

Abstract
Rationale
Cartilaginous metaplasia of vascular smooth muscle (VSM) is characteristic for arterial calcification in diabetes and uremia and in the background of genetic alterations in matrix Gla protein (MGP). A better understanding of the molecular details of this process is critical for the development of novel therapeutic approaches to VSM transformation and arterial calcification.
Objective
This study aimed to identify the effects of bioflavonoid quercetin on chondrogenic transformation and calcification of VSM in the MGP-null mouse model and upon TGF-β3 stimulation in vitro, and to characterize the associated alterations in cell signaling.
Methods and Results
Molecular analysis revealed activation of β-catenin signaling in cartilaginous metaplasia in Mgp-/- aortae in vivo and during chondrogenic transformation of VSMCs in vitro. Quercetin intercepted chondrogenic transformation of VSM and blocked activation of β-catenin both in vivo and in vitro. Although dietary quercetin drastically attenuated calcifying cartilaginous metaplasia in Mgp-/- animals, approximately one-half of total vascular calcium mineral remained as depositions along elastic lamellae.
Conclusion
Quercetin is potent in preventing VSM chondrogenic transformation caused by diverse stimuli. Combined with the demonstrated efficiency of dietary quercetin in preventing ectopic chondrogenesis in the MGP-null vasculature, these findings indicate a potentially broad therapeutic applicability of this safe for human consumption bioflavonoid in the therapy of cardiovascular conditions linked to cartilaginous metaplasia of VSM. Elastocalcinosis is a major component of MGP-null vascular disease and is controlled by a mechanism different from chondrogenic transformation of VSM and not sensitive to quercetin.
Introduction
In the normal vasculature, the principal function of smooth muscle cells is contraction to regulate blood pressure and blood flow throughout the body [1]. In contrast to the other terminally-differentiated muscle cells, cardiac and skeletal, vascular smooth muscle cells (VSMCs) undergo a unique phenotypic modulation during normal development, and in the physiologic response to blood vessel remodeling or injury. This is associated with decreased expression of VSMC-specific contractile genes and increased cell proliferation and migration. However, there is growing evidence that this phenotypic plasticity of VSMCs contributes to vascular disease by allowing for differentiation to inappropriate lineages [1–3]. For example, in diabetic arteriosclerosis VSMCs undergo osteogenic transformation, losing contractile gene expression and depositing a calcified bone-like matrix. Together this results in increased vessel stiffness and decreased compliance, a strong predictor of cardiovascular risk [4].
Recent mechanistic studies reveal the complexity of phenotypic instability in VSMCs. The literature in this field is extensive and identifies diverse regulators including, but not limited to, growth factors, inflammatory cytokines, calcium-phosphate homeostasis, oxidized phospholipids, retinoic acid and mechanical stress, and involves multiple signaling pathways including MAPK kinases, Rho, Notch, BMP and β-catenin signaling (reviewed in [5–7]).
Lineage tracing studies have demonstrated that VSMC-derived hyperproliferative cells play a role in neointimal formation induced by either injury [8] or atherosclerosis [9], and that osteochondrogenic transformation of VSMCs associates with calcification of the arterial media [10–13] as well as atherosclerotic calcification [14]. Vascular calcification (or pathologic calcium phosphate deposition in the blood vessels) is a common complication associated with diabetes, hypercholesterolemia, chronic renal insufficiency and osteoporosis, and is highly correlated with cardiovascular disease mortality. Its clinical consequences include stroke, amputation, ischemic heart disease, and complications during artery stenting. In addition, vascular calcification is now recognized as a marker of atherosclerotic plaque burden [15,16]. The incidence of vascular calcification also correlates with age, putting a growing percentage of our population at risk as life expectancy increases.
A decrease in circulating and local inhibitors of mineralization, especially in dialysis patients with chronic kidney disease, appears to significantly contribute to calcification. One of the major anti-calcific factors is matrix Gla protein (MGP), a 10 kDa polypeptide containing calcium-binding γ-carboxyglutamic acid (Gla) residues and phosphoserine residues. In the vasculature, MGP is expressed in vascular smooth muscle (VSM) and endothelium [17,18]. MGP polymorphisms have been linked to coronary artery calcification in patients [19,20], while in mice genetic loss of MGP leads to cartilaginous metaplasia and extensive calcification of the tunica media [21]. MGP-null arterial disease has been described primarily as a product of chondrogenic transformation of Mgp-/- VSM resulting in the ectopic formation of cartilage in the vessel wall, extensive calcification, and ultimately rupture of the blood vessels soon after birth [21]. While mechanisms underlying the osteogenic transformation of VSM induced by warfarin [10–13] and by hyperphosphatemia [22,23] are emerging [7,24,25], the chondrogenic transformation of vascular cells that is prominent in uremia- and diabetes-associated medial calcification [26,27] is not well understood. The Mgp-/- vasculature is an excellent model for mechanistic analysis of chondrogenic transformation in VSM because 97% of the chondrocyte-like cells in arterial cartilaginous metaplasia originate from VSM [13] suggesting a minimal (if any) contribution by circulating or resident multipotent mesenchymal progenitors.
With a growing mechanistic understanding, treatment options for vascular diseases are rapidly developing. In particular, emerging interest has been focused on the protective effects of flavonoid-rich diets in cardiovascular disease. The reported beneficial effects of the major bioflavonoid, dietary quercetin, in humans include lower blood pressure and LDL levels and overall reduced cardiovascular disease-related mortality [26–29]. In animal models, quercetin effectively alleviated atherosclerosis [30,31] and attenuated warfarin-induced hypertension and elastocalcinosis [12]. Quercetin has anti-inflammatory, anti-oxidative and anti-proliferation effects. In addition, we have found that in VSMCs quercetin inhibits β-catenin signaling, central for osteogenic transformation of these cells, and vascular calcification induced by warfarin [12,25]. β-catenin is a multi-tasking molecule regulating developmental and homeostatic processes. It is an integral part of adherent junctions and the key mediator of the canonical Wnt/β-catenin signaling cascade, and has been shown to cross-talk directly with non-Wnt pathways [32]. During development Wnt/β-catenin signaling plays a manifold role and promotes physiological chondrocyte maturation [33] as well as vascular remodeling and differentiation [34]. In adult vessels, the β-catenin pathway is usually dormant but activates in disease [35]. In particular, a critical role for β-catenin signaling has been shown in warfarin-induced calcification [36,37]. Further, Wnt/β-catenin signaling has also been implicated in BMP2-induced aortic mineralization in the diabetic LDLR-/- mice [38] and in calcification of heart valves [39]. Here, we investigated β-catenin signaling associated with chondrogenic transformation of Mgp-/- VSM, and examined the efficiency of quercetin in alleviating the MGP-null vascular disease.
Materials and Methods
Animals
4.5 to 5 week-old C57BL/6J wild-type mice (Mgp+/+) and mice deficient in MGP (Mgp-/-
) were used in this study. MGP-deficient mice were a kind gift from Dr. Karsenty, Columbia University, New York. Mice were genotyped using standard PCR for Mgp. All procedures were approved by the institutional animal care and use committees at the University of Maryland Medical School (protocol #: 0912010) and the University of North Carolina School of Medicine, and were conducted in compliance with National Institutes of Health guidelines for the care and use of laboratory animals.
Quercetin treatment
Quercetin (Quercegen, Newton, MA) was administered as a dietary supplement (0.02% w/w in drinking water) to weaned pups for 2 weeks. Alternatively, quercetin was given to lactating Mgp+/- females immediately after they gave birth to the progeny and continued throughout the 3-week breastfeeding period. Weaned Mgp-/- pups continued to receive quercetin in drinking water for an additional 2 weeks.
Immunohistochemistry and Histological Staining
Frozen 10 µm sections of freshly-dissected aortas fixed in 4% paraformaldehyde were stained for proteoglycan deposition and calcified matrix using the alcian blue and von Kossa silver nitrate methods, respectively, according to standard protocols [40].
Immunofluorescence was performed using the standard protocol. Briefly, tissue was blocked with 10% goat serum for 30 minutes at room temperature. Tissue sections were incubated with primary antibodies diluted in 1% goat serum overnight at 4°C and secondary antibodies diluted in PBS for 1.5 hours at room temperature. Nuclei were counterstained with 4',6-diamidino-2-phenylindole (DAPI). Primary antibodies used were rabbit anti-β-catenin (1:80; Santa Cruz Biotechnology) and rabbit anti-Ki67 (1:100; Abcam), detected with a secondary Dylight 549-conjugated goat anti-rabbit antibody (1:400; Jackson ImmunoResearch); mouse anti-type II collagen antibody (a kind gift of Dr. Thomas Linsenmayer), detected with a secondary Alexa 488-conjugated goat anti-mouse antibody; and mouse anti-smooth muscle actin antibody directly conjugated to fluorescein isothiocyanate (FITC). Images were collected using a Leica DMIL inverted microscope equipped with a SPOT RT3 real-time CCD camera (Diagnostic Instruments).
Morphologic Analysis
For morphologic analyses, serial sections spaced 100 µm apart along a 1 mm length of descending aorta from heterozygous (Mgp+/-
), Mgp-/-, and quercetin-treated Mgp-/- (Mgp-/- +Querc) animals were analyzed for chondrogenic lesions and average thickness of tunica media. For each animal, the average value of 4-5 serial sections was used. Mean and standard error values used for graphical display were calculated from the average values of each animal. All values were normalized to wild-type animals (set at 100%).
Cell culture and Luciferase Analysis
Murine aortic smooth muscle cells were obtained by a modification of the explant method described by Ross [41]. Briefly, medial tissue was isolated from segments of thoracic aorta from wild-type C57BL/6J (Mgp+/+) mice. Small pieces of tissue (1 to 2 mm) were loosened by a 1-hour incubation at 37°C in medium supplemented with 165 U/mL collagenase type I. Partially digested tissues were placed in 6-well plates and cultured for 10 days in DMEM supplemented with 20% FBS at 37°C in a humidified atmosphere containing 5% CO2. Cells that migrated from the explants were collected and maintained in growth medium (DMEM containing 10% FBS and 100 U/mL of penicillin and 100 µg/mL of streptomycin). To confirm that the cells isolated were VSMCs, α-smooth muscle actin expression was confirmed by immunofluorescence (data not shown). For experiments, cells were used at passages 2 and 3.
The A10 clonal embryonic rat aortic smooth muscle cell line (ATCC, Manassas VA) was also maintained in growth medium. A stable β-catenin-dependent luciferase reporter cell line was established by transducing A10 VSMCs with the Cignal Lentiviral TCF/LEF luciferase reporter (SA Biosciences), according to manufacturer’s protocol, followed by a 2-week selection with puromycin (10 µg/mL). Luciferase activity in whole cell lysates was measured in a 96-well plate luminometer (Harta Instruments, Bethesda MD) using the Promega Luciferase Assay Kit and was normalized to the total lactate dehydrogenase (LDH) present in whole cell lysates, measured using a commercial LDH activity kit (BioVision, San Francisco CA). LDH activity in the culture medium was also measured to determine cell viability.
To induce chondrogenic transformation, VSMCs were seeded at high density (2.5x105 cells in 10 µL volume) and cultured in chondrogenic medium (ChoM) [DMEM–high glucose supplemented with 10−7 mmol/L dexamethasone, 0.1 mmol/L ascorbic acid (Wako Chemicals, VA), 1% ITS premix (BD Biosciences, NJ), 1 mmol/L sodium pyruvate, 0.35 mmol/L L-proline, 4 mmol/L L-Glutamine (Invitrogen), 1% Pencillin–Streptomycin (Invitrogen), and 10 ng/mL TGF-β3 (ProSpec, NJ)] in the presence or absence of quercetin (50 µmol/L) or recombinant Dkk1 (0.5 µg/mL; R&D Systems) at 37°C, 5% CO2. Medium was changed twice a week for up to 14 days. Sulfated glycosaminoglycan (GAG) synthesized by cultured VSMCs was detected by fixing them in 4% PFA and staining with 1% Alcian blue (8GX) dissolved in 0.1 N HCl. For quantitative analysis, Alcian blue was extracted with 4 M guanidine hydrochloride and absorbance at 590 nm was measured using an Optima spectrophotometer (PolarStar). Cell density was estimated using WST substrate (Dojindo).
qRT-PCR
mRNA was isolated using the RNeasy micro kit (Qiagen) and reverse transcription was performed with MaximaRT kit (Thermo, Fisher). Quantitative real-time PCR was performed according to the standard protocol with EVA green chemistry in a CFX96 thermocycler (Bio-Rad) using the primers in Table 1. Relative change in gene expression was calculated by the ΔΔCt method using Microsoft Excel.
Table 1. Primer sequences for mouse genes analyzed by real-time PCR.
| Gene | Accession # | Forward primer | Reverse primer | |
|---|---|---|---|---|
| β-catenin targets | ||||
| Lrp6 | NM_008514 | ggtgtcaaagaagcctctgc | gctcgaggactgtcaaggtc | |
| Tcf4 | NM_013685 | cgaaaagttcctccgggtttg | cgtagccgggctgattcat | |
| Numb | NM_010949 | cttcccagttaagtacctcggc | cccgtttttccaaagaagcct | |
| CyclinD1 (cycD1) | NM_007631 | gcgtaccctgacaccaatctc | ctcctcttcgcacttctgctc | |
| Chondrogenic markers | ||||
| Sox9 | NM_011448 | gttgtaacaccagcagcgtcaag | tgacatactccactttggccacct | |
| Type II collagen (Col II) | NM_031163 | caccgaaagtttaagcacaccca | aaataaccctgcccacactcttg | |
| Aggrecan (Agg) | NM_007424 | atgccacaagtcacagaaaccacg | aaggcagtcacagcattgttgagc | |
| Indian hedgehog (Ihh) | NM_010544 | cgtgaccgaaataagtatggact | gctgctggttctgtatgattg | |
| MMP13 | NM_008607 | tggagtgcctgatgtgggtgaata | tggtgtcacatcagaccagacctt | |
| TG2 | NM_009373 | aggtgtccctgaagaacccacttt | ttccacagacttctgctccttggt | |
| Housekeeping genes | ||||
| Ribosomal protein L19 (Rpl19) | NM_009078 | aagaggaagggtactgccaatgct | tgaccttcaggtacaggctgtgat | |
| beta-actin | NM_007393 | taatttctgaatggcccaggtct | ctggctgcctcaacacctcaa | |
Calcium Determination
Aortas were dried at 55°C overnight and then dissolved in 0.1N hydrochloric acid (HCl) overnight. Calcium content was determined biochemically using the Calcium (CPC) LiquiColor kit as measured with o-Cresolphthalein (Stanbio). Calcium content of the aorta was normalized to dry weight of the tissue prior to HCl extraction.
Tissue Preparation for Gene Expression Analysis using RT2 Profiler PCR Arrays
Aortic tissue from 5 week old mice was disrupted by rapid agitation in the presence of a stainless steel bead (5 mm mean diameter) and lysis buffer using a TissueLyser (2 min at 20Hz, twice; Qiagen). Total RNA was isolated following the RNeasy Fibrous Tissue micro kit protocol (Qiagen) according to the to the manufacturer’s instructions and quantified with NanoDrop spectrophotometer (Thermo Scientific) prior to storage at −80°C. Reverse transcription was performed using 1 µg of RNA according to SA Biosciences’ recommendations using the RT2 First Strand kit. Gene expression analysis was done using the WNT Signaling Pathway PCR Array (PAMM-043), WNT Signaling Targets PCR Array (PAMM-243) and Osteogenesis PCR Array (PAMM-026). ΔΔCt based fold-change calculations were performed using the RT² Profiler PCR Array Data Analysis Template v3.2. In this software standard deviation is calculated for Ct values and not for final fold-changes in expression.
Statistical Analysis
The data are presented as mean ± standard error. For experiments containing only 2 groups, significance was determined by comparison using Wilcoxon-Mann-Whitney test. For experiments containing more than 2 groups, Levene’s test was used to determine equality of variance (homoscedasticity) followed by 1-way analysis of variance (ANOVA) and Tukey-Kramer post-hoc analysis for comparison between groups. A p-value of < 0.05 was considered to be statistically significant. *, p <0.05; **, p < 0.01; ***, p<0.001.
Results
Analysis of proliferation in chondroplastic areas of Mgp-/- aortae
Mgp-/- mice are characterized by the presence of ectopic cartilage in the tunica media [21]. To identify potential mechanisms of the formation of cartilaginous lesions, we first analyzed the possible contribution of increased cell proliferation. Foci of chondrogenic metaplasia were readily detected as areas with the characteristic appearance of rounded chondrocyte-like cells embedded in glycoprotein-rich matrix positive for Alcian Blue staining and cartilaginous collagen type II (Figure 1A), in agreement with previous studies [13,21,42]. We then performed immunohistochemistry for Ki67, a marker of cell proliferation, (Figure 1B) and determined the proportion of the proliferating cells positive for Ki67, compared to total nuclei visible by DAPI nuclear counterstain, in wild-type and Mgp-/- aortae. Aortic tissue isolated from newborn, 7- and 30-day old animals was examined. 3 random cross-sections of aortae from each animal were analyzed. The percentage of Ki67-positive proliferating cells was similar between wild-type and Mgp-/- aortae at all ages examined (Figure 1C), suggesting that the formation of cartilaginous metaplasia is driven by phenotypic transformation, rather than proliferation, of VSMCs.
Figure 1. Cartilaginous metaplasia and cell proliferation in the Mgp-/- aorta.
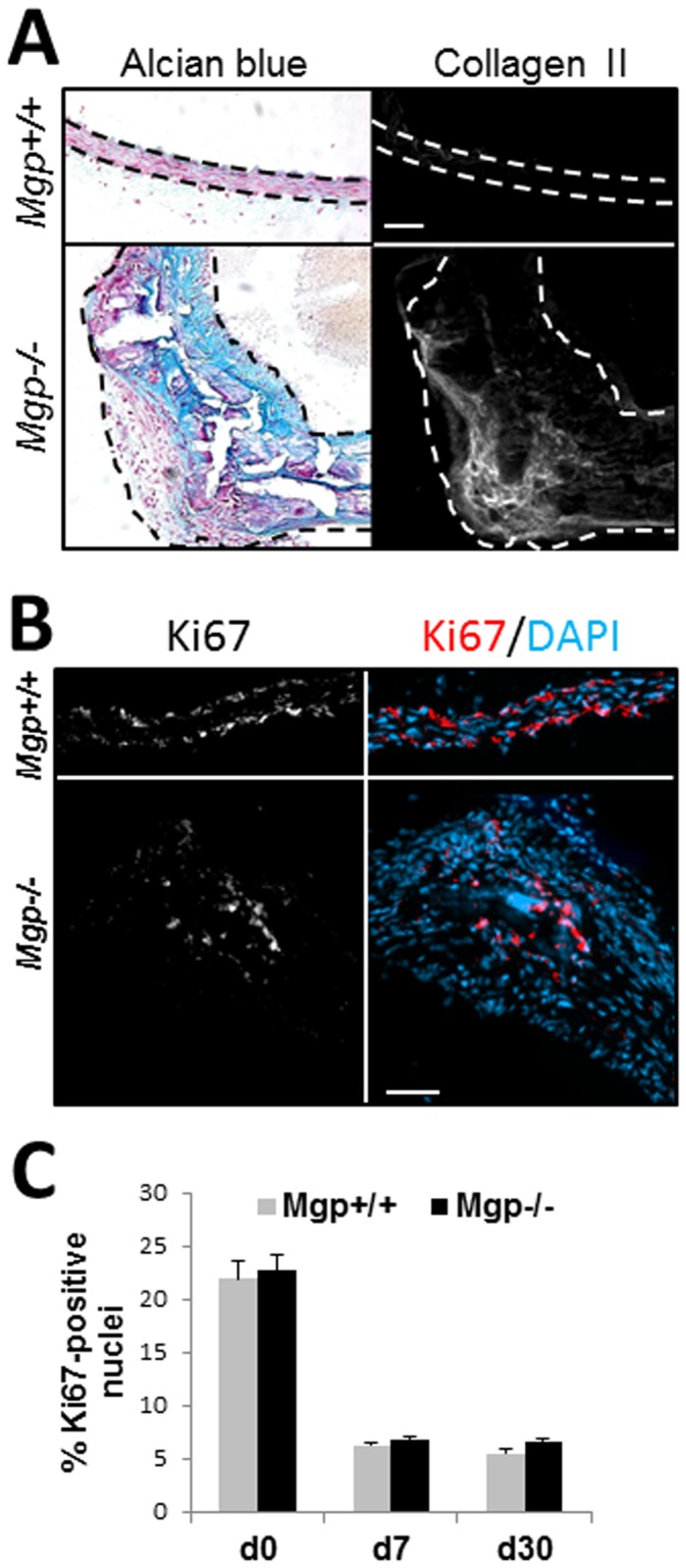
A, Detection of cartilage matrix using Alcian blue staining for GAG deposition and immunostaining for Collagen type II (Collagen II) protein on adjacent sections of aorta from 4.5 week old wild-type (Mgp+/+) and Mgp-/- animals. Scale = 50 µm. Dashed lines denote internal and external elastic lamina. B, Representative immunostaining for cell proliferation marker, Ki67 (white, red) [nuclei counterstained with DAPI (blue)] in 30 day old Mgp+/+ and Mgp-/- aortae. Scale = 50 µm. C, Quantitation of the percentage of Ki67-positive nuclei compared to total nuclei in Mgp+/+ and Mgp-/- aortic tissue from animals at birth (d0, N=3), at 7 days old (d7, N=4), and at 30 days old (d30, N=4). Four sections per animal were analyzed.
Activation of β-catenin signaling in Mgp-/- aortae
Canonical β-catenin signaling regulates normal physiologic chondrogenesis [33]. Here, we investigated the association between activation of this signaling cascade and chondrogenic transformation in Mgp-/- aortae using a qRT-PCR-based microarray of 84 components and regulators of Wnt/β-catenin signaling (4.5 week old Mgp-/- and wild-type mice, N=6). A number of activating Wnt ligands and transcriptional co-activators of β-catenin signaling such as Ccnd3, Pitx2 and Lef1 are induced in the Mgp-/- aortae (Table 2), although arterial expression levels of the canonical Wnts detected by the qRT-PCR were very low in both wild-type and Mgp-/- tissue (with only Wnt3a threshold cycle number being less than 35). In addition, the drastic down-regulation of sclerostin, an inhibitor of Wnt signaling, further indicates activation of this pathway in Mgp-/- arteries (Table 2).
Table 2. Changes in expression of genes in the Wnt signaling pathway in Mgp-/- compared to wild-type aortae.
| Gene | Gene product | Fold change | p-value | Role in Wnt/β-catenin signaling |
|---|---|---|---|---|
| Ccnd3 | Cyclin D3 | 2.32* | 0.021 | Co-activator of nuclear β-catenin [56] |
| Lef1 | Lymphoid enhancer binding factor 1 | 3.63* | 0.036 | Co-activator of nuclear β-catenin [57] |
| Pitx2 | Paired-like homeodomain transcription factor 2 | 2.72** | 0.009 | Co-activator of nuclear β-catenin [58] |
| Tle2 | Transducin-like enhancer of split 2, homolog of Drosophila E(spl) | 2.62* | 0.014 | possible link between Wnt and Notch signaling [59] |
| Wnt3a | Wingless-related MMTV integration site 3A | 2.70* | 0.020 | |
| Wnt4 | Wingless-related MMTV integration site 4 | 3.23* | 0.028 | |
| Up-regulated, low abundance (Ct > 35) | ||||
| Fshb | Follicle stimulating hormone beta | 2.73** | 0.005 | transcriptional co-activator for aromatase [60] |
| Wnt1 | Wingless-related MMTV integration site 1 | 2.35** | 0.005 | |
| Wnt10a | Wingless related MMTV integration site 10a | 2.35** | 0.005 | |
| Wnt3 | Wingless-related MMTV integration site 3 | 2.41* | 0.031 | |
| Wnt7a | Wingless-related MMTV integration site 7A | 2.35** | 0.005 | |
| Wnt7b | Wingless-related MMTV integration site 7B | 2.73** | 0.005 | |
| Wnt8a | Wingless-related MMTV integration site 8A | 3.08* | 0.031 | |
| Down-regulated genes | ||||
| Fzd2 | Frizzled homolog 2 (Drosophila) | -2.85* | 0.011 | |
| Sost | Sclerostin | -165.46*** | 0.006 | Negative regulator of Wnt signaling [61] |
| Wif1 | Wnt inhibitory factor 1 | -8.56* | 0.013 | |
| Wnt16 | Wingless-related MMTV integration site 16 | -17.98*** | 0.001 | non-canonical Wnt signaling, possible link to Notch signaling [62] |
Aortic tissue from 4.5 week old animals was analyzed using a qRT-PCR-based microarray N=6. * p<0.05; ** p<0.01; *** p<0.001.
To address this possibility, we used the qRT-PCR-based array approach to study expression of 84 β-catenin target genes in Mgp-/- and wild-type aortae (N=6). This analysis revealed a higher than 2-fold induction in 50% of the β-catenin target genes in the Mgp-/- aortae with 28 genes showing a statistically significant change (Table 3; p<0.05) and 16 additional genes showing a trend towards induction (p>0.05). Importantly, many of the β-catenin target genes significantly upregulated in the Mgp-/- arterial tissue are related to osteochondrogenic transformation or VSMC de-differentiation (Table 3). These results indicate β-catenin signaling is activated in the absence of MGP and that its activation may contribute to chondrogenic transformation in VSM.
Table 3. Changes in expression of Wnt/β-catenin target genes in Mgp-/- compared to wild-type aortic tissue.
| Gene | Protein | Fold change | p-value | Function |
|---|---|---|---|---|
| Abcb1a | Transmembrane glycoprotein | 6.41** | 0.005 | ATP-binding cassette |
| Abr | Aryl-hydrocarbon receptor | 3.04* | 0.043 | Transcription factor |
| Birc5 | Survivin | 3.67*** | 0.001 | Inhibitor of apoptosis |
| Cacna2d3 | 3.02* | 0.035 | Calcium channel | |
| Cd44 | 2.93** | 0.008 | Receptor for ECM | |
| Cdkn2a | Cyclin-dependent kinase inhibitor | 5.32* | 0.016 | Cell cycle |
| Cdon | Cell adhesion molecule | 2.47** | 0.007 | Shh receptor |
| Cubn | Cobalamin and vitamin B12 receptor | 2.58* | 0.013 | |
| Enpp2 | Autotaxin | 5.32*** | 0.001 | Lysophospholipase |
| Foxn1 | Forkhead box N1 | 6.30* | 0.026 | Cell growth and proliferation |
| Mmp9 | Matrix metalloproteinase 9 | 4.16* | 0.036 | VSMC migration/proliferation [63] |
| Nanog | 13.94* | 0.041 | “stem-like” phenotype | |
| Tcf4 | 3.89*** | 0.001 | Transcription factor | |
| Chondro/Osteogenic differentiation | ||||
| Dkk1 | Dickkopf 1 | 9.85** | 0.007 | Wnt inhibitor, negative regulator of osteogenesis [64] |
| Fgf9 | Fibroblast growth factor 9 | 4.14* | 0.036 | Chondrocyte hypertrophy [65,66] |
| Gdf5 | Growth differentiation factor 5 | 5.02* | 0.046 | Osteogenic differentiation [67,68] |
| Met | Receptor for hepatocyte growth factor | 2.00* | 0.028 | Osteogenic transformation in VSMC, bone/cartilage formation [69,70] |
| Mmp2 | Matrix metalloproteinase 2 | 3.48** | 0.010 | Required for mineralization in VSMC [71] |
| Plaur | Plasminogen activator, urokinase (uPAR) receptor | 3.84* | 0.015 | VSMC de-differentiation in [72] |
| T | Brachyury | 3.38* | 0.020 | Chondrogenic differentiation [73] |
| Wisp1 | WNT1-inducible signaling protein 1 | 4.30** | 0.008 | Osteoblast differentiation, represses chondrogenesis [74] |
| Endothelial genes | ||||
| Cdh1 | E-cadherin | 6.19*** | 0.001 | |
| Dlk1 | Delta-like 1 homolog | 5.84* | 0.038 | Non-canonical Notch ligand, inhibits angiogenesis [75] |
| Nrcam | Neuron-glia-CAM-related cell adhesion molecule | 16.14*** | 0.001 | Cell communication, angiogenesis [76] |
| Down-regulated genes | ||||
| Ccnd2 | Cyclin D2 | -1.39* | 0.040 | Proliferation |
| Fzd7 | Frizzled 7 | -4.05* | 0.043 | Non-canonical β-catenin signaling [77] |
| Gja1 | Connexin 43 | -2.99* | 0.013 | Supports bone formation [78–80] |
| Igf2 | Insulin-like growth factor 2 | -3.49* | 0.047 | Enhances osteogenic capacity of hMSCs [81] |
Aortic tissue from 4.5 week old animals was analyzed using a qRT-PCR-based microarray. N=6. * p<0.05; ** p<0.01; *** p<0.001.
Chondrogenic transformation of VSMCs in vitro: activation of β-catenin signaling
To determine whether activation of β-catenin signaling directly associates with chondrogenic transformation of VSM, in contrast to being potentially induced by systemic factors in the Mgp-/- mice, we employed an in vitro system in which the A10 rat aortic VSMC line or primary mouse VSMCs (P2-3) were induced to undergo chondrogenic transformation in high-density micromass cultures stimulated by dexamethasone and TGF-β3 [43]. Within two weeks in culture, both rat and mouse VSMCs underwent chondrogenic transformation, evidenced by the formation of characteristic glycosaminoglycan (GAG)-rich cartilaginous nodules (Figure 2A). This was accompanied by activation of β-catenin signaling, detected by increased expression of a β-catenin-dependent luciferase transgene in A10 cells (Figure 2B, 8.16 ±2.02-fold, p<0.05).
Figure 2. Inhibition of β-catenin signaling attenuates chondrogenic transformation of cultured VSMCs.

Rat A10 VSMCs stably-expressing a β-catenin-responsive luciferase transgene were induced to undergo chondrogenesis in high-density micromass culture in chondrogenic medium (ChoM) containing TGF-β3. A, VSMCs form cartilaginous GAG-positive nodules as detected by Alcian blue stain, in ChoM (- Querc). In the presence of 50 µmol/L Quercetin (ChoM + Querc) both the number and size of the nodules is reduced. B-E, Expression of β-catenin-dependent luciferase reporter (B,D) and deposition of GAG-rich matrix, quantified by extraction of Alcian blue stain normalized to cell number (C,E), in VSMCs cultured in ChoM in the presence or absence of quercetin (B,C) or the β-catenin signaling pathway inhibitor Dkk1 (D,E). N=4.
Chondrogenic transformation of VSMCs in vitro: attenuation by quercetin
Quercetin has been characterized as a potent inhibitor of β-catenin signaling, and in VSMCs quercetin stabilizes the contractile phenotype [25]. Here we show that quercetin significantly attenuated the activation of β-catenin signaling in chondrogenic micromass cultures of A10 cells (Figure 2B) and also significantly reduced chondrogenic differentiation in these cells as evidenced by a reduction in GAG-positive matrix detected with Alcian Blue stain (Figure 2C). To determine whether the observed inhibition of β-catenin signaling caused the reduction in GAG synthesis, we treated chondrogenic micromass cultures with recombinant Dkk1, a known inhibitor of the canonical β-catenin pathway [44]. Dkk1 prevented both increased expression of the β-catenin-dependent luciferase reporter (Figure 2D) and deposition of cartilaginous GAG matrix by A10 VSMCs (Figure 2E), demonstrating a critical role for activation of β-catenin signaling in the chondrogenic transformation of VSMCs in vitro.
Similar to its effects on the A10 VSMC line, quercetin significantly reduced GAG deposition by primary mouse aortic VSMCs treated with chondrogenic medium in micromass culture without affecting cell survival as evidenced by unchanged levels of LDH activity in the culture medium (Figure 3A). Analysis of stage-specific markers of chondrogenesis using qRT-PCR showed that quercetin prevented induction of both early (Sox9, collagen type II, and aggrecan) and late (Ihh, MMP13, and TG2) markers (Figure 3B), identifying this flavonoid as a potent inhibitor of chondrogenic transformation in VSMCs and suggesting the potential therapeutic value of quercetin in preventing chondrogenic transformation of VSM in vivo. In addition, qRT-PCR analysis revealed that the expression of four endogenous β-catenin target genes induced during chondrogenic transformation of VSMCs was prevented by quercetin indicating inhibition of β-catenin signaling (Figure 3C). Further, treatment of the primary VSMC micromasses with the β-catenin pathway inhibitor Dkk1 blocked GAG-rich matrix deposition (Figure 3D), mimicking the effect of quercetin and implicating the β-catenin signaling pathway as a major target of quercetin-mediated inhibition of chondrogenic transformation in cultured VSMCs.
Figure 3. Quercetin attenuates chondrogenic transformation and β-catenin activation in primary VSMCs.

A, Quercetin reduces deposition of GAG-positive matrix in VSMCs induced to undergo chondrogensis in high-density micromass culture in chondrogenic medium (ChoM). Quantitation of GAG deposition (left graph) and cell death, detected by LDH release into the culture medium (right graph), in the presence or absence of 50 µmol/L quercetin. N=4. B-C, Real-time PCR analysis of markers of chondrogenesis (B) and β-catenin target genes (C) in VSMC micromass cultures treated with ChoM and quercetin as indicated. N=4. D, Quantitation of GAG deposition by VSMC micromass cultures treated with ChoM in the presence or absence of 0.5 µg/mL Dkk1. N=4.
Quercetin reduces calcium accrual in Mgp-/- aortae
Taking into consideration that extensive calcification of chondrogenic metaplasia is characteristic for MGP-null vascular disease, we examined whether quercetin treatment alleviates calcification in MGP-deficient animals. After weaning, we treated Mgp-/- and Mgp+/- pups for 2 weeks with dietary quercetin (0.02% w/w in drinking water [30]) and analyzed total calcium content in the aortae biochemically by the o- cresolphthalein complexone method. We found that quercetin treatment associated with a significant 20.0±2.1% (p<0.001) reduction in total calcium content of Mgp-/- aortic tissue (Figure 4A), although aortic calcium levels in the quercetin-treated Mgp-/- animals remained significantly higher than those in heterozygous littermates. In addition, quercetin treatment associated with a significant reduction in the expression of osteogenic genes in Mgp-/- aortic tissue (Table 4).
Figure 4. Quercetin reduces calcium accrual in Mgp-/- mice.

A-B, Total calcium in aortic tissue from 5 week old untreated Mgp-/- mice (N=8) or Mgp-/- mice treated with 0.02% w/w dietary quercetin for 2 weeks after weaning (A; N=16) or for 5 weeks from birth through weaning (B; N=9). Heterzygous (Mgp+/-) mice served as control (N=6). C, Von Kossa stain for calcified matrix deposition in aortic tissue from Mgp+/-, untreated Mgp-/-, and quercetin-treated Mgp-/- animals. Sections from 2 representative animals are shown. Arrows denote chondrogenic metaplasia. Scale = 50 µm.
Table 4. Changes in osteogenic gene expression in aortas from Mgp-/- mice treated with quercetin compared to untreated Mgp-/- aortic tissue.
| Gene | Protein name | Fold Down-Regulation | p-value |
|---|---|---|---|
| AlpI | Alkaline phosphatase | -2.09** | 0.032 |
| Anxa5 | Annexin 5 | -1.90** | 0.010 |
| Bmpr1b | BMP receptor, type 1B | -2.94** | 0.006 |
| Col14a1 | Collagen type XIV, alpha 1 | -4.28** | 0.008 |
| Col1a2 | Collagen type I, alpha 2 | -3.18** | 0.043 |
| Col4a2 | Collagen type IV, alpha 2 | -2.63** | 0.035 |
| Col6a1 | Collagen type VI, alpha 1 | -2.53** | 0.048 |
| Col6a2 | Collagen type VI, alpha 2 | -3.76** | 0.015 |
| Fgf3 | Fibroblast growth factor 3 | -6.78** | 0.002 |
| Fgfr1 | Fibroblast growth factor receptor 1 | -2.19** | 0.014 |
| Itga3 | Integrin alpha 3 | -2.03** | 0.027 |
| Itgav | Integrin alpha V | -3.17** | 0.002 |
| Mmp8 | Matrix metalloproteinase 8 | -4.20** | 0.051 |
| Smad1 | MAD homolog 1 | -1.48** | 0.017 |
| Tgfb3 | Transforming growth factor, beta 3 | -2.77** | 0.001 |
| Tgfbr1 | Transforming growth factor receptor, beta 1 | -3.19** | 0.043 |
| Col10a1 | Collagen type X, alpha 1 | -8.07** | 0.105 |
| Mmp10 | Matrix metallopeptidase 10 | -7.06** | 0.145 |
Tissue from 4.5 week old animals was analyzed using a qRT-PCR-based microarray N=5. *, p<0.05; ** p<0.01.
We hypothesized that quercetin may prevent the de novo formation of calcifying cartilaginous metaplasia but not reverse calcification already present in 3 week old animals at weaning. Therefore, to affect younger animals we treated newborn Mgp-/- mice with quercetin for 5 weeks (via the lactating moms until weaning and then in the drinking water as described above) and analyzed calcium accrual in these mice compared to Mgp+/- littermates. Quercetin treatment from birth produced a 36.3±1.7% (p<0.01) reduction in total calcium content of Mgp-/- aortic tissue (Figure 4B), almost twice what was observed in animals treated with quercetin for only 2 weeks. However, calcium levels in the quercetin-treated Mgp-/- aortae still remained significantly elevated. To examine this phenomenon further, localization of calcium phosphate deposits in the arterial walls was detected with von Kossa staining (Figure 4C). Excessive calcium phosphate deposits aligning with elastic lamellae were present in both untreated and quercetin-treated Mgp-/- mice, suggesting that elastocalcinosis accounts for the remaining 64% of calcium deposits in quercetin-treated mice thus identifying elastocalcinosis as a major contributor to MGP null vascular disease. In contrast, the massive calcified zones corresponding to cartilaginous metaplasia were absent in quercetin-treated Mgp-/- mice, indicating that in vivo chondrogenic transformation of VSM is sensitive to quercetin treatment.
Dietary quercetin alleviates cartilaginous metaplasia
We next analyzed the reduction of cartilaginous metaplasia by quercetin in more detail. Aortae were collected from Mgp-/- mice treated with quercetin for 4.5 weeks and vessel wall morphology was analyzed along a 1mm long aortic segment using serial tissue sections collected 100 µm apart (Figure 5A). Quercetin diet prevented cartilaginous metaplasia (indicated by dashed lines) and improved gross morphology of the Mgp-/- aortic wall (Figure 5B). Quantitatively, quercetin treatment significantly reduced the proportion of cartilaginous lesions in the circumference of vessel wall from 59.6±7.3% in the untreated Mgp-/- mice (N=5) to 13.3±3.1% in the quercetin treated animals (N=6, p<0.01) (Figure 5C). Accordingly, we detected an approximately 30% reduction in arterial wall thickness (64.2±5.0 µm in quercetin-treated Mgp-/- mice compared to 90.1±9.9 µm in untreated Mgp-/- animals), rendering this parameter similar to that of the control aortae (Mgp+/-, N=3) (Figure 5D).
Figure 5. Quercetin prevents chondroplasia and improves vessel morphology in Mgp-/- mice.

A, Serial sections spaced 100 µm apart through a 1 mm segment of descending aorta from each animal were analyzed. B, Representative images of serial sections from Mgp+/- (N=3), untreated Mgp-/- (N=5), and quercetin-treated Mgp-/- (N=6) animals stained with Alcian blue. Dashed lines denote the proportional presence of chondroplastic lesions in the vessel wall. Scale = 50 µm. C-D, Quantitative analysis of the serial sections of aortae show that quercetin diet reduces the percentage of vessel circumference occupied by chondrogenic lesions (C) and the cross-sectional thickness of the medial aorta (D).
Further, qRT-PCR analysis revealed that expression of cartilaginous extracellular matrix proteins and transcription factors associated with chondrogenesis [45,46] were increased in Mgp-/- animals, and this increase was significantly attenuated in quercetin-treated Mgp-/- animals (Table 5). Of note, both early and late markers of chondrogenesis were attenuated by quercetin, implying that the prevention of chondrogenic transformation of VSM by this flavonoid in vivo is similar to its action in vitro.
Table 5. Changes in expression of genes related to chondrogenic differentiation in Mgp-/- versus wild-type and in quercetin-treated Mgp-/- (Mgp-/- +Q) versus untreated Mgp-/- aortic tissue from 4.5 week old animals analyzed by qRT-PCR-based microarray.
| Gene | Protein | Fold change in expression | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mgp-/- to wild-type | p-value | Mgp-/- +Q to Mgp-/- | p-value | ||||||
| Extracellular matrix proteins | |||||||||
| Col11a1 | Collagen, type XI | 4.34*** | 0.005 | -3.79*** | 0.006 | ||||
| Col12a1 | Collagen, type XII | 9.86*** | 0.029 | -3.94*** | 0.031 | ||||
| Col1a1 | Collagen, type I | 7.28*** | 0.034 | -2.58*** | 0.143 | ||||
| Col2a1 | Collagen, type II | 1152.89*** | 0.015 | -22.74*** | 0.020 | ||||
| Comp | Cartilage oligomeric matrix protein | 75.92*** | 0.031 | -3.26*** | 0.058 | ||||
| Dmp1 | Dentin matrix protein 1 | 12.19*** | 0.048 | -4.52*** | 0.070 | ||||
| Fn1 | Fibronectin 1 | 4.31*** | 0.031 | -1.94*** | 0.138 | ||||
| Vcam1 | Vascular cell adhesion molecule 1 | 11.62*** | 0.015 | -2.17*** | 0.073 | ||||
| Mmp2 | Matrix metalloproteinase 2 | 3.48*** | 0.010 | -2.01*** | 0.028 | ||||
| Transcription factors | |||||||||
| Msx1 | Homeobox, msh-like 1 | 2.85*** | 0.003 | -3.66*** | <0.001 | ||||
| Runx2 | Runt related transcription factor 2 | 8.81*** | <0.001 | -2.40*** | 0.002 | ||||
| Sox9 | SRY-box containing gene 9 | 7.95*** | 0.002 | -2.24*** | 0.028 | ||||
| Twist1 | Twist homolog 1 (Drosophila) | 1.69*** | 0.040 | -2.19*** | 0.027 | ||||
| Vdr | Vitamin D receptor | 5.38*** | 0.008 | -1.03*** | 0.740 | ||||
N=5. *, p<0.05; ** p<0.01; *** p<0.001.
Dietary quercetin blocks nuclear accumulation of β-catenin protein in Mgp-/- aortae
Elaborating on our findings that the inhibition of chondrogenesis in cultured VSMCs associates with inhibition of β-catenin signaling, we tested whether quercetin similarly affects this signaling conduit in MGP null arterial tissue. Accumulation and nuclear localization of β-catenin protein, indicative of activation of this pathway [47], were examined in wild-type and Mgp-/- aortic tissue. In wild-type aortic tissue with normally inactive β-catenin signaling [48], β-catenin protein was not observed (Figure 6, Mgp+/+). In contrast, β-catenin protein was readily detected by immunofluorescence in the aortae of 5 week old Mgp-/- mice in which foci of alcian blue-positive cartilaginous metaplasia develop in the tunica media (Figure 6, Mgp-/-, - Querc). Further, in the quercetin-treated Mgp-/- mice we did not detect any accumulation of β-catenin protein in the vessel media (Figure 6, Mgp-/- + Querc), suggesting that the in vivo morphological effects of quercetin correlated with attenuation of β-catenin signaling. To further clarify the relationship between β-catenin activation and calcifying chondrogenic metaplasia in vivo, we determined the location of cells with active β-catenin within arterial tissue. Nuclear β-catenin protein was detected in chondrocyte-like cells, which appear as rounded cells surrounded by alcian blue positive GAG-rich matrix, in the Mgp-/- aortae (Figure 6, insets), indicating that activation of the β-catenin signaling associates with chondrogenic transformation in VSM in vivo.
Figure 6. Quercetin blocks accumulation and nuclear localization of β-catenin protein in Mgp-/- aortae.

Immunostaining for β-catenin (white, red) [nuclei counterstained with DAPI (blue)] and Alcian blue staining for GAG deposition on adjacent sections of aortae from Mgp+/+, untreated Mgp-/-, and quercetin-treated Mgp-/- animals shows that β-catenin protein is detected only in untreated Mgp-/- arterial tissue. Scale = 30 µm. Inset panels show higher magnification of representative nuclei, demonstrating co-localization of β-catenin with DAPI in the Mgp-/- aorta in rounded chondrocyte-like cells surrounded by cartilaginous GAG-rich matrix.
Discussion
Data from this study demonstrate that MGP null vascular disease associates primarily with chondrogenic but not osteogenic transformation of VSM as we did not detect expression of the key regulator of osteogenesis osterix or induction of Msx2 in calcified Mgp-/- aortae. Similarly, a previous study suggested chondrogenic transformation as a major cell fate of the Mgp-/- VSM [13]. Of note, stimulation of the β-catenin signaling pathway can promote chondrogenic differentiation of mesenchymal cells in a Sox-9-dependent manner [49] and in our system we also detected an increase in both β-catenin activity and Sox-9 expression. Although activation of canonical β-catenin signaling is broadly linked to cardiovascular disorders [35,48,50], our study is the first to our knowledge to demonstrate activation of this signaling pathway in VSM undergoing chondrogenic transformation in vitro and in vivo.
The origin of increased β-catenin activity in the Mgp-/- aortae deserves further inquiry. The canonical Wnt ligands expressed in the Mgp-/- aortae are present at very low levels, in contrast to the robust induction of Wnt3a and Wnt7a reported in the diabetic model. The observed dramatic reduction in expression of the β-catenin pathway antagonist sclerostin may contribute to activation of the pathway in the Mgp-/- model, and non-canonical agonists of this signaling may also play a role. For example, enzyme transglutaminase 2 (TG2), an important regulator of β-catenin in calcifying VSM [24,51], accumulates in Mgp-/- vessels in vivo [42] and is induced in VSMCs undergoing chondrogenic differentiation in vitro (Figure 3B). Taking into account that quercetin inhibits TG2 via direct binding [12] it is possible that modulation of TG2 enzymatic activity is central in chondrogenic metaplasia of VSM and its prevention. Current studies are underway to address this possibility.
Although quercetin drastically attenuated chondrogenic metaplasia and its calcification, and intercepted chondrogenic transformation of VSM as evidenced by attenuated expression of pro-chondrogenic master regulators Sox9, Twist1, and Runx2, the overall calcium accrual in Mgp-/- blood vessels was only partially alleviated by this flavonoid. About one-half of total mineral remained as deposits along the elastic lamellae, supporting the model that elastocalcinosis may precede chondrocyte-like transformation in VSM [52] and consistent with the model in which MGP acts as a direct inhibitor of tissue calcification by binding to small calcium phosphate precipitates and/or the elastic lamellae, thereby preventing nucleation and growth of mineral crystals (reviewed in [53]) This finding challenges the perception of MGP-null arterial disease as primarily a product of ectopic chondrogenesis resulting in calcification [13], and emphasizes the importance of elastocalcinosis in this pathology.
Attenuation of chondrogenic transformation in cultured VSMCs by quercetin associated with inhibition of β-catenin signaling, and in the Mgp-/- aortae quercetin treatment blocked accumulation and nuclear localization of β-catenin protein, supporting a role for this pathway in the phenotypic instability of VSM. While inhibition of β-catenin signaling may be one of the major mechanisms of quercetin action in this system, the possible contribution of other known quercetin activities including anti-inflammatory and anti-oxidative effects cannot be excluded. However, the equally low cell proliferation in Mgp-/- and wild type vessels demonstrated in this study and earlier [13] indicate that cartilaginous metaplasia is not linked to hyperproliferation and suggest a limited role for the anti-proliferative activity of quercetin. While the exact mechanisms whereby quercetin achieves a reduction in cartilaginous metaplasia are not definitively proven, the data support a role for inhibition of β-catenin signaling as a significant part of the action.
In conclusion, this study adds to the growing understanding of the complexity of MGP-mediated effects on vascular tissue by adding the β-catenin signaling pathway to the MGP-modulated cellular network that already includes BMP [54] and Notch [55] conduits, and by showing that MGP may regulate elastocalcinosis even when chondrogenic transformation is suppressed. In addition, the presented studies contribute to the mechanistic understanding of the beneficial effects of quercetin in cardiovascular disease and identify a potential therapeutic approach to the calcifying cartilaginous metaplasia of VSM with quercetin that stands out as a pharmacological tool with remarkably wide safety margin. Of note, the effects of quercetin delivered in drinking water or via breast milk are similar in Mgp-/- mice indicating that dietary supplement of quercetin in breastfeeding females should be considered carefully for possible effects on infants.
Acknowledgments
We thank Quercegen Pharma, Newton, MA for the generous gift of quercetin, and Dr. Shobana Shanmugasundaram and Dr. Beatrice Milon for technical assistance.
Funding Statement
This work was supported by the National Institutes of Health (NIH) grants R01HL099305 (to M.N.), EY13126 (to T.B.) and T32AR007592 (to K.B.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM (2006) Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med 260: 192-210. doi:10.1111/j.1365-2796.2006.01692.x. PubMed: 16918817. [DOI] [PubMed] [Google Scholar]
- 2. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB (2012) The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 95: 194-204. doi:10.1093/cvr/cvs135. PubMed: 22467316. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW et al. (2013) Smooth muscle cell plasticity: fact or fiction? Circ Res 112: 17-22. doi:10.1161/CIRCRESAHA.112.281048. PubMed: 23093573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ng K, Hildreth CM, Phillips JK, Avolio AP (2011) Aortic stiffness is associated with vascular calcification and remodeling in a chronic kidney disease rat model. Am J Physiol Renal Physiol 300: F1431-F1436. doi:10.1152/ajprenal.00079.2011. PubMed: 21478483. [DOI] [PubMed] [Google Scholar]
- 5. Spin JM, Maegdefessel L, Tsao PS (2012) Vascular smooth muscle cell phenotypic plasticity: focus on chromatin remodelling. Cardiovasc Res 95: 147-155. doi:10.1093/cvr/cvs098. PubMed: 22362814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM (2011) Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109: 697-711. doi:10.1161/CIRCRESAHA.110.234914. PubMed: 21885837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bostrom KI, Rajamannan NM, Towler DA (2011) The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res %19;109: 564-577. [DOI] [PMC free article] [PubMed]
- 8. Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, etl. al (2011) SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol 3 Yanvar: 1300-1308. doi:10.1161/ATVBAHA.111.223701. PubMed: 21415388. [DOI] [PMC free article] [PubMed]
- 9. Wamhoff BR, Hoofnagle MH, Burns A, Sinha S, McDonald OG et al. (2004) A G/C element mediates repression of the SM22alpha promoter within phenotypically modulated smooth muscle cells in experimental atherosclerosis. Circ Res 95: 981-988. doi:10.1161/01.RES.0000147961.09840.fb. PubMed: 15486317. [DOI] [PubMed] [Google Scholar]
- 10. Price PA, Faus SA, Williamson MK (1998) Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol 18: 1400-1407. doi:10.1161/01.ATV.18.9.1400. PubMed: 9743228. [DOI] [PubMed] [Google Scholar]
- 11. Essalihi R, Dao HH, Yamaguchi N, Moreau P (2003) A new model of isolated systolic hypertension induced by chronic warfarin and vitamin K1 treatment. Am J Hypertens 16: 103-110. doi:10.1016/S0895-7061(02)03204-1. PubMed: 12559675. [DOI] [PubMed] [Google Scholar]
- 12. Beazley KE, Banyard D, Lima F, Deasey SC, Nurminsky DI et al. (2012) Transglutaminase Inhibitors Attenuate Vascular Calcification in a Preclinical Model. Arterioscler Thromb Vasc Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Speer MY, Yang HY, Brabb T, Leaf E, Look A et al. (2009) Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 104: 733-741. doi:10.1161/CIRCRESAHA.108.183053. PubMed: 19197075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naik V, Leaf EM, Hu JH, Yang HY, Nguyen NB et al. (2012) Sources of cells that contribute to atherosclerotic intimal calcification: an in vivo genetic fate mapping study. Cardiovasc Res 94: 545-554. doi:10.1093/cvr/cvs126. PubMed: 22436847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abedin M, Tintut Y, Demer LL (2004) Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 24: 1161-1170. doi:10.1161/01.ATV.0000133194.94939.42. PubMed: 15155384. [DOI] [PubMed] [Google Scholar]
- 16. Johnson RC, Leopold JA, Loscalzo J (2006) Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res 99: 1044-1059. doi:10.1161/01.RES.0000249379.55535.21. PubMed: 17095733. [DOI] [PubMed] [Google Scholar]
- 17. Shanahan CM, Weissberg PL, Metcalfe JC (1993) Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res 73: 193-204. doi:10.1161/01.RES.73.1.193. PubMed: 8508530. [DOI] [PubMed] [Google Scholar]
- 18. Boström K, Zebboudj AF, Yao Y, Lin TS, Torres A (2004) Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-beta1 activity in endothelial cells. J Biol Chem 279: 52904-52913. doi:10.1074/jbc.M406868200. PubMed: 15456771. [DOI] [PubMed] [Google Scholar]
- 19. Crosier MD, Booth SL, Peter I, Dawson-Hughes B, Price PA et al. (2009) Matrix Gla protein polymorphisms are associated with coronary artery calcification in men. J Nutr Sci Vitaminol (Tokyo) 55: 59-65. doi:10.3177/jnsv.55.59. PubMed: 19352064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herrmann SM, Whatling C, Brand E, Nicaud V, Gariepy J et al. (2000) Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler Thromb Vasc Biol 20: 2386-2393. doi:10.1161/01.ATV.20.11.2386. PubMed: 11073842. [DOI] [PubMed] [Google Scholar]
- 21. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E et al. (1997) Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 386: 78-81. doi:10.1038/386078a0. PubMed: 9052783. [DOI] [PubMed] [Google Scholar]
- 22. Giachelli CM (2009) The emerging role of phosphate in vascular calcification. Kidney Int 75: 890-897. doi:10.1038/ki.2008.644. PubMed: 19145240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sage AP, Tintut Y, Demer LL (2010) Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 7: 528-536. doi:10.1038/nrcardio.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beazley KE, Deasey S, Lima F, Nurminskaya MV (2012) Transglutaminase 2-Mediated Activation of beta-Catenin Signaling Has a Critical Role in Warfarin-Induced Vascular Calcification. Arterioscler Thromb Vasc Biol 32: 123-130. doi:10.1161/ATVBAHA.111.237834. PubMed: 22034513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beazley KE, Eghtesad S, Nurminskaya MV (2012) Quercetin attenuates warfarin-induced vascular calcification in vitro independently from Matrix Gla protein. J Biol Chem 288: 2632-2640. PubMed: 23223575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harwood M, Danielewska-Nikiel B, Borzelleca JF, Flamm GW, Williams GM et al. (2007) A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem Toxicol 45: 2179-2205. doi:10.1016/j.fct.2007.05.015. PubMed: 17698276. [DOI] [PubMed] [Google Scholar]
- 27. Huxley RR, Neil HA (2003) The relation between dietary flavonol intake and coronary heart disease mortality: a meta-analysis of prospective cohort studies. Eur J Clin Nutr 57: 904-908. doi:10.1038/sj.ejcn.1601624. PubMed: 12879084. [DOI] [PubMed] [Google Scholar]
- 28. Egert S, Bosy-Westphal A, Seiberl J, Kürbitz C, Settler U et al. (2009) Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. Br J Nutr 102: 1065-1074. doi:10.1017/S0007114509359127. PubMed: 19402938. [DOI] [PubMed] [Google Scholar]
- 29. Edwards RL, Lyon T, Litwin SE, Rabovsky A, Symons JD et al. (2007) Quercetin reduces blood pressure in hypertensive subjects. J Nutr 137: 2405-2411. PubMed: 17951477. [DOI] [PubMed] [Google Scholar]
- 30. Kleemann R, Verschuren L, Morrison M, Zadelaar S, van Erk MJ et al. (2011) Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis 218: 44-52. doi:10.1016/j.atherosclerosis.2011.04.023. PubMed: 21601209. [DOI] [PubMed] [Google Scholar]
- 31. Bhaskar S, Kumar KS, Krishnan K, Antony H (2013) Quercetin alleviates hypercholesterolemic diet induced inflammation during progression and regression of atherosclerosis in rabbits. Nutrition 29: 219-229. doi:10.1016/j.nut.2012.01.019. PubMed: 22595451. [DOI] [PubMed] [Google Scholar]
- 32. Valenta T, Hausmann G, Basler K (2012) The many faces and functions of beta-catenin. EMBO J 31: 2714-2736. doi:10.1038/emboj.2012.150. PubMed: 22617422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chun JS, Oh H, Yang S, Park M (2008) Wnt signaling in cartilage development and degeneration. BMB Rep 41: 485-494. doi:10.5483/BMBRep.2008.41.7.485. PubMed: 18682032. [DOI] [PubMed] [Google Scholar]
- 34. Reis M, Liebner S (2013) Wnt signaling in the vasculature. Exp Cell Res 10: 1317–23. PubMed: 23291327. [DOI] [PubMed] [Google Scholar]
- 35. Goodwin AM, D’Amore PA (2002) Wnt signaling in the vasculature. Angiogenesis 5: 1-9. doi:10.1023/A:1021563510866. PubMed: 12549854. [DOI] [PubMed] [Google Scholar]
- 36. Beazley KE, Zhang T, Lima F, Pozharskaya T, Niger C et al. (2012) Implication for transglutaminase 2-mediated activation of beta-catenin signaling in neointimal vascular smooth muscle cells in chronic cardiac allograft rejection. J Heart Lung Transplant 31: 1009-1017. doi:10.1016/j.healun.2012.04.009. PubMed: 22694852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shao JS, Cai J, Towler DA (2006) Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol 26: 1423-1430. doi:10.1161/01.ATV.0000220441.42041.20. PubMed: 16601233. [DOI] [PubMed] [Google Scholar]
- 38. Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP et al. (2005) Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest 115: 1210-1220. doi:10.1172/JCI24140. PubMed: 15841209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caira FC, Stock SR, Gleason TG, McGee EC, Huang J et al. (2006) Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol 47: 1707-1712. doi:10.1016/j.jacc.2006.02.040. PubMed: 16631011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ishizeki K, Saito H, Shinagawa T, Fujiwara N, Nawa T (1999) Histochemical and immunohistochemical analysis of the mechanism of calcification of Meckel’s cartilage during mandible development in rodents. J Anat 194: 265-277. doi:10.1046/j.1469-7580.1999.19420265.x. PubMed: 10337959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ross R (1971) The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol 50: 172-186. doi:10.1083/jcb.50.1.172. PubMed: 4327464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaartinen MT, Murshed M, Karsenty G, McKee MD (2007) Osteopontin upregulation and polymerization by transglutaminase 2 in calcified arteries of Matrix Gla protein-deficient mice. J Histochem Cytochem 55: 375-386. PubMed: 17189522. [DOI] [PubMed] [Google Scholar]
- 43. Nurminsky D, Shanmugasundaram S, Deasey S, Michaud C, Allen S et al. (2011) Transglutaminase 2 regulates early chondrogenesis and glycosaminoglycan synthesis. Mech Dev 128: 234-245. doi:10.1016/j.mod.2010.11.007. PubMed: 21129482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moon RT, Kohn AD, De Ferrari GV, Kaykas A (2004) WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet 5: 691-701. doi:10.1038/nrg1427. PubMed: 15372092. [DOI] [PubMed] [Google Scholar]
- 45. Karsenty G, Wagner EF (2002) Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2: 389-406. doi:10.1016/S1534-5807(02)00157-0. PubMed: 11970890. [DOI] [PubMed] [Google Scholar]
- 46. Tchetina E, Mwale F, Poole AR (2003) Distinct phases of coordinated early and late gene expression in growth plate chondrocytes in relationship to cell proliferation, matrix assembly, remodeling, and cell differentiation. J Bone Miner Res 18: 844-851. doi:10.1359/jbmr.2003.18.5.844. PubMed: 12733723. [DOI] [PubMed] [Google Scholar]
- 47. Hendrickx M, Leyns L (2008) Non-conventional Frizzled ligands and Wnt receptors. Dev Growth Differ 50: 229-243. doi:10.1111/j.1440-169X.2008.01016.x. PubMed: 18366384. [DOI] [PubMed] [Google Scholar]
- 48. van Gijn ME, Daemen MJ, Smits JF, Blankesteijn WM (2002) The wnt-frizzled cascade in cardiovascular disease. Cardiovasc Res 55: 16-24. doi:10.1016/S0008-6363(02)00221-3. PubMed: 12062705. [DOI] [PubMed] [Google Scholar]
- 49. Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H et al. (2005) The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun 333: 1300-1308. doi:10.1016/j.bbrc.2005.06.041. PubMed: 15979579. [DOI] [PubMed] [Google Scholar]
- 50. Wang X, Adhikari N, Li Q, Hall JL (2004) LDL receptor-related protein LRP6 regulates proliferation and survival through the Wnt cascade in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 287: H2376-H2383. doi:10.1152/ajpheart.01173.2003. PubMed: 15271658. [DOI] [PubMed] [Google Scholar]
- 51. Faverman L, Mikhaylova L, Malmquist J, Nurminskaya M (2008) Extracellular transglutaminase 2 activates beta-catenin signaling in calcifying vascular smooth muscle cells. FEBS Lett 582: 1552-1557. doi:10.1016/j.febslet.2008.03.053. PubMed: 18405667. [DOI] [PubMed] [Google Scholar]
- 52. El-Maadawy S, Kaartinen MT, Schinke T, Murshed M, Karsenty G et al. (2003) Cartilage formation and calcification in arteries of mice lacking matrix Gla protein. Connect Tissue Res 44 Suppl 1: 272-278. doi:10.1080/713713617. PubMed: 12952208. [PubMed] [Google Scholar]
- 53. Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY et al. (2002) Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J Exp Med 196: 1047-1055. doi:10.1084/jem.20020911. PubMed: 12391016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boström KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y (2011) Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res 108: 446-457. doi:10.1161/CIRCRESAHA.110.236596. PubMed: 21193740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharma B, Albig AR (2012) Matrix Gla protein reinforces angiogenic resolution. Microvasc Res 10: 24–33. PubMed: 23110920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wong SC, Chan AT, Lo ES, Lo YM (2005) Nuclear beta-catenin expression is rare and its potential association with short survival in colorectal signet-ring cell carcinoma. Appl Immunohistochem Mol Morphol 13: 248-251. doi:10.1097/01.pai.0000142845.91862.c8. PubMed: 16082250. [DOI] [PubMed] [Google Scholar]
- 57. Vadlamudi U, Espinoza HM, Ganga M, Martin DM, Liu X et al. (2005) PITX2, beta-catenin and LEF-1 interact to synergistically regulate the LEF-1 promoter. J Cell Sci 118: 1129-1137. doi:10.1242/jcs.01706. PubMed: 15728254. [DOI] [PubMed] [Google Scholar]
- 58. Amen M, Liu X, Vadlamudi U, Elizondo G, Diamond E et al. (2007) PITX2 and beta-catenin interactions regulate Lef-1 isoform expression. Mol Cell Biol 27: 7560-7573. doi:10.1128/MCB.00315-07. PubMed: 17785445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM et al. (2005) Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res 65: 10214-10222. doi:10.1158/0008-5472.CAN-05-0884. PubMed: 16288009. [DOI] [PubMed] [Google Scholar]
- 60. González-Fernández R, Peña O, Hernández J, Martín-Vasallo P, Palumbo A et al. (2010) FSH receptor, KL1/2, P450, and PAPP genes in granulosa-lutein cells from in vitro fertilization patients show a different expression pattern depending on the infertility diagnosis. Fertil Steril 94: 99-104. doi:10.1016/j.fertnstert.2010.07.412. PubMed: 19342032. [DOI] [PubMed] [Google Scholar]
- 61. Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N et al. (2006) Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res 21: 1738-1749. doi:10.1359/jbmr.060810. PubMed: 17002572. [DOI] [PubMed] [Google Scholar]
- 62. Clements WK, Kim AD, Ong KG, Moore JC, Lawson ND et al. (2011) A somitic Wnt16/Notch pathway specifies haematopoietic stem cells. Nature 474: 220-224. doi:10.1038/nature10107. PubMed: 21654806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mason DP, Kenagy RD, Hasenstab D, Bowen-Pope DF, Seifert RA et al. (1999) Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ Res 85: 1179-1185. doi:10.1161/01.RES.85.12.1179. PubMed: 10590245. [DOI] [PubMed] [Google Scholar]
- 64. Morvan F, Boulukos K, Clément-Lacroix P, Roman RS, Suc-Royer I et al. (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21: 934-945. doi:10.1359/jbmr.060311. PubMed: 16753024. [DOI] [PubMed] [Google Scholar]
- 65. Agrotis A, Kanellakis P, Kostolias G, Di Vitto G, Wei C et al. (2004) Proliferation of neointimal smooth muscle cells after arterial injury. Dependence on interactions between fibroblast growth factor receptor-2 and fibroblast growth factor-9. J Biol Chem 279: 42221-42229. doi:10.1074/jbc.M408121200. PubMed: 15292181. [DOI] [PubMed] [Google Scholar]
- 66. Hung IH, Yu K, Lavine KJ, Ornitz DM (2007) FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol 307: 300-313. doi:10.1016/j.ydbio.2007.04.048. PubMed: 17544391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Buxton P, Edwards C, Archer CW, Francis-West P (2001) Growth/differentiation factor-5 (GDF-5) and skeletal development. J Bone Joint Surg Am 83-A Suppl 1: S23-S30. PubMed: 11263662. [PubMed] [Google Scholar]
- 68. Zeng Q, Li X, Beck G, Balian G, Shen FH (2007) Growth and differentiation factor-5 (GDF-5) stimulates osteogenic differentiation and increases vascular endothelial growth factor (VEGF) levels in fat-derived stromal cells in vitro. Bone 40: 374-381. doi:10.1016/j.bone.2006.09.022. PubMed: 17070126. [DOI] [PubMed] [Google Scholar]
- 69. Liu Y, Wang T, Yan J, Jiagbogu N, Heideman DA et al. (2011) HGF/c-Met signalling promotes Notch3 activation and human vascular smooth muscle cell osteogenic differentiation in vitro. Atherosclerosis 219: 440-447. doi:10.1016/j.atherosclerosis.2011.08.033. PubMed: 21920521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Amano O, Koshimizu U, Nakamura T, Iseki S (1999) Enhancement by hepatocyte growth factor of bone and cartilage formation during embryonic mouse mandibular development in vitro. Arch Oral Biol 44: 935-946. doi:10.1016/S0003-9969(99)00086-2. PubMed: 10580541. [DOI] [PubMed] [Google Scholar]
- 71. Mosig RA, Dowling O, DiFeo A, Ramirez MC, Parker IC et al. (2007) Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet 16: 1113-1123. doi:10.1093/hmg/ddm060. PubMed: 17400654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Furlan F, Galbiati C, Jorgensen NR, Jensen JE, Mrak E et al. (2007) Urokinase plasminogen activator receptor affects bone homeostasis by regulating osteoblast and osteoclast function. J Bone Miner Res 22: 1387-1396. doi:10.1359/jbmr.070516. PubMed: 17539736. [DOI] [PubMed] [Google Scholar]
- 73. Otto WR, Rao J (2004) Tomorrow’s skeleton staff: mesenchymal stem cells and the repair of bone and cartilage. Cell Prolif 37: 97-110. doi:10.1111/j.1365-2184.2004.00303.x. PubMed: 14871240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. French DM, Kaul RJ, D’Souza AL, Crowley CW, Bao M et al. (2004) WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. Am J Pathol 165: 855-867. doi:10.1016/S0002-9440(10)63348-2. PubMed: 15331410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Miceli-Libby L, Johnson MJ, Harrington A, Hara-Kaonga B, Ng AK et al. (2008) Widespread delta-like-1 expression in normal adult mouse tissue and injured endothelium is reflected by expression of the Dll1LacZ locus. J Vasc Res 45: 1-9. doi:10.1159/000113929. PubMed: 17898542. [DOI] [PubMed] [Google Scholar]
- 76. Aitkenhead M, Wang SJ, Nakatsu MN, Mestas J, Heard C et al. (2002) Identification of endothelial cell genes expressed in an in vitro model of angiogenesis: induction of ESM-1, (beta)ig-h3, and NrCAM. Microvasc Res 63: 159-171. doi:10.1006/mvre.2001.2380. PubMed: 11866539. [DOI] [PubMed] [Google Scholar]
- 77. Dufourcq P, Leroux L, Ezan J, Descamps B, Lamazière JM et al. (2008) Regulation of endothelial cell cytoskeletal reorganization by a secreted frizzled-related protein-1 and frizzled 4- and frizzled 7-dependent pathway: role in neovessel formation. Am J Pathol 172: 37-49. doi:10.2353/ajpath.2008.070130. PubMed: 18156211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Johnstone SR, Kroncke BM, Straub AC, Best AK, Dunn CA et al. (2012) MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ Res 111: 201-211. doi:10.1161/CIRCRESAHA.112.272302. PubMed: 22652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liao Y, Regan CP, Manabe I, Owens GK, Day KH et al. (2007) Smooth muscle-targeted knockout of connexin 43 enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol 27: 1037-1042. doi:10.1161/ATVBAHA.106.137182. PubMed: 17332489. [DOI] [PubMed] [Google Scholar]
- 80. Loiselle AE, Jiang JX, Donahue HJ (2012) Gap junction and hemichannel functions in osteocytes. Bone 10: 205–12. PubMed: 23069374. [DOI] [PubMed] [Google Scholar]
- 81. Hamidouche Z, Fromigué O, Ringe J, Häupl T, Marie PJ (2010) Crosstalks between integrin alpha 5 and IGF2/IGFBP2 signalling trigger human bone marrow-derived mesenchymal stromal osteogenic differentiation. BMC Cell Biol 11: 44. doi:10.1186/1471-2121-11-44. PubMed: 20573191.: 44-11 [DOI] [PMC free article] [PubMed] [Google Scholar]