

Abstract
Angiotensin II (Ang II) infusion into rats elevates local angiotensin II levels through an AT1 receptor–dependent pathway in the kidney. We examined whether treatment with an angio-tensin-converting enzyme (ACE) inhibitor, temocapril, or an AT1-receptor blocker, olmesartan, prevented elevation of Ang II levels in the kidney of angiotensin I (Ang I)-infused rats. Rats were infused with Ang I (100 ng/min) and treated with temocapril (30 mg/kg per day, n = 10) or olmesartan (10 mg/kg per day, n = 9) for 4 weeks. Ang I infusion significantly elevated blood pressure compared with vehicle-infused rats (n = 6). Treatment with temocapril or olmesartan suppressed Ang I–induced hypertension. Temocapril suppressed both plasma and renal ACE activity. Ang I infusion increased Ang II content in the kidney. Interestingly, temocapril failed to reduce the level of Ang II in the kidney, while olmesartan markedly suppressed an increase in renal Ang II levels. These results suggest a limitation of temocapril and a benefit of olmesartan to inhibit the renal renin–angiotensin system and suggest the possible existence of an ACE inhibitor–insensitive pathway that increases Ang II levels in rat kidney.
Keywords: angiotensin-converting enzyme (ACE) inhibitor, angiotensin II, kidney
Introduction
Hypertension increases the risk of cardiovascular and renal events (1, 2), and lowering blood pressure lowers the risk. In addition, the prevention of target organ damage should be a goal with antihypertensive therapy. The renin-angiotensin system (RAS) regulates blood pressure (3 – 5), reduces sodium excretion (6, 7), and causes kidney damage (8 – 10), and RAS inhibitors are important drugs for treating hypertension or organ damage.
RAS in the kidney is independently regulated from that in the systemic circulation (11, 12), and the increase in renal angiotensin II (Ang II) is implicated in renal pathophysiology as well as the development of hypertension. Blood pressure and renal Ang II increased in mice expressing angiotensin converting enzyme (ACE) in the kidney only, compared with systemic ACE null mice (13), indicating the importance of ACE-dependent angiotensin I (Ang I) formation in the kidney on systemic hypertension. AT1 receptor–dependent Ang II internalization in the apical membrane of proximal tubules is thought to be one mechanism for Ang II accumulation in the kidney (14, 15). Indeed, intrarenal Ang II accumulation in Ang II–infused animals was strongly suppressed by antagonists of the AT1 receptor (ARB), regardless of Ang II plasma levels (16, 17). The reduction of renal Ang II by ARB is accompanied by well-established renal-protective effects of ARB (18, 19). Conversely, there are relatively few data regarding the effects of ACE inhibitors compared with that of ARB. We previously reported that renal interstitial fluid contains much higher concentration of Ang I (> 5 ×) and Ang II (> 30 ×) compared with plasma (20). Surprisingly, neither intra-arterial infusion nor renal interstitial infusion of enalaprilat, an ACE inhibitor, affected Ang II levels in renal interstitial fluid, although it reduced plasma Ang II levels. We speculated that renal interstitial Ang II levels may maintain renal Ang II levels, but might be insensitive to drugs that inhibit ACE. Therefore, in the present study, we investigated whether the augmentation of renal Ang II levels was affected by oral treatment with an ACE inhibitor in Ang I–infused rats.
Materials and Methods
Animals
All experimental procedures were performed according to the guidelines for the care and use of animals as established by Kagawa University. Male Sprague Dawley rats were purchased from SLC (Shizuoka). Animals were divided into four experimental groups as follows: Group 1: vehicle (n = 10); Group 2: Ang I + control (Ang I, n = 10); Group 3: Ang I + temocapril (30 mg/kg per day, p.o., n = 10); and Group 4: Ang I + olmesartan (10 mg/kg per day, p.o., n = 10). The drugs were mixed in their diet.
Systolic blood pressure (SBP) was measured in conscious rats by tail-cuff plethysmography (Softron, Tokyo) and 24-h urine samples were collected every week. All animals underwent a 24-h acclimatization period in metabolic cages prior to urine collection. Blood and kidney samples were harvested at the end of the experimental protocol. The wet weight of the left kidney was measured. The right kidney was snap-frozen in liquid nitrogen for measurement of renal Ang II content and ACE activity.
Ang II and ACE activity assay
For Ang II measurements, blood was collected in a glass tube containing EDTA and protease inhibitor cocktail. The right kidney was homogenized in methanol immediately after extraction. Ang II contents were measured by radioimmunoassay. The blood samples were collected in 100% methanol, evaporated to dryness in a vacuum centrifuge, reconstituted in buffer, and assayed. As ACE activity could not be detected in blood containing EDTA, a blood sample collected in a separate tube containing heparin instead of EDTA was prepared for plasma ACE activity measurement. ACE activity in the homogenate was measured in duplicate with a commercial kit (ACEcolor; Fujirebio, Malvern, PA, USA) containing p-hydroxybenzoyl-glycyl-L-histidyl-L-leucine as synthetic substrate (21, 22). Tissue samples were ultracentrifuged at 15,000 × g for 1 h. Pellets were suspended in PBS containing Triton-X and assayed.
Real-Time PCR
The mRNA expression of glyceraldehydes-3-phosphate dehydrogenase (GAPDH), angiotensinogen (AGT), and ACE2 were analyzed by real-time PCR using a LightCycler FastStart DNA Master SYBR Green I kit or TaqMan Gene Expression Assay kits (Applied Bio-systems, Foster City, CA, USA). The oligonucleotide primer sequences of GAPDH and AGT and PCR conditions were as described previously (23, 24). The primer sequences of ACE2 were as follows: ACE2 forward, 5′-CCCAGAGAACAGTGGACCAAAA-3′ and reverse, 5′-GCTCCACCACACCAACGAT-3′. All data were expressed as the relative differences between the vehicle and other groups after normalization to GAPDH expression.
Other analytical procedures
Plasma aldosterone concentration was analyzed by a commercially available kit (SPACK-S aldosterone kit; TFB, Tokyo). Plasma renin activity (PRA) was measured by the ability to generate Ang I and expressed as ng/mL per hour of Ang I (25). Urinary protein excretion was determined using a protein assay kit (microTP-test; Wako, Osaka). Creatinine concentration in urine was measured using an assay kit (Creatinine-test, Wako).
Statistical analyses
Statistical comparisons of the differences between treatments for other parameters were performed using one-way ANOVA combined with the Tukey–Kramer post-hoc test. A P-value < 0.05 was considered statistically significant.
Results
Blood pressure, body weight, kidney weight, and urinary protein excretion
The effects of Ang I infusion and treatment with temocapril or olmesartan on SBP are shown in Fig. 1. SBP increased significantly in Ang I–infused rats, compared with vehicle. Both temocapril and olmesartan suppressed the blood pressure elevation induced by Ang I and the effect was stronger with olmesartan.
Fig. 1.
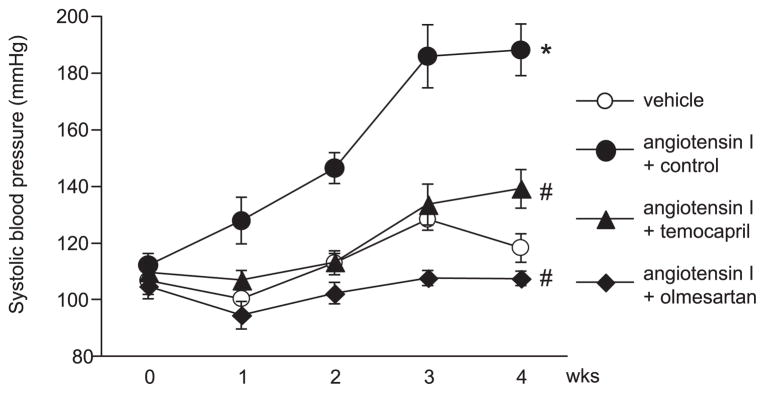
Changes in blood pressure in Ang I–infused rats. Tail cuff-blood pressure was increased by Ang I infusion, but the increase was suppressed by temocapril or olmesartan treatment. *P < 0.05 vs. vehicle-infused group, #P < 0.05 vs. Ang I + control group.
There was no statistically significant difference in the changes in body weight between the groups at week 4 (Table 1). Kidney weight / body weight ratio was significantly greater in Ang I–infused rats. Both temocapril and olmesartan suppressed the increase in kidney weight / body weight ratio induced by Ang I. Ang I infusion also increased urinary excretion of protein/ creatinine ratio. The urinary protein/creatinine ratio of temocapril- or olmesartan-treated rats was slightly greater than that of vehicle-infused rats and significantly milder than that of Ang I–infused control rats.
Table 1.
Body weight, kidney weight, kidney/body weight ratio, and urinary protein/creatinine ratio
| vehicle | Angiotensin I
|
||||
|---|---|---|---|---|---|
| control | temocapril | olmesartan | |||
| Body weight (g) | baseline | 180 ± 1 | 179 ± 1 | 180 ± 1 | 177 ± 2 |
| week 4 | 315 ± 7 | 320 ± 8 | 297 ± 5 | 310 ± 5 | |
| Kidney weight (g) | 2.2 ± 0.1 | 2.7 ± 0.1* | 2.3 ± 0.1# | 2.2 ± 0.1# | |
| Kidney weight / body weight ratio (× 10−3) | 7.1 ± 0.1 | 8.5 ± 0.2* | 7.6 ± 0.1# | 7.1 ± 0.2# | |
| Urinary protein/creatinine ratio | 3.3 ± 0.4 | 14.3 ± 2.2* | 6.2 ± 0.6# | 5.7 ± 1.1# | |
Plasma or renal ACE activity
We did not detect a significant difference between basal plasma and renal ACE activity between vehicle and Ang I–infused rats (Fig. 2). Plasma ACE activity of animals treated with temocapril was reduced to almost half compared with Ang I–infused rats (Fig. 2A). Similarly, renal ACE activity in temocapril-treated rats was reduced to 40% of that in Ang I–infused rats. In contrast, olmesartan did not change plasma or renal ACE activity (Fig. 2B).
Fig. 2.

Plasma (A) and renal (B) ACE activities, plasma renin activity (PRA) (C), and plasma aldosterone concentration (D). A and B: Plasma ACE activity in plasma and the kidney were unchanged by Ang I infusion. Temocapril, but not olmesartan, reduced ACE activity in both plasma and kidney. C: PRA was undetectable following Ang I infusion. Temocapril and olmesartan treatment increased PRA. D: Plasma aldosterone levels were increased by Ang I infusion. Either temocapril or olmesartan treatment suppressed the increase in plasma aldosterone level. *P < 0.05 vs. vehicle-infused group, #P < 0.05 vs. Ang I + control group, n.d.: not detectable.
PRA and plasma aldosterone level
PRA and plasma aldosterone levels are shown in Fig. 2. Ang I infusion for 4 weeks decreased PRA to undetectable levels and increased plasma aldosterone levels. Both temocapril and olmesartan increased PRA to a level significantly greater than that in vehicle-infused animals and decreased plasma aldosterone levels, confirming their inhibition efficacy on plasma Ang I and Ang II generation.
Plasma and renal Ang II
Plasma and renal Ang II levels in Ang I–infused rats with temocapril or olmesartan treatment are summarized in Fig. 3. Plasma Ang II was not increased significantly in Ang I controls or in temocapril-treated animals. Olmesartan increased plasma Ang II levels significantly. In contrast, Ang I infusion increased renal Ang II levels at both week 2 and 4. Either temocapril or olmesartan suppressed the increase in renal Ang II levels at week 2. Surprisingly, temocapril failed to suppress increased renal Ang II levels, while olmesartan lowered levels strongly at week 4.
Fig. 3.

Plasma (A) and kidney (B) Ang II levels. A: Plasma Ang II did not significantly respond to Ang I infusion with or without temocapril treatment. Olmesartan increased plasma Ang II levels. B: Kidney Ang II levels increased at weeks 2 and 4 following Ang I infusion. Temocapril suppresses the increase in renal Ang II at week 2, but not at week 4. Olmesartan suppressed renal Ang II levels both at weeks 2 and 4. *P < 0.05 vs. vehicle-infused group, #P < 0.05 vs. Ang I + control group.
ACE2 mRNA
Renal ACE mRNA levels increased in Ang I–infused rats compared with those in vehicle-infused rats (1.9 ± 0.4 vs. 1.0 ± 0.1 fold, respectively, P < 0.05). Temocapril further increased renal ACE2 expression (2.6 ± 0.5 fold). The increased ACE2 expression by Ang I infusion was abolished by olmesartan treatment (0.6 ± 0.1 fold).
Discussion
We and others have previously shown that Ang II, which theoretically may be filtered from glomeruli unlike AGT (24), is internalized by tubular cells, especially proximal tubular cells via AT1 receptor–mediated endocytosis (26, 27). Ang II stimulates transcription of AGT (26, 27) and may induce Ang II content in the kidney and subsequent renal injury (28, 29). Thus, it may be that the decrease in renal Ang II following olmesartan treatment, consistent with our previous studies (30 – 32), resulted from the inhibition of AT1 receptor–mediated Ang II internalization and subsequent accumulation of Ang II in the kidney. Interestingly, kidney Ang II in temocapril-treated rats increased at week 4, in contrast with a previous report by Gonzalez et al. (33) who observed a suppression of Ang II accumulation by lisinopril in the kidney of Ang II–infused mice. Several mechanisms may suppress the increase in renal Ang II. We have demonstrated that PRA increases with ACE inhibition during Ang I infusion. Kobori et al. (27) showed that PRA was undetectable in Ang II–infused rats and increased following treatment with AT1-receptor antagonists. Furthermore, several studies have reported that renin secretion and PRA increase in response to ACE inhibitor treatment (30, 31). We also observed increased PRA in ACE inhibitor–treated animals compared with untreated animals. The greater PRA should increase Ang I synthesis since renin is the rate-limiting enzyme in the renin–Ang I–aldosterone system. It is possible that treatment with ACE inhibitor at the dosage used is insufficient to inhibit Ang II production under the influence of up-regulated PRA, even if up-regulation results from the ACE inhibition itself. Second, this may result from insufficient inhibition of renal ACE activity relative to that of plasma ACE activity with temocapril treatment. Nishiyama et al. (20) has shown that intra-arterial infusion of enalaprilat reduced plasma ACE activity to undetectable levels, whereas renal ACE activity decreased only 50%. Gonzalez et al. (33) showed renal ACE activity was not reduced to the same level as plasma ACE activity in Ang II–infused mice treated with lisinopril. These results suggest that renal ACE may have lower sensitivity against ACE inhibitors, which were administered orally in the present study, than the plasma enzyme. The pharmacokinetic/dynamic character of ACE inhibitors may influence the accumulation of drugs in the kidney and their efficacy (ACE inhibition). Although 30% of orally administered temocapril is excreted in the urine as temocaprilat, an active metabolite of temocapril, we could not find any reports in the literature that has examined kidney retention and renal ACE inhibition. The drug may have poor distribution inside kidney cells, and compensatory increases in PRA and Ang I may contribute to increased Ang II generation in the kidney. These potential mechanisms may explain the time-course changes of renal Ang II levels in temocapril-treated rats, which were suppressed at week 2 and increased at week 4.
The current results might be due to the increase in the production of ACE-independent Ang II or to the decrease in catalyzed Ang II in the kidney of Ang I–infused rats. We previously showed that chymase contributed to the Ang II formation in the fibrotic kidneys of hamsters (18). Several recent studies have detected the chymase-like activity in the isolated tissues from rats (34 – 38). Thus, Ang I infusion for 4 weeks may increase the chymase-dependent Ang II formation and this Ang II may promote the temocapril-insensitive Ang II accumulation in the kidney in the present study. On the other hand, ACE2, an enzyme that catalyzes the conversion of Ang II into Ang-(1-7), was increased in temocapril-treated rats. Thus, this mechanism seems unlikely.
Temocapril showed significant antihypertensive effects even with elevated renal Ang II, possibly due to the effect on plasma ACE. It is also possible that an ACE inhibitor–induced increase in bradykinin and subsequent NO release could contribute to the effects.
Several studies have suggested that the increase in renal Ang II level contributes to the development of renal disease (23, 39 – 41). However, there was no difference in proteinuria at week 4 of Ang I infusion between temocapril- and olmesartan-treated groups. This might be due to the strong antihypertensive effects of these drugs: the average SBP was suppressed below 140 mmHg throughout the experimental period in both temocapril- and olmesartan-treated groups. Also, importantly, temocapril could suppress renal Ang II in the first 2 weeks of Ang I infusion, but failed to suppress it at week 4, indicating that there was a threshold point for suppressing renal Ang II by temocapril between week 2 and 4. Therefore, the elevated level of renal Ang II for less than 2 weeks in the temocapril-treated groups might not be sufficient to induce a significant level of renal damage. There may be a worse insult in the temocapril-treated groups than the olmesartan-treated group in a study with a longer experimental period.
In summary, the present experiments demonstrate that treatment with temocapril, which reduces blood pressure, failed to inhibit accumulation of Ang II in the kidney of Ang I–infused rats. In contrast, olmesartan treatment prevents accumulation of Ang II in the kidney. These results suggest a limitation of temocapril and a benefit of olmesartan to inhibit the renal renin–angiotensin system and a possible ACE inhibitor-insensitive pathway for intrarenal Ang II augmentation in Ang I–infused rats.
Acknowledgments
We are grateful to Daiichi-Sankyo Co. for supplying temocapril and olmesartan.
References
- 1.Wilson PW. Established risk factors and coronary artery disease: the framingham study. Am J Hypertens. 1994;7:7S–12S. doi: 10.1093/ajh/7.7.7s. [DOI] [PubMed] [Google Scholar]
- 2.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 3.Mori T, Ogawa S, Cowely AW, Jr, Ito S. Role of renal medullary oxidative and/or carbonyl stress in salt-sensitive hypertension and diabetes. Clin Exp Pharmacol Physiol. 2012;39:125–131. doi: 10.1111/j.1440-1681.2011.05653.x. [DOI] [PubMed] [Google Scholar]
- 4.Oki K, Gomez-Sanchez EP, Gomez-Sanchez CE. Role of mineralocorticoid action in the brain in salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2012;39:90–95. doi: 10.1111/j.1440-1681.2011.05538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosaka S, Pelisch N, Rahman M, Nakano D, Hitomi H, Kobori H, et al. Effects of angiotensin II AT1-receptor blockade on high fat diet-induced vascular oxidative stress and endothelial dysfunction in dahl salt-sensitive rats. J Pharmacol Sci. 2013;121:95–102. doi: 10.1254/jphs.12169fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang CT, Chin SY, Navar LG. Impairment of pressure-natriuresis and renal autoregulation in ang ii-infused hypertensive rats. Am J Physiol Renal Physiol. 2000;279:F319–F325. doi: 10.1152/ajprenal.2000.279.2.F319. [DOI] [PubMed] [Google Scholar]
- 7.Kittikulsuth W, Pollock JS, Pollock DM. Loss of renal medullary endothelin b receptor function during salt deprivation is regulated by angiotensin ii. Am J Physiol Renal Physiol. 2012;303:F659–F666. doi: 10.1152/ajprenal.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitada K, Nakano D, Hitomi H, Kobori H, Deguchi K, Mori H, et al. Aldosterone induces p21-regulated apoptosis via increased synthesis and secretion of tumour necrosis factor-alpha in human proximal tubular cells. Clin Exp Pharmacol Physiol. 2012;39:858–863. doi: 10.1111/1440-1681.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitada K, Nakano D, Liu Y, Fujisawa Y, Hitomi H, Shibayama Y, et al. Oxidative stress-induced glomerular mineralocorticoid receptor activation limits the benefit of salt reduction in dahl salt-sensitive rats. PLoS One. 2012;7:e41896. doi: 10.1371/journal.pone.0041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Yamamoto T, Misaki T, Suzuki H, Togawa A, Ohashi N, et al. Enhanced intrarenal receptor-mediated prorenin activation in chronic progressive anti-thymocyte serum nephritis rats on high salt intake. Am J Physiol Renal Physiol. 2012;303:F130–F138. doi: 10.1152/ajprenal.00275.2011. [DOI] [PubMed] [Google Scholar]
- 11.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 12.Shao W, Seth DM, Navar LG. Angiotensin ii type 1 receptor-mediated augmentation of urinary excretion of endogenous angiotensin ii in val5-angiotensin ii-infused rats. Hypertension. 2010;56:378–383. doi: 10.1161/HYPERTENSIONAHA.110.153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Villalobos RA, Billet S, Kim C, Satou R, Fuchs S, Bernstein KE, et al. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin i infusion. J Am Soc Nephrol. 2011;22:449–459. doi: 10.1681/ASN.2010060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou JX, Luciw PA. The transcriptional transactivator of simian foamy virus 1 binds to a DNA target element in the viral internal promoter. Proc Natl Acad Sci U S A. 1996;93:326–330. doi: 10.1073/pnas.93.1.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang ii accumulation in rat renal endosomes during ang ii-induced hypertension: Role of at(1) receptor. Hypertension. 2002;39:116–121. doi: 10.1161/hy0102.100780. [DOI] [PubMed] [Google Scholar]
- 16.Zou LX, Imig JD, von Thun AM, Hymel A, Ono H, Navar LG. Receptor-mediated intrarenal angiotensin ii augmentation in angiotensin ii-infused rats. Hypertension. 1996;28:669–677. doi: 10.1161/01.hyp.28.4.669. [DOI] [PubMed] [Google Scholar]
- 17.Zou LX, Imig JD, Hymel A, Navar LG. Renal uptake of circulating angiotensin ii in val5-angiotensin ii infused rats is mediated by at1 receptor. Am J Hypertens. 1998;11:570–578. doi: 10.1016/s0895-7061(97)00410-x. [DOI] [PubMed] [Google Scholar]
- 18.Fan YY, Nishiyama A, Fujisawa Y, Kobori H, Nakano D, Matsuura J, et al. Contribution of chymase-dependent angiotensin ii formation to the progression of tubulointerstitial fibrosis in obstructed kidneys in hamsters. J Pharmacol Sci. 2009;111:82–90. doi: 10.1254/jphs.09152fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingelfinger JR. Selective, isolated proximal tubular angiotensinogen overexpression and salt-sensitive hypertension. Am J Hypertens. 2012;25:628. doi: 10.1038/ajh.2012.23. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins i and ii in anesthetized rats. Hypertension. 2002;39:129–134. doi: 10.1161/hy0102.100536. [DOI] [PubMed] [Google Scholar]
- 21.Danser AH, Schalekamp MA, Bax WA, van den Brink AM, Saxena PR, Riegger GA, et al. Angiotensin-converting enzyme in the human heart. Effect of the deletion/insertion polymorphism. Circulation. 1995;92:1387–1388. doi: 10.1161/01.cir.92.6.1387. [DOI] [PubMed] [Google Scholar]
- 22.Shibahara H, Kamata M, Hu J, Nakagawa H, Obara H, Kondoh N, et al. Activity of testis angiotensin converting enzyme (ace) in ejaculated human spermatozoa. Int J Androl. 2001;24:295–299. doi: 10.1046/j.1365-2605.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- 23.Lei B, Nakano D, Fujisawa Y, Liu Y, Hitomi H, Kobori H, et al. N-type calcium channel inhibition with cilnidipine elicits glomerular podocyte protection independent of sympathetic nerve inhibition. J Pharmacol Sci. 2012;119:359–367. doi: 10.1254/jphs.12075fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakano D, Kobori H, Burford JL, Gevorgyan H, Seidel S, Hitomi H, et al. Multiphoton imaging of the glomerular permeability of angiotensinogen. J Am Soc Nephrol. 2012;23:1847–1856. doi: 10.1681/ASN.2012010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeifer CA, Suzuki F, Jackson EK. Selective a1 adenosine receptor antagonism augments beta-adrenergic-induced renin release in vivo. Am J Physiol. 1995;269:F469–F479. doi: 10.1152/ajprenal.1995.269.4.F469. [DOI] [PubMed] [Google Scholar]
- 26.van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin ii type 1 (at1) receptor-mediated accumulation of angiotensin ii in tissues and its intracellular half-life in vivo. Hypertension. 1997;30:42–49. doi: 10.1161/01.hyp.30.1.42. [DOI] [PubMed] [Google Scholar]
- 27.Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. At1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin ii-dependent hypertension. Hypertension. 2004;43:1126–1132. doi: 10.1161/01.HYP.0000122875.91100.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishiyama A, Yoshizumi M, Rahman M, Kobori H, Seth DM, Miyatake A, et al. Effects of at1 receptor blockade on renal injury and mitogen-activated protein activity in dahl salt-sensitive rats. Kidney Int. 2004;65:972–981. doi: 10.1111/j.1523-1755.2004.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei B, Nakano D, Fan YY, Kitada K, Hitomi H, Kobori H, et al. Add-on aliskiren elicits stronger renoprotection than high-dose valsartan in type 2 diabetic KKAy mice that do not respond to low-dose valsartan. J Pharmacol Sci. 2012;119:131–138. doi: 10.1254/jphs.12031fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ihara G, Kiyomoto H, Kobori H, Nagai Y, Ohashi N, Hitomi H, et al. Regression of superficial glomerular podocyte injury in type 2 diabetic rats with overt albuminuria: effect of angiotensin ii blockade. J Hypertens. 2010;28:2289–2298. doi: 10.1097/HJH.0b013e32833dfcda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sofue T, Kiyomoto H, Kobori H, Urushihara M, Nishijima Y, Kaifu K, et al. Early treatment with olmesartan prevents juxta-medullary glomerular podocyte injury and the onset of micro-albuminuria in type 2 diabetic rats. Am J Hypertens. 2012;25:604–611. doi: 10.1038/ajh.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AH, et al. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation. 2012;125:1402–1413. doi: 10.1161/CIRCULATIONAHA.111.064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Villalobos RA, Satou R, Seth DM, Semprun-Prieto LC, Katsurada A, Kobori H, et al. Angiotensin-converting enzyme-derived angiotensin ii formation during angiotensin ii-induced hypertension. Hypertension. 2009;53:351–355. doi: 10.1161/HYPERTENSIONAHA.108.124511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kakimoto K, Takai S, Murano M, Ishida K, Yoda Y, Inoue T, et al. Significance of chymase-dependent matrix metallo-proteinase-9 activation on indomethacin-induced small intestinal damages in rats. J Pharmacol Exp Ther. 2010;332:684–689. doi: 10.1124/jpet.109.162933. [DOI] [PubMed] [Google Scholar]
- 35.Kanemitsu H, Takai S, Tsuneyoshi H, Nishina T, Yoshikawa K, Miyazaki M, et al. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens Res. 2006;29:57–64. doi: 10.1291/hypres.29.57. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad S, Varagic J, Westwood BM, Chappell MC, Ferrario CM. Uptake and metabolism of the novel peptide angiotensin-(1-12) by neonatal cardiac myocytes. PLoS One. 2011;6:e15759. doi: 10.1371/journal.pone.0015759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin ii from angiotensin-(1-12) in human atrial tissue. PLoS One. 2011;6:e28501. doi: 10.1371/journal.pone.0028501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Chen J, Xu T, Tian W, Li Y, Zhang Z, et al. Qiliqiangxin improves cardiac function in spontaneously hypertensive rats through the inhibition of cardiac chymase. Am J Hypertens. 2012;25:250–260. doi: 10.1038/ajh.2011.219. [DOI] [PubMed] [Google Scholar]
- 39.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin ii causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramkumar N, Stuart D, Ying J, Kohan DE. A possible interaction between systemic and renal angiotensinogen in the control of blood pressure. Am J Hypertens. 2013;26:473–480. doi: 10.1093/ajh/hps078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibrahim HN, Jackson S, Connaire J, Matas A, Ney A, Najafian B, et al. Angiotensin ii blockade in kidney transplant recipients. J Am Soc Nephrol. 2013;24:320–327. doi: 10.1681/ASN.2012080777. [DOI] [PMC free article] [PubMed] [Google Scholar]