathway. Consistently, Bcl10-deficient B cells were impaired in BAFF-induced IκBα phosphorylation and formation of nuclear p50:c-Rel complexes. Bcl10-deficient B cells also displayed reduced expression of NF-κB2/p100, severely reducing BAFF-induced nuclear accumulation of noncanonical p52:RelB complexes. Consequently, Bcl10-deficient B cells failed to express Bcl-xL, a BAFF-induced NF-κB target gene. Taken together, these data demonstrate that Bcl10 controls BAFF-induced canonical NF-κB activation directly and noncanonical NF-κB activation indirectly. The BAFF-R/Bcl10/NF-κB signaling axis plays a critical role in peripheral B cell tolerance by regulating the survival of self-reactive anergic B cells.
Keywords: BAFF, Bcl10, NF-κB, apoptosis, anergic B cells, tolerance
Newly produced B cells, 50-75% of which are estimated to be self-reactive B cells (1, 2), undergo negative selection by three distinct mechanisms in the bone marrow, receptor editing, clonal deletion and anergy (3-8). The signaling threshold of a self-reactive B cell receptor (BCR) is long thought to determine the negative selection mechanism. A highly self-reactive BCR initiates new rearrangements of V genes to change receptor specificity by receptor editing (9-13), or drives B cells to undergo apoptosis and be eliminated by clonal deletion (14, 15). A moderately self-reactive BCR drives B cells to become unresponsive to antigens, a state termed anergy (16, 17). After emerging from the bone marrow, transitional B cells undergo further maturation and selection in the periphery. Highly self-reactive B cells are removed by clonal deletion whereas moderately self-reactive B cells become anergic (11, 18). As much as 5-10% of peripheral B cells are anergic, and given the fast turnover rate of these cells, anergy likely represents a major physiological mechanism for establishing immune tolerance (19). In the periphery, although BCR signaling is required for the maintenance of normal and self-reactive anergic B cells (20, 21), TNF family member B cell activating factor (BAFF) is essential for the survival of these B cells (22-24). Importantly, self-reactive anergic B cells have increased dependence on BAFF for survival compared to normal B cells (24, 25). Excess BAFF mainly rescues self-reactive anergic B cells from competitive elimination (24, 25). Thus, in addition to BCR, BAFF plays a crucial role in sustaining the survival of anergic B cells in the periphery.
BAFF supports the survival of B cells through its cognate receptor. Although BAFF binds to three receptors, BAFF receptor (BAFF-R), B cell maturation antigen (BCMA), and transmembrane activator and cyclophilin ligand interactor (TACI), BAFF-R plays the predominate role in BAFF function (22, 23). Deficiency of BAFF-R impairs the survival of peripheral B cells in a same way as deficiency of BAFF, resulting in nearly complete loss of peripheral B cells (22, 26, 27). In contrast, deficiency of BCMA impairs the survival of long-lived plasma cells (28) whereas lack of TACI leads to a two- to three-fold increase in mature B cell number (29).
One important signaling event of BAFF-R that leads to B cell survival is the NF-κB-dependent up-regulation of Bcl-2 family pro-survival members (22, 30). NF-κB/Rel family of transcription factors consists of five members, RelA/p65, cRel, RelB, NF-κB1 (p105/p50) and NF-κB2 (p100/p52), which can form homo- and heterodimers (31, 32). NF-κB dimers exist as inactive complexes bound to the inhibitor of NF-κB (IκB), such as IκBα or IκBβ, in resting cells (33). There are two major signaling pathways that mediate the activation of NF-κB, known as the canonical and noncanonical pathways (34). It is known that BAFF-R is able to induce NF-κB activation through both pathways (35-40).
Upon BAFF binding, BAFF-R induces a cellular inhibitor of apoptosis 1/2 (cIAP1/2) and TNF receptor-associated factor 2 (TRAF2) ubiquitin ligase complex-dependent degradation of TRAF3, preventing NF-κB-inducing kinase (NIK) degradation and resulting in the subsequent activation of the noncanonical NF-κB pathway (37, 38). NIK activates IκB kinase α (IKKα) and activated IKKα phosphorylates p100, leading to processing of NF-κB2 from a p100 precursor to a p52 product. Then p52 dimerizes with RelB to form the p52:RelB active heterodimer that translocates to the nucleus and regulates gene expression (35, 36). In addition, BAFF-R can activate NF-κB through the canonical pathway that involves the activation of the IKK complex consisting of IKKα, IKKβ and NEMO (39, 40). Activated IKK complex induces the phosphorylation and subsequent degradation of IκBs. Removal of IκBs results in the nuclear translocation of p50-containing NF-κB heterodimers and subsequent initiation of gene transcription. Despite progress in this area of research, the mechanisms by which BAFF-R mediates the activation of NF-κB in both the canonical and noncanonical pathways are not fully understood.
Bcl10 is an adaptor protein characterized by an N-terminal caspase recruitment domain (CARD) and a C-terminal Ser/Thr-rich region (41-45). Bcl10 is essential for BCR-mediated NF-κB activation (46, 47). Bcl10, along with CARD-membrane-associated guanylate kinase (MAGUK) protein 1 (CARMA1) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), forms a three-component complex that couples BCR-induced protein kinase C (PKC) activity to IKK complex activation (48-51). Bcl10 deficiency impairs BCR-induced NF-κB activation (47). In addition, Bcl10 plays a critical role in T cell receptor (TCR), FcεR and Toll-like receptor 4 (TLR4)-induced NF-κB activation and its deficiency impairs NF-κB activation by these receptors (46). Although Bcl10 plays a critical role in NF-κB activation by multiple receptors, its role in BAFF-R-mediated NF-κB activation is not known.
Our current studies find that Bcl10 directly controls BAFF-mediated canonical NF-κB activation, and induces the expression of NF-κB2/p100, thus indirectly regulating BAFF-mediated noncanonical NF-κB activation. This BAFF-R/Bcl10/NF-κB signaling axis specifically supports the survival of self-reactive anergic B cells in vivo. Impairment of this axis by Bcl10 deficiency alters the peripheral tolerance mechanism for self-reactive B cells, switching from anergy to deletion.
Materials and Methods
Mice
Bcl10-deficient mice have been previously described (47). Heterozygous Bcl10-deficient mice were bred to generate wild-type control and Bcl10-deficient mice. IgHEL transgenic mice (C57BL/6 MD4) and sHEL transgenic mice (C57BL/6 ML5) were obtained from Jackson Laboratory. These transgenic mice were bred with heterozygous Bcl10+/- mice to generate wild-type IgHEL, Bcl10-deficient IgHEL, wild-type IgHELsHEL, and Bcl10-deficient IgHELsHEL mice. Mice used for the experiment were generally 2-4 months old except where specifically indicated. All mouse procedures were approved by the Institutional Animal Care and Use Committee (IACUC).
Flow cytometry
Single-cell suspensions from the spleen of the experimental and control mice were treated with Gey’s solution to lyse red blood cells and resuspended in phosphate-buffered saline (PBS) with 2% fetal bovine serum (FBS). The cells were then stained with a combination of fluorescence-conjugated antibodies. PE-Cy7-conjugated anti-CD19 (25-0193), APC-conjugated anti-IgM (17-5790), FITC-conjugated anti-BAFF-R (11-5943), Cychrome-conjugated anti-B220 (15-0452), PE-conjugated anti-B220 (12-0452), biotin-conjugated anti-AA4.1 (CD93) (13-5892), PE-conjugated anti-CD23 (12-0232), and PECy7-conjugated Streptavidin (25-4317) were purchased from eBioscience. PE-conjugated anti-IgD (1120-09) and FITC-conjugated anti-IgM (1140-02) were purchased from Southern Biotechnology. APC-conjugated Streptavidin (554067) was purchased from BD Pharmingen. Stained cells were analyzed on a BD LSR II cytometer with BD FACSDiva software.
B cell isolation
The splenic CD93+ immature (also known as AA4.1+) and CD93- (AA4.1-) mature B cells from were isolated from wild-type and Bcl10-deficient mice as previously described (52). Briefly, splenic B cells were isolated by negative selection using anti-CD4-, anti-CD8-, and anti-CD11b-coated MACS microbeads (Miltenyi Biotec), and then stained with biotin-conjugated anti-CD93 (eBioscience). Immature B cells (CD93+) were isolated using Streptavidin-conjugated microbeads with the flow through constituting the mature B cells (CD93-). For NF-κB2/p100 mRNA expression studies, mature B cells were also isolated from CD93-depleted splenocytes by positive selection using biotin-conjugated anti-CD23 (eBioscience) antibodies and Streptavidin-conjugated microbeads. Purity of CD93+ and CD93-CD23+ B cells was about 90% and 95%, respectively, as determined by anti-B220 antibody staining followed by flow cytometry.
Immunoprecipitation and Western blotting analysis
Splenic AA4.1- mature B cells (1 × 106) from wild-type and Bcl10-deficient mice were stimulated with BAFF (100 ng/ml) at 37°C for the indicated times and then lysed in lysis buffer (10 mM Tris-HCl pH7.5, 150 mM NaCl, and 1% NP-40, 3 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin). Cell lysates were subjected to Western blotting analysis with the indicated antibodies. For detecting IκB phosphorylation, splenic mature B cells were pre-treated with MG132 for 30 min before BAFF stimulation. For co-immunoprecipitation, wild-type AA4.1- mature B cells (2 × 107) were stimulated with BAFF (200 ng/ml) or anti-IgM (10 μg/ml) for the indicated times. Cell lysates were pre-cleared with Sepharose beads and subsequently incubated with anti-Bcl10 antibody-conjugated Sepharose beads (sc-5273 AC, Santa Cruz) at 4°C overnight. After washing five times with cold lysis buffer, bound proteins were eluted in 2 × Laemmli buffer at 95°C for 5 min and subjected to 10% SDS-PAGE, followed by Western blotting analysis with the indicated antibodies.
Rabbit polyclonal anti-ERK (sc-093), anti-p50 (sc-114), anti-p65 (sc-109), anti-c-Rel (sc-071) and anti-RelB (sc-226), and mouse monoclonal anti-p52 (sc-7386), anti-phospho-ERK (pThr202/pTyr204, sc-7383), anti-Bcl10 (sc-5273) and anti-YY1 (sc-7341) were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-MALT1 (SHEKER: 2427) was purchased from Genentech. Mouse monoclonal anti-phospho–IκBα (Ser32/36, 9246), and rabbit polyclonal anti-phospho-Akt (phospho-Ser473, 9271) and anti-IκBα (9242), and rabbit monoclonal anti-IKKβ (2C8, 2370) were purchased from Cell Signaling Technology. Rabbit polyclonal anti–Bcl-xL (B22630) was purchased from Transduction Laboratories. Hamster monoclonal anti-Bcl2 (554218) was purchased from BD Biosciences. Mouse monoclonal anti-GADPH (MAB374), anti-Bcl-10 (AB16506), and anti-Actin (MAB1501R) were purchased from Chemicon Int, Millipore.
NF-κB gel mobility shift assay
Splenic CD93- mature B cells (1 × 106) from wild-type and Bcl10-deficient mice were stimulated with BAFF (200 ng/ml) for 3 or 16 hours and then lysed in the lysis buffer (20 mM HEPES pH 7.9, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 20% glycerol, 1% NP-40). Cell lysates were incubated with 32P-labeled NF-κB probe (5’-AGTTGAGGGGACTTTCCCAGGC-3’; Santa Cruz Biotechnology) for 15 min at room temperature, resolved on a 4%polyacrylamide gel at 4°C, and exposed to X-ray film. For supershift, cell lysates were incubated with the indicated antibodies for 15 minutes before adding the probes.
Preparation of cytoplasmic and nuclear extracts
Splenic CD93- mature B cells (2 × 106) from wild-type and Bcl10-deficient mice were stimulated with BAFF (100 ng/ml) for 16 hours. The cells were collected and suspended in 100 μl of cold buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.2 mM EDTA, 1 mM DTT, 3 μg/ml aprotinin, 2 μg/ml pepstain, and 1 μg/ml leupeptin). After incubation on ice for 15 min, NP-40 was added to a final concentration of 0.5%. The mixtures were vortexed for 10 seconds and spun at 16,000 g for 30 seconds. The supernatants were collected as the cytoplasmic extracts. The pellets were washed with buffer A once and resuspended in 50 μl of buffer B (20 mM HEPES pH 7.9, 400 mM NaCl, 2 mM EDTA, 1 mM DTT, 3 μg/ml aprotinin, 2 μg/ml pepstain, and 1 μg/ml leupeptin), followed by incubation on ice for 15 minutes. The mixtures were spun at 16,000 g for 5 minutes and the supernatants were collected as the nuclear extracts.
Quantitative RT-PCR
Splenic CD93-CD23+ mature or CD93+ immature B cells from wild-type and Bcl10-deficient mice were stimulated with BAFF (100 ng/ml) for 4 or 16 hours. RNA was extracted from the cells using RNeasy Mini kit (Qiagen) and quantified. Equal amounts of mRNA from were used to generate cDNA using QuantiTect Reverse Transcription kit (Qiagen). Real-time PCR was performed with NF-κB2/p100 primers (Mm00479807), other FAM-labeled probes and TaqMan Universal Master mix using Step One Real-Time PCR System (Applied Biosystems). The relative p100 mRNA fold induction was calculated relative to 18S ribosomal RNA.
PI staining assay
Splenic AA4.1- mature or AA4.1+ immature B cells from wild-type or Bcl10-deficient mice were cultured at a density of 2 × 106 cells/ml with or without BAFF (100 ng/ml) for the indicated times, followed by PI staining and FACS analysis.
BrdU incorporation assay
The in vivo BrdU labeling assay was performed as described (53). In brief, mice were injected intraperitoneally with 1 mg of BrdU (Sigmal-Aldrich) in 0.2 ml PBS at 12-hour intervals for 4 days. Splenocytes from BrdU-treated mice were stained with anti-B220. Cells were then fixed and stained with anti-BrdU according to the manufacturer’s instructions of FITC BrdU Flow Kit (559619, BD Pharmingen). The degree of BrdU-positivity in the gated B cells was analyzed by FACS.
TUNEL assay
Splenocytes were stained with a combination of fluorescence-conjugated antibodies to B220, CD93, CD23, and IgM. Then, cells were fixed, permeabilized and labeled with fluorescein-conjugated dUTP according to the manufacturer’s instructions (In Situ Cell Death Detection Kit; 11-684-795-910, Roche). The degree of TUNEL positivity in the gated B cell subpopulations was analyzed by FACS.
Results
Bcl10 deficiency severely impairs BAFF-mediated B cell survival
All peripheral B cells require signals from both the BCR and BAFF-R for their survival (20-25). Self-reactive anergic B cells display even greater dependence on BAFF-R signaling for their survival (25). In current study, we examined the role of Bcl10 in BAFF-mediated B cell survival. Splenic CD93+ B cells, which contain T1, T2 and An1 anergic subpopulations, and CD93- B cells, which are mature FO and MZ B cells, were isolated from wild-type and Bcl10-deficient mice. The cells were cultured in the absence or presence of BAFF and cell viability was determined at various time points. In the absence of BAFF, both wild-type and Bcl10-deficient CD93+ and CD93- B cells underwent apoptosis over time, although more mutant cells than corresponding wild-type cells died after the initial time point (Fig. 1). As expected, addition of BAFF markedly rescued both wild-type CD93+ and CD93- B cells from apoptosis (Fig. 1). Importantly, CD93+ B cells from Bcl10-deficient mice displayed no survival response to BAFF (Fig. 1, right). Although BAFF increased viability of Bcl10-deficient CD93- B cells, however, this was markedly reduced compared to wild-type CD93- B cells (Fig. 1, left). Thus, BAFF-mediated immature B cell survival is entirely Bcl10-dependent whereas mature B cells only partially depend on Bcl10 for their survival.
Figure 1.
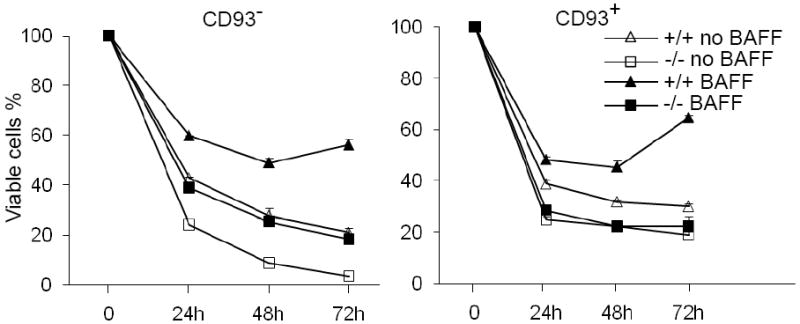
Bcl10 deficiency impairs the survival of peripheral B cells in response to BAFF. CD93+ (AA4.1+) and CD93- (AA4.1-) B cells from wild-type (+/+) and Bcl10-deficient (-/-) mice were cultured in the absence (no BAFF) or presence (BAFF) of BAFF. At the indicated time points, cell survival rates were determined by PI staining. Data are representative of 5 independent experiments.
Bcl10 deficiency causes drastic reduction in anergic B cells
Prior studies have shown that the survival function of BAFF is particularly important for self-reactive anergic B cells (22-25). Based on our finding that Bcl10-deficient B cells survived poorly in response to BAFF, we examined the effects of loss of Bcl10 on the survival of anergic B cell population. In the spleen, B220+CD93- cells are largely mature follicular (FO) B cells (B220+CD93-CD23+IgMlo) whereas B220+CD93+ cells contain T1 (B220+CD93+CD23-IgMhi), T2 (B220+CD93+CD23+IgMhi) and T3-type An1 anergic (B220+CD93+CD23+ IgMlo) populations (19). Previous studies have shown that FO B cells were reduced in the spleens derived from Bcl10-deficient relative to wild-type mice (47). Here we found that T1 B cells were slightly increased and T2 B cells were markedly increased in the spleens derived from Bcl10-deficient mice relative to those from wild-type animals (Fig. 2A-B). In contrast, the percentage and number of splenic An1 anergic B cells (B220+CD93+CD23+IgMlo) was drastically reduced in Bcl10-deficient relative to wild-type mice (Fig. 2A-C). Thus, Bcl10 deficiency results in a drastic reduction in self-reactive anergic B cells.
Figure 2.
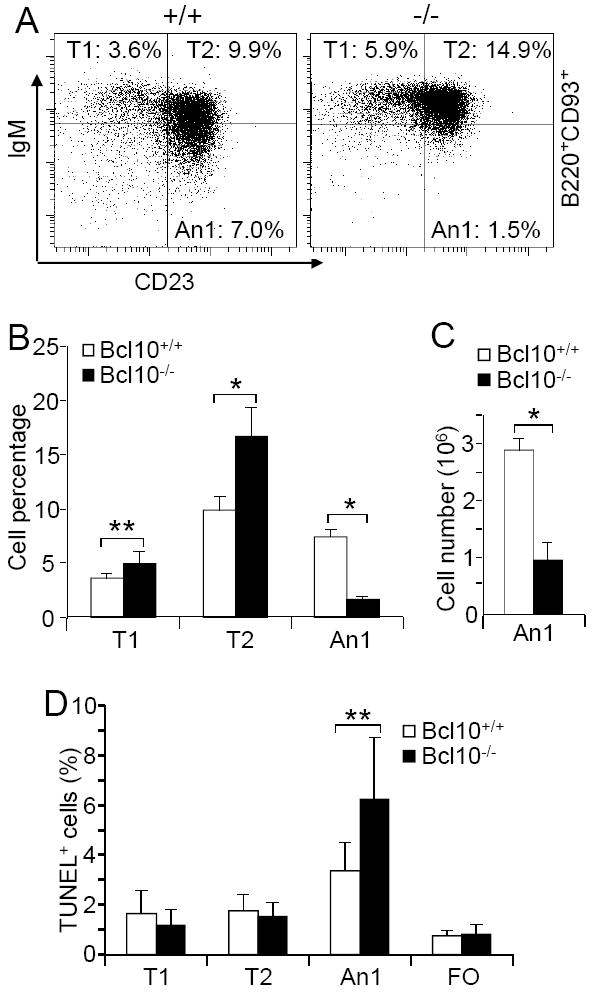
Marked reduction of anergic B cells in Bcl10-deficient mice and increased apoptosis of the mutant anergic B cells. A, Splenic B cell subpopulations. Splenocytes from wild-type and Bcl10-deficient mice were stained with antibodies to B220, CD93, IgM, and CD23. In B220+CD93+-gated cells, T1 (CD23-IgMhi), T2 (CD23+IgMhi) and An1 anergic (CD23+ IgMlo) B cells are shown. Percentages indicate B cells in the gated B220+ population. B, Bar graphs show the percentages of T1, T2 and An1 anergic B cells in the gated B220+ population. C, Bar graphs show the numbers of An1 anergic B cells in the spleens of wild-type and Bcl10-deficient mice. D, Apoptosis of splenic B cell subpopulations. Bar graphs show the degree of TUNEL labeling in T1, T2, An1 anergic and FO B cells of wild-type and Bcl10-deficient mice. Data shown are obtained from at least 7 (A, B and D) or 4 (C) mice in each group. Error bars show ± SD. *, P < 0.01; **, P = 0.02.
Bcl10 deficiency specifically increases anergic B cell apoptosis in vivo
Next, we examined the rate of apoptosis in mutant An1 anergic B cells. We stained splenocytes from wild-type or Bcl10-deficient mice with anti-B220, anti-CD93, anti-IgM and anti-CD23, and then analyzed them for apoptosis by TUNEL assay. Bcl10-deficient An1 anergic B cells had an increased rate of apoptosis relative to that of the corresponding wild-type B cells (Fig. 2D). In contrast, Bcl10-deficient T1, T2, and FO B cells had similarly low rates of apoptosis relative to those of the corresponding wild-type B cell subpopulations as previously reported (Fig. 2D) (47). These data demonstrate that Bcl10 deficiency specifically impairs the survival of An1 anergic B cells in vivo.
Reduced Bcl10-deficient anergic B cells in IgHELsHEL mouse model
To further investigate a role of Bcl10 in BAFF-mediated survival of An1 anergic B cells, Bcl10-deficient mice were crossed with IgHEL transgenic mice, which bear rearranged heavy (H) and Igκ L chain genes encoding a BCR that specifically recognizes hen egg lysozyme (HEL) (16). Soluble HEL (sHEL) induces wild-type IgHEL transgenic B cells to become anergic (16, 19). In the absence of self-antigen sHEL, the spleens from Bcl10-deficient IgHEL transgenic mice displayed a slight reduction of total splenic B cell compared to wild-type controls (Fig. 3A-B). FACS analysis of splenocytes with B220, CD93, IgM and CD23 staining showed that the spleens from both wild-type and Bcl10-deficient IgHEL transgenic mice had a large population of FO mature B cells, small population of T1 B cells, moderate population of T2 B cells, and no An1 anergic B cells (Fig. 3C). As expected, chronic exposure of wild-type IgHEL B cells to sHEL induced self-reactive transgenic B cells into An1 anergic B cells (Fig 3C, 2nd row) and reduced the population of splenic B cells in wild-type IgHELsHEL relative to wild-type IgHEL transgenic mice (Fig. 3A-B). In contrast, in the absence of Bcl10, sHEL drastically reduced splenic B cells, especially An1 anergic B cells, in the spleens of Bcl10-deficient relative to wild-type IgHELsHEL mice (Fig. 3). These results confirm that Bcl10 deficiency results in a drastic reduction of An1 anergic B cells in the well-defined IgHELsHEL transgenic model of B cell anergy.
Figure 3.
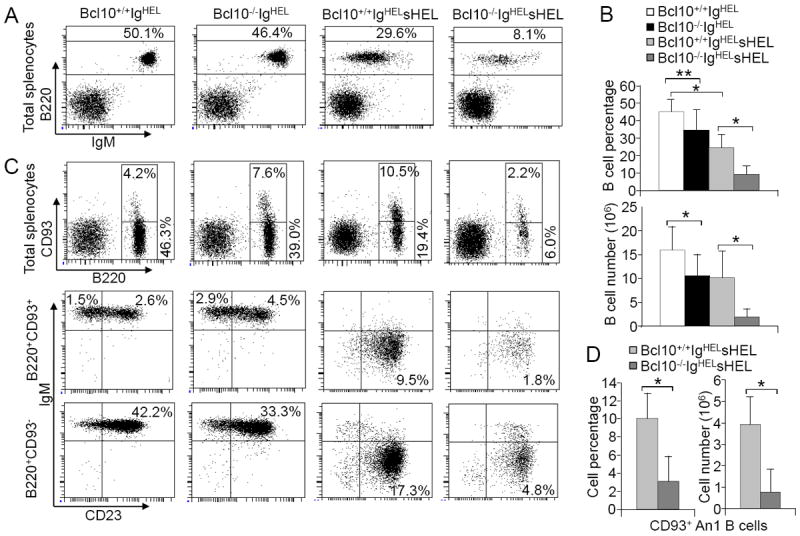
Dramatic reduction of Bcl10-deficient anergic B cells in IgHELsHEL mouse model. Splenocytes from Bcl10+/+IgHEL, Bcl10-/-IgHEL, Bcl10+/+IgHELsHEL and Bcl10-/-IgHEL sHEL transgenic mice were stained with antibodies to B220, IgM, CD93 and CD23. A, FACS analysis with B220 and IgM staining of lymphocytes. Percentages indicate B220+ cells in the gated lymphoid populations. B, Bar graphs show the percentages (upper) and numbers (lower) of total splenic B cells in the spleens of the indicated mice. C, Splenic B cell subpopulations. Upper: FACS analysis with B220 and CD93 staining of lymphocytes. Middle: FACS analysis with IgM and CD23 staining of B220+CD93+-gated cells. Lower: FACS analysis with IgM and CD23 staining of B220+CD93--gated cells. Percentages indicate cells in the gated lymphoid populations. D, Bar graphs show the numbers of CD93+ An1 anergic B cells in the spleens of the indicated mice. Data shown are obtained from 11 (A and B), 3 (C) or 4 (D) mice in each group. Error bars show ± SD. *, P < 0.01; **, P = 0.01.
The severe reduction of the An1 anergic B cells in Bcl10-deficient IgHELsHEL mice led us to examine whether the reduction is due to increased apoptosis of the mutant cells. TUNEL assay demonstrated that the apoptosis rates of splenic B cells were markedly increased in Bcl10-deficient relative to wild-type IgHELsHEL double-transgenic mice (Fig. 4A-B). Of note, the apoptosis rates of splenic B cells from both wild-type and mutant IgHEL single-transgenic mice were equally low (Fig. 4A-B).
Figure 4.
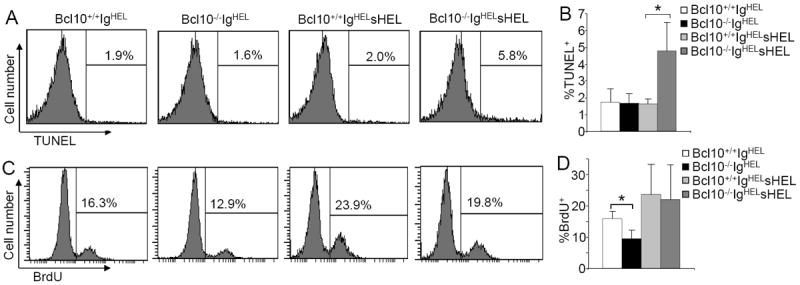
Increased apoptosis and normal proliferation of anergic B cells in Bcl10-deficient IgHELsHEL mice. A, TUNEL labeling of splenic B cells. Splenocytes from Bcl10+/+IgHEL, Bcl10-/-IgHEL, Bcl10+/+IgHELsHEL and Bcl10-/-IgHELsHEL transgenic mice were stained with anti-B220 antibodies. Then, the degree of TUNEL labeling in B220+ cells were determined by FACS analysis. Percentages indicate TUNEL+ cells in the gated B220+ cells. B, Statistical analysis of the percentages of TUNEL+ cells from panel A. C, BrdU incorporation in splenic B cells. BrdU was injected into Bcl10+/+IgHEL, Bcl10-/-IgHEL, Bcl10+/+ IgHELsHEL and Bcl10-/-IgHELsHEL transgenic mice. Splenocytes from the mice were stained with anti-B220 antibodies, followed by BrdU staining. Percentages indicate BrdU+ cells in the gated B220+ cells. D, Statistical analysis of the percentages of BrdU+ cells from panel C. Data shown are obtained from at least 5 (A and B) or 7 (C and D) mice in each group. Error bars show ± SD. *, P < 0.01.
We also examined whether impaired cell proliferation contributes to the drastic reduction of anergic B cells in mutant IgHELsHEL mice. In vivo bromodeoxyuridine (BrdU) labeling assay demonstrated that the BrdU-labeling rates of splenic B cells were reduced in Bcl10-deficient relative to wild-type IgHEL single-transgenic mice (Fig. 4C-D). However, the in vivo BrdU-labeling rates of splenic B cells were comparable between Bcl10-deficient and wild-type IgHELsHEL double-transgenic mice (Fig. 4C-D). Of note, splenic B cells in Bcl10-sufficient and Bcl10-deficient IgHELsHEL double-transgenic mice were mainly anergic (Fig. 3C). Compared to wild-type double-transgenic mice, mutant double-transgenic mice had a dramatic reduction of these anergic cells (Fig. 3A & C). Thus, Bcl10 deficiency mainly impairs the in vivo survival of anergic B cells in IgHELsHEL transgenic mice. Overall, these data demonstrate that in the absence of self-antigen, Bcl10-deficient peripheral B cells can usually survive in vivo whereas in the presence of self-antigen, self-reactive Bcl10-deficient B cells undergo apoptosis instead of anergy.
Bcl10 deficiency impairs BAFF-induced canonical NF-κB activation
Due to a critical role for BAFF in anergic B cell survival, Bcl10-dependent mechanisms that contribute to BAFF-R signaling were investigated. Bcl10 deficiency could reduce the expression of BAFF-R on B cells, resulting in an impairment of BAFF’s ability to support B cell survival. To examine this possibility, we compared BAFF-R expression on wild-type and Bcl10-deficient splenic B cells at different maturation stages. Based on expression of IgD and IgM, splenic B cells can be divided into IgMhiIgD- (T1), IgMhiIgDhi (T2), and IgMloIgDhi (FO and An1) B cells (54). FACS analysis demonstrated that subpopulations of Bcl10-deficient B cells expressed comparable levels of BAFF-R relative to corresponding subsets of wild-type B cells (Fig. 5A). Thus, Bcl10 deficiency has no apparent effect on the expression of BAFF-R.
Figure 5.
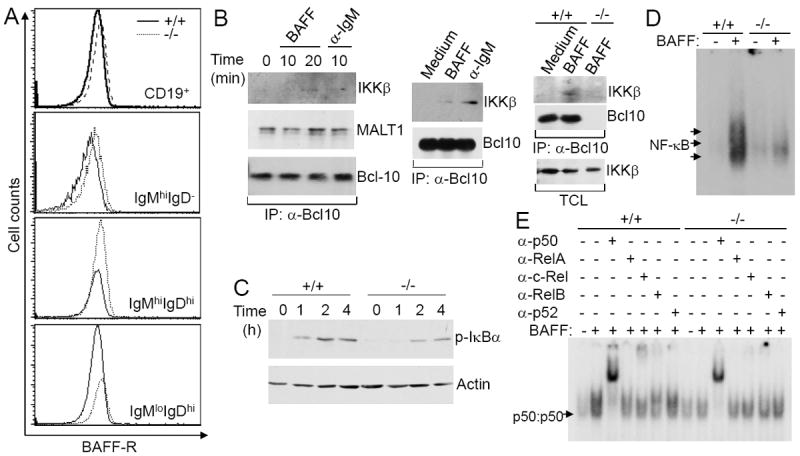
Bcl10 deficiency impairs BAFF-induced canonical NF-κB activation. A, Expression of BAFF-R. Splenocytes from wild-type and Bcl10-deficient mice were stained with antibodies to CD19, IgM, IgD and BAFF-R. Expression levels of BAFF-R on total splenic (CD19+), T1 (IgMhiIgD-), T2 (IgMhiIgDhi), and FO (IgMloIgDhi) B cells were measured by FACS analysis. (B-E) Splenic mature B cells (CD93-) were isolated from wild-type and Bcl10-deficient mice. B, BAFF-induced association of Bcl10 with IKKβ. Wild-type or Bcl10-deficient mature B cells were stimulated with BAFF or anti-IgM for the indicated times. Cell lysates were immunoprecipitated with anti-Bcl10-conjugated Sepharose beads, followed by Western blotting analysis with the indicated antibodies. C, BAFF-induced IκBα phosphorylation. Cells were pre-treated with MG132, and subsequently stimulated with BAFF for the indicated times. Cell lysates were subjected to direct Western blotting analysis with anti-phospho-IκBα or anti-Actin antibodies. D and E, BAFF-induced canonical activation of NF-κB. Cells were stimulated with (+) or without (-) BAFF for 3 hrs and total cell lysates were subjected to NF-κB gel mobility shift (D) and antibody supershift (E) analysis. Arrows indicate the position of NF-κB complexes (D) and p50:p50 homodimers (E). Data are representative of 3 (A, C-E) or 2 (B) independent experiments.
The other possible explanation for the impaired BAFF’s ability to support the survival of Bcl10-deficient B cells is that Bcl10 is required for BAFF-R signaling. NF-κB activation by BAFF-R is essential for B cell survival (22, 30). BAFF-R can induce canonical NF-κB activation through IKKβ-dependent degradation of IκBα (39, 40). To study the potential role of Bcl10 in BAFF-induced canonical NF-κB activation, we first examined whether Bcl10 interacts with IKKβ upon BAFF stimulation. Three independent co-immunoprecipitation analyses demonstrated that BAFF stimulation induced the association of Bcl10 with IKKβ in wild-type splenic B cells, similar to anti-IgM-induced BCR engagement (Fig. 5B). In contrast, BAFF-induced association of Bcl10 with IKKβ was absent in Bcl10-deficient splenic B cells (Fig. 5B, right). Of note, Bcl10 constitutively associated with MALT1 without stimulation as previously described (55) (Fig. 5B, left). Thus, Bcl10 signaling complex recruits IKKβ upon BAFF stimulation.
Next, we examined whether Bcl10 is involved in BAFF-induced canonical NF-κB activation by assessing IKKβ-dependent IκBα phosphorylation at Ser32/36. We compared IκBα phosphorylation in wild-type and Bcl10-deficient splenic mature B cells following BAFF stimulation. BAFF-induced IκBα phosphorylation was markedly reduced in Bcl10-deficient relative to wild-type cells (Fig. 5C). Thus, Bcl10 deficiency impairs BAFF-induced IKKβ activation.
Further, we investigated whether Bcl10 is required for BAFF-induced activation of the canonical NF-κB by the gel mobility shift assays. As previously reported (39, 40), canonical NF-κB activation by BAFF stimulation occurred at an early time point (Fig. 5D) and involved the formation of p50:c-Rel heterodimer (Fig. 5E) in wild-type mature B cells. However, this canonical NF-κB activation by BAFF was severely impaired in Bcl10-deficient mature B cells (Fig. 5D-E). Mutant B cells only displayed basal levels of transcriptionally inactive p50:p50 homodimers, which were not elevated by BAFF stimulation (Fig. 5E). Taken together, these data demonstrate that Bcl10 is critical for BAFF-induced canonical NF-κB activation in B cells.
Bcl10 deficiency indirectly impairs BAFF-induced noncanonical NF-κB activation
Previous studies have shown that BCR-mediated canonical NF-κB activity contributes to the basal as well as induced expression of NF-κB2/p100, which is a critical substrate in BAFF-mediated noncanonical RelB:p52 activation and B cell survival (56, 57). Bcl10 deficiency disrupts BCR-mediated NF-κB activation (46, 47). Our Western blot analysis of the expression of the NF-κB family members revealed that the basal level of NF-κB2/p100, but not other NF-κB members, was reduced in Bcl10-deficient relative to wild-type splenic mature B cells (Fig. 6A). However, the NF-κB2/p100 protein was efficiently processed into p52 in the presence of BAFF in both wild-type and Bcl10-deficient mature B cells, as indicated by reduction in NF-κB2/p100 protein (Fig. 6B). Consistent with reduced NF-κB2/p100 protein in the Bcl10-deficient B cells, BAFF-induced NF-κB2/p100 mRNA expression was also reduced in Bcl10-deficient relative to wild-type immature and mature B cells (Fig. 6C). Thus, lack of Bcl10 reduces basal and BAFF-induced expression of NF-κB2/p100 in B cells. This limits the p100 substrate necessary for the optimal activation of noncanonical NF-κB in Bcl10-deficient B cells.
Figure 6.
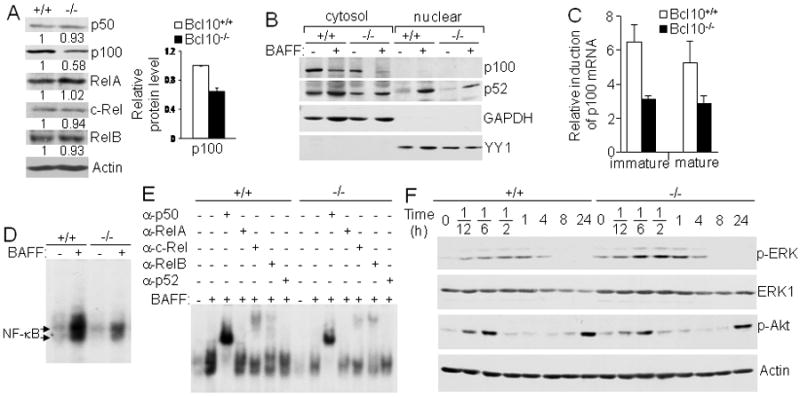
Bcl10-deficient splenic B cells express reduced levels of p100, indirectly affecting BAFF-induced noncanonical NF-κB activation. A, Expression of NF-κB/Rel family members. Protein levels of NF-κB/Rel family members in total cellular extracts from splenic mature B cells were determined by direct Western blotting analysis with the indicated antibodies. Densitometric analysis of protein bands was performed and normalized to actin loading control. Levels of each NF-κB protein in Bcl10-deficient and wild-type B cells were compared by giving wild-type an arbitrary value of 1. The bar graph shows the relative p100 protein levels calculated from 3 independent experiments. p < 0.01. B, BAFF-induced NF-κB2/p100 processing and nuclear translocation. Following 16 hours of culture of splenic mature B cells with or without BAFF, cytoplasmic and nuclear extracts were prepared and subjected to Western blotting with the indicated antibodies. GAPDH was used as cytoplasmic protein loading control and YY1 as nuclear protein loading control. C, BAFF-induced NF-κB2/p100 mRNA expression. Splenic immature (CD93+) and mature (CD93-CD23+) B cells were either stimulated with BAFF or left non-stimulated for 16 hrs. NF-κB2/p100 mRNA expression was determined by quantitative real-time PCR. Relative fold induction in response to BAFF was normalized to 18S rRNA and calibrated to non-stimulated sample for the corresponding genotype. D and E, BAFF-induced noncanonical activation of NF-κB. Splenic mature B cells were constantly stimulated with BAFF for 16 hrs and total cell lysates were subjected to NF-κB gel mobility shift (D) and antibody supershift (E) analysis. Arrows indicate the position of NF-κB complexes. F, BAFF-induced ERK and Akt activation. Splenic mature B cells were stimulated with BAFF for the indicated times and cell lysates were subjected to direct Western blotting analysis with the indicated antibodies. Data are representative of 3 (A, B, D and E) or 2 (F) independent experiments, or data is representative of at least 3 experiments that used one animal from each genotype per experiment (C).
It is well established that constant BAFF exposure induces noncanonical NF-κB activation through a relatively well-defined pathway that requires processing of NF-κB2/p100 into p52. The p52 then dimerizes with RelB to form p52:RelB heterodimer (35, 36). Our results showed that induction of NF-κB DNA binding activity by constant BAFF exposure was severely reduced in Bcl10-deficient relative to wild-type B cells (Fig. 6D).
These NF-κB DNA-binding complexes in wild-type B cells were supershifted strongly by anti-p50 and slightly by anti-c-Rel or anti-RelB whereas the supershift with anti-RelA/p65 or anti-p52 was least prominent (Fig. 6E). However, anti-RelA/p65 and anti-p52 reduced intensity of BAFF-induced NF-κB bands (Fig. 6E). These results suggest that in addition to the expected p52:RelB heterodimer (35, 36), p50:RelA and p50:c-Rel heterodimers were also formed in response to BAFF.
In contrast, the NF-κB DNA-binding complexes in Bcl10-deficient B cells were supershifted or reduced in intensities by all antibodies used except anti-p52 (Fig. 6E). These results are consistent with reduced canonical NF-κB activation (p50:RelA and p50:c-Rel heterodimers) in Bcl10-deficient B cells. Further, a severe reduction in the formation of p52:RelB heterodimer was in agreement with reduced basal p100 protein levels combined with an inability to transcriptionally upregulate p100. Nonetheless, the limiting amounts of NF-κB2/p100 available in Bcl10-deficient B cells, were efficiently processed into p52 and subsequently translocated into nucleus (Fig. 6B). Taken together, these data demonstrate that Bcl10 deficiency severely impairs the formation of not only p50:RelA and p50:c-Rel complexes, typically activated by the canonical NF-κB pathway, but also p52:RelB complexes, typically activated by the noncanonical NF-κB pathway. The impairment of noncanonical NF-κB activation in Bcl10-deficient B cells is likely due to the reduction of NF-κB2/ p100, which is in part regulated by the canonical NF-κB pathway.
In addition to the NF-κB pathway, BAFF stimulation promotes B cell survival by down-regulating the proapoptotic molecule Bim via ERK activation as well as by activating the prosurvival kinase Akt (58, 59). To address the role of Bcl10 plays in these pathways, we examined whether Bcl10 is involved in BAFF-induced activation of ERK and Akt. Splenic mature B cells were isolated from wild-type and Bcl10-deficient mice and then stimulated with BAFF. The activation of ERK was evaluated by immunoblotting with antibodies that detect phosphorylation of Thr202//Tyr204 within ERK1 and Thr185//Tyr187 within ERK2. BAFF-induced activation of ERK was comparable in Bcl10-deficient and wild-type B cells (Fig. 6F). In addition, the activation of Akt was evaluated by immunoblotting with antibodies that detect phosphorylation of Ser473 within Akt. BAFF-induced activation of Akt was biphasic. The initial activation, which occurred 5-10 min post-stimulation, was reduced to basal level 30 min later, followed by a second phase of activation at 24 h (Fig. 6F). This biphasic activation of Akt by BAFF was not affected in Bcl10-deficient mature B cells (Fig. 6F). Thus, Bcl10 is not required for BAFF-induced activation of ERK and Akt.
Bcl10 deficiency impairs BAFF-dependent NF-κB target gene expression
The activation of the NF-κB pathway by BAFF stimulation ultimately up-regulates the expression of anti-apoptotic genes, such as Bcl-xL (22, 30). To confirm that Bcl10 plays an important role in BAFF-mediated survival of B cells through the NF-κB pathway, we examined BAFF-induced up-regulation of the pro-survival NF-κB target gene, Bcl-xL, in Bcl10-deficient B cells. Following BAFF stimulation, the level of Bcl-xL proteins was markedly up-regulated in wild-type B cells (Fig. 7). In contrast, the up-regulation of Bcl-xL proteins was abolished in Bcl10-deficient B cells (Fig. 7). Of note, the amount of Bcl-2 proteins remained unaltered before and after BAFF stimulation in both wild-type and Bcl10-deficient B cells (Fig. 7). Therefore, Bcl10 is required for BAFF-induced expression of Bcl-xL, a pro-survival gene of the Bcl-2 family that is regulated by NF-κB.
Figure 7.
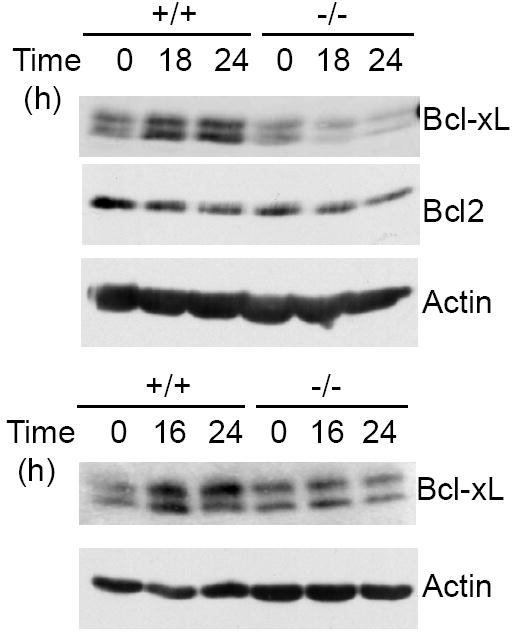
Bcl10 deficiency impairs BAFF-induced expression of NF-κB target gene Bcl-xL. Splenic mature B cells were isolated from wild-type and Bcl10-deficient mice and stimulated with BAFF for the indicated times. Cell lysates were subjected to direct Western blotting analysis with the indicated antibodies. Data from 2 independent experiments are shown.
Discussion
Previous studies have demonstrated that the survival of all peripheral B cells as well as maintenance of anergic B cells depends on signals from both the BCR and BAFF-R (20-24). Our findings demonstrate that Bcl10 was essential for anergic B cell survival. This Bcl10 function was apparent in the diverse repertoire as well as IgHEL system. Our findings further suggest that this was likely consequence of a failure to active both canonical and non-canonical NF-κB pathways; Bcl10 directly contributed to BAFF-mediated activation of canonical NF-κB pathway and indirectly disrupted BAFF-mediated noncanonical NF-κB pathway by reducing the availability of p100. Thus, Bcl10 plays a critical role in the peripheral B cell anergy in part by mediating BAFF-R signaling.
Our data demonstrate a dramatic reduction of anergic B cells in Bcl10-deficient mice relative to wild-type controls. Consistently, Bcl10-deficient An1 anergic B cells displayed increased rate of apoptosis in vivo relative to the corresponding wild-type B cells, whereas rates of apoptosis were similar when T1, T2, or FO B cell populations were compared. The finding that Bcl10 deficiency primarily affected the survival of anergic B cells with diverse repertoire was recapitulated in the Ig transgenic IgHELsHEL model of anergy. The IgHEL single transgene did not appear to affect T1 and T2 B cells and only slightly reduced mature FO B cells in Bcl10-deficient relative to wild-type control mice, indicating that Bcl10 function was not critical for the survival of peripheral B cells in the absence of self-antigen. In IgHELsHEL double-transgenic model, soluble self-antigen (sHEL) drove self-reactive B cells into anergy in both wild-type and mutant mice. Anergic B cells that depend more heavily on BAFF underwent more apoptosis in the absence of Bcl10. Thus, BAFF-R/Bcl10/NF-κB signaling axis contributes to the establishment of peripheral B cell tolerance by maintaining anergic B cells and preventing these B cells from self-antigen-induced elimination.
BAFF can induce canonical and noncanonical NF-κB activation pathways, both of which are required for B cell survival (35, 60). While BAFF-R-induced activation of the noncanonical NF-κB pathway is relatively well studied, however, the membrane proximal signaling cascades upstream of the canonical pathway remain unclear. Our current studies demonstrate that Bcl10 directly participated in BAFF-induced activation of canonical NF-κB via mechanisms involving direct association of Bcl10 with IKKβ. This results in the activation of the IKK complex consisting of IKKα, IKKβ and NEMO. Our observation that noncanonical NF-κB activation by BAFF was impaired in Bcl10-deficient mice suggests a role for Bcl10 in the regulation of this NF-κB pathway as well. A direct role for Bcl10 in the activation of noncanonical pathway was ruled out by our data showing that Bcl10-deficiency did not affect NF-κB2/p100 to p52 processing. Instead, this defect was found to be an indirect effect of Bcl10-deficiency on the expression of NF-κB2/p100. Thus our results suggest that reduced availability of NF-κB2/p100 limited the extent of noncanonical NF-κB activation in Bcl10-deficient B cells.
It is well known that Bcl10 forms a ternary complex with CARMA1 and MALT1 (CBM) to couple PKC activity to IKK complex activation during BCR-induced canonical NF-κB activation (48-51). Our data demonstrating a requirement for Bcl10 in the activation of the canonical pathway suggest that CBM complex is required for BAFF-induced activation of the IKK complex. This hypothesis is supported by our finding that Bcl10 is constitutively associated with MALT1, suggesting that Bcl10/MALT1 partner with CARMA1 to form the CBM complex to activate IKK complex during BAFF-R signaling. This possibility remains to be investigated and the role of MALT1 in the activation of the canonical NF-κB pathway remains unknown. However, a recent study has demonstrated that MALT1 regulates BAFF-induced noncanonical NF-κB activation in B cells. MALT1 deficiency impairs BAFF-induced phosphorylation and processing of NF-κB2/p100, as well as RelB nuclear translocation, suggesting a direct role for MALT1 in the noncanonical NF-κB activation (61). In addition, the role of CARMA1 in BAFF-induced canonical NF-κB activation is also unknown. Our findings suggest Bcl10 containing CBM complex mediates canonical NF-κB pathway. Our results lay the foundation for further studies to determine the contribution of CARMA1 and MALT1 to BAFF-induced canonical NF-κB activation.
The membrane proximal steps of the BCR signaling cascade that leads to the formation of CBM complex have been well studied. Upon BCR ligation, protein tyrosine kinases Lyn, Syk and Btk are sequentially activated, and B cell linker protein (BLNK) and CD19 are recruited to the receptor complex (62-65). The BCR complex subsequently activates phosphatidylinositol 3-kinase (PI3K) and phospholipase Cγ2 (PLCγ2) (66, 67). Activated PLCγ2 hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and IP3 (68, 69). In turn, DAG plus IP3-induced Ca2+ flux activate PKC, a critical step for the formation of CBM ternary complex (48-51). Prior studies have shown that BAFF stimulation activates PI3K and PKCβ (59). PKCβ deficiency impairs BAFF-R-mediated survival of B cells (59). A recent study has demonstrated that Btk plays a critical role in BAFF-mediated activation of both canonical and noncanonical NF-κB activation. Like Bcl10, Btk also directly regulates BAFF-induced activation of IKK complex for canonical NF-κB activation and affects the noncanonical NF-κB pathway due to its role in the expression of NF-κB2/p100 (70). Moreover, previous studies have found that PLCγ2 plays an important role in BAFF-mediated survival of B cells (70, 71). Our findings have placed Bcl10 in the BAFF-R signaling to canonical NF-κB pathway together with multiple signaling components discussed above, however, additional studies will be required to define the precise mechanisms of the membrane proximal signaling events that relay BAFF-R to canonical NF-κB activation.
In summary, our studies have revealed a novel function for Bcl10 in the activation of canonical NF-κB pathway and its significance in B cell survival, particularly of anergic B cells. Our studies further suggest that loss of Bcl10 also severely reduces the extent of noncanonical NF-κB pathway. In this regard, Bcl10 is also integral to the BCR mediated NF-κB activation (53), which has also been implicated in the regulation of NF-κB2/p100 expression (56, 57). Taken together, these data suggest that BCR and BAFF-R signaling, which orchestrate appropriate levels of canonical and non-canonical NF-κB pathways, contribute to the survival of peripheral B cells. The BAFF-R/Bcl10/NF-κB signaling axis plays an important role in the establishment of peripheral B cell tolerance by maintaining anergic B cells and preventing these B cells from self-antigen-induced elimination.
Acknowledgments
We thank Jennifer Idsvoog for helping maintain our mouse colony.
This work is supported in part by National Institutes of Health grants R01 AI079087 (D. W.), PO1 HL44612 (D. W.), R21 AI088511 (W. N. K.), by Scholar Award from the Leukemia & Lymphoma Society (D. W.), by a grant from Advancing a Healthier Wisconsin endowment fund (D. W.), and by ARRA supplement R01 AI060729-06S1 (W. N. K. to support J. A. W.).
References
- 1.Nemazee D. Antigen receptor ‘capacity’ and the sensitivity of self-tolerance. Immunol Today. 1996;17:25–29. doi: 10.1016/0167-5699(96)80565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 3.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 4.Nemazee D. Receptor selection in B and T lymphocytes. Annu Rev Immunol. 2000;18:19–51. doi: 10.1146/annurev.immunol.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Healy JI, Goodnow CC. Positive versus negative signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1998;16:645–670. doi: 10.1146/annurev.immunol.16.1.645. [DOI] [PubMed] [Google Scholar]
- 6.Radic MZ, Zouali M. Receptor editing, immune diversification, and self-tolerance. Immunity. 1996;5:505–511. doi: 10.1016/s1074-7613(00)80266-6. [DOI] [PubMed] [Google Scholar]
- 7.Sandel PC, Monroe JG. Negative selection of immature B cells by receptor editing or deletion is determined by site of antigen encounter. Immunity. 1999;10:289–299. doi: 10.1016/s1074-7613(00)80029-1. [DOI] [PubMed] [Google Scholar]
- 8.Nossal GJ. Negative selection of lymphocytes. Cell. 1994;76:229–239. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 9.Radic MZ, Erikson J, Litwin S, Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J Exp Med. 1993;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casellas R, Shih TA, Kleinewietfeld M, Rakonjac J, Nemazee D, Rajewsky K, Nussenzweig MC. Contribution of receptor editing to the antibody repertoire. Science. 2001;291:1541–1544. doi: 10.1126/science.1056600. [DOI] [PubMed] [Google Scholar]
- 13.Edry E, Melamed D. Receptor editing in positive and negative selection of B lymphopoiesis. J Immunol. 2004;173:4265–4271. doi: 10.4049/jimmunol.173.7.4265. [DOI] [PubMed] [Google Scholar]
- 14.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 15.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 16.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 17.Nossal GJ, Pike BL. Clonal anergy: persistence in tolerant mice of antigen-binding B lymphocytes incapable of responding to antigen or mitogen. Proc Natl Acad Sci U S A. 1980;77:1602–1606. doi: 10.1073/pnas.77.3.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cyster JG, Goodnow CC. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 1995;3:691–701. doi: 10.1016/1074-7613(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 19.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, Cambier JC. Identification of anergic B cells within a wild-type repertoire. Immunity. 2006;25:953–962. doi: 10.1016/j.immuni.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Lam KP, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 21.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6:1160–1167. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- 22.Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, Soppet D, Charters M, Gentz R, Parmelee D, Li Y, Galperina O, Giri J, Roschke V, Nardelli B, Carrell J, Sosnovtseva S, Greenfield W, Ruben SM, Olsen HS, Fikes J, Hilbert DM. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 24.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, Brink R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 26.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 27.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 28.O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 30.Hsu BL, Harless SM, Lindsley RC, Hilbert DM, Cancro MP. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J Immunol. 2002;168:5993–5996. doi: 10.4049/jimmunol.168.12.5993. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Verma IM. NF-kappaB regulation in the immune system. Annu Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 33.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 34.Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 35.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 36.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J, Starovasnik MA, Dixit VM. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 37.Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Hatada EN, Do RK, Orlofsky A, Liou HC, Prystowsky M, MacLennan IC, Caamano J, Chen-Kiang S. NF-kappa B1 p50 is required for BLyS attenuation of apoptosis but dispensable for processing of NF-kappa B2 p100 to p52 in quiescent mature B cells. J Immunol. 2003;171:761–768. doi: 10.4049/jimmunol.171.2.761. [DOI] [PubMed] [Google Scholar]
- 40.Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Thome M, Martinon F, Hofmann K, Rubio V, Steiner V, Schneider P, Mattmann C, Tschopp J. Equine herpesvirus-2 E10 gene product, but not its cellular homologue, activates NF-kappaB transcription factor and c-Jun N-terminal kinase. J Biol Chem. 1999;274:9962–9968. doi: 10.1074/jbc.274.15.9962. [DOI] [PubMed] [Google Scholar]
- 42.Srinivasula SM, Ahmad M, Lin JH, Poyet JL, Fernandes-Alnemri T, Tsichlis PN, Alnemri ES. CLAP, a novel caspase recruitment domain-containing protein in the tumor necrosis factor receptor pathway, regulates NF-kappaB activation and apoptosis. J Biol Chem. 1999;274:17946–17954. doi: 10.1074/jbc.274.25.17946. [DOI] [PubMed] [Google Scholar]
- 43.Costanzo A, Guiet C, Vito P. c-E10 is a caspase-recruiting domain-containing protein that interacts with components of death receptors signaling pathway and activates nuclear factor-kappaB. J Biol Chem. 1999;274:20127–20132. doi: 10.1074/jbc.274.29.20127. [DOI] [PubMed] [Google Scholar]
- 44.Yan M, Lee J, Schilbach S, Goddard A, Dixit V. mE10, a novel caspase recruitment domain-containing proapoptotic molecule. J Biol Chem. 1999;274:10287–10292. doi: 10.1074/jbc.274.15.10287. [DOI] [PubMed] [Google Scholar]
- 45.Koseki T, Inohara N, Chen S, Carrio R, Merino J, Hottiger MO, Nabel GJ, Nunez G. CIPER, a novel NF kappaB-activating protein containing a caspase recruitment domain with homology to Herpesvirus-2 protein E10. J Biol Chem. 1999;274:9955–9961. doi: 10.1074/jbc.274.15.9955. [DOI] [PubMed] [Google Scholar]
- 46.Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, Mak TW. Bcl10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell. 2001;104:33–42. doi: 10.1016/s0092-8674(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 47.Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, Wang D. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol. 2003;4:857–865. doi: 10.1038/ni963. [DOI] [PubMed] [Google Scholar]
- 48.Lin X, Wang D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin Immunol. 2004;16:429–435. doi: 10.1016/j.smim.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 49.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–359. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 50.Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Wang D, Lin X. Phosphorylation of CARMA1 Plays a Critical Role in T Cell Receptor-Mediated NF-kappaB Activation. Immunity. 2005;23:575–585. doi: 10.1016/j.immuni.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. Phosphorylation of the CARMA1 Linker Controls NF-kappaB Activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Schuman J, Chen Y, Podd A, Yu M, Liu HH, Wen R, Chen ZJ, Wang D. A critical role of TAK1 in B-cell receptor-mediated nuclear factor kappaB activation. Blood. 2009;113:4566–4574. doi: 10.1182/blood-2008-08-176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue L, Morris SW, Orihuela C, Tuomanen E, Cui X, Wen R, Wang D. Defective development and function of Bcl10-deficient follicular, marginal zone and B1 B cells. Nat Immunol. 2003;4:857–865. doi: 10.1038/ni963. [DOI] [PubMed] [Google Scholar]
- 54.Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med. 1999;190:75–89. doi: 10.1084/jem.190.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell. 2000;6:961–967. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 56.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ, 3rd, Brezski RJ, Treml LS, Jordan KA, Monroe JG, Sen R, Cancro MP. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol. 2008;9:1379–1387. doi: 10.1038/ni.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castro I, Wright JA, Damdinsuren B, Hoek KL, Carlesso G, Shinners NP, Gerstein RM, Woodland RT, Sen R, Khan WN. B cell receptor-mediated sustained c-Rel activation facilitates late transitional B cell survival through control of B cell activating factor receptor and NF-kappaB2. J Immunol. 2009;182:7729–7737. doi: 10.4049/jimmunol.0803281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J Exp Med. 2005;202:1363–1374. doi: 10.1084/jem.20051283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patke A, Mecklenbrauker I, Erdjument-Bromage H, Tempst P, Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKC beta-and Akt-dependent mechanism. J Exp Med. 2006;203:2551–2562. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K, Fujita S, Matsumoto M. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-kappa B-inducing kinase. J Immunol. 2000;165:804–812. doi: 10.4049/jimmunol.165.2.804. [DOI] [PubMed] [Google Scholar]
- 61.Tusche MW, Ward LA, Vu F, McCarthy D, Quintela-Fandino M, Ruland J, Gommerman JL, Mak TW. Differential requirement of MALT1 for BAFF-induced outcomes in B cell subsets. J Exp Med. 2009;206:2671–2683. doi: 10.1084/jem.20091802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurosaki T. Genetic analysis of B cell antigen receptor signaling. Annu Rev Immunol. 1999;17:555–592. doi: 10.1146/annurev.immunol.17.1.555. [DOI] [PubMed] [Google Scholar]
- 63.Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009;228:132–148. doi: 10.1111/j.1600-065X.2008.00748.x. [DOI] [PubMed] [Google Scholar]
- 64.Fu C, Turck CW, Kurosaki T, Chan AC. BLNK: a central linker protein in B cell activation. Immunity. 1998;9:93–103. doi: 10.1016/s1074-7613(00)80591-9. [DOI] [PubMed] [Google Scholar]
- 65.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–107. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- 66.Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, Cantley LC. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283:393–397. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- 67.Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, Ihle JN. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- 68.Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–15048. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 69.Rhee SG, Choi KD. Regulation of inositol phospholipid-specific phospholipase C isozymes. J Biol Chem. 1992;267:12393–12396. [PubMed] [Google Scholar]
- 70.Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, Scott ML, Wang D, Khan WN. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179:3872–3880. doi: 10.4049/jimmunol.179.6.3872. [DOI] [PubMed] [Google Scholar]
- 71.Hikida M, Johmura S, Hashimoto A, Takezaki M, Kurosaki T. Coupling between B cell receptor and phospholipase C-gamma2 is essential for mature B cell development. J Exp Med. 2003;198:581–589. doi: 10.1084/jem.20030280. [DOI] [PMC free article] [PubMed] [Google Scholar]