

Abstract
CYP19A1, also known as aromatase or estrogen synthetase, is the rate-limiting enzyme in the biosynthesis of estrogens from their corresponding androgens. Several clinically used breast cancer therapies target aromatase. In this work, explicitly solvated all-atom molecular dynamics simulations of aromatase with a model of the lipid bilayer and the transmembrane helix are performed. The dynamics of aromatase and the role of titration of an important amino acid residue involved in aromatization of androgens are investigated via two 250-ns long simulations. One simulation treats the protonated form of the catalytic aspartate 309, which appears more consistent with crystallographic data for the active site, while the simulation of the deprotonated form shows some notable conformational shifts. Ensemble-based computational solvent mapping experiments indicate possible novel druggable binding sites that could be utilized by next-generation inhibitors. In addition, the effects of protonation on the ligand positioning and channel dynamics are investigated using geometrical models that estimate the opening width of critical channels. Significant differences in channel dynamics between the protonated and deprotonated trajectories are exhibited, suggesting that the mechanism for substrate and product entry and the aromatization process may be coupled to a “locking” mechanism and channel opening. Our results may be particularly relevant in the design of novel drugs, which may be useful therapeutic treatments of cancers such as those of the breast and prostate.
INTRODUCTION
The mammalian cytochrome P450s (CYPs) are a diverse group of enzymes that catalyze various versatile reactions in the cell,1 ranging from metabolism of xenobiotics to the synthesis of sex steroids. In particular, CYPs involved with the synthesis and metabolism of sex steroids may play a major role in both development and progression of certain cancers, such as those of the breasts and prostate. CYP19A1, also known as aromatase or estrogen synthetase, is the rate-limiting enzyme in the biosynthesis of estrogens from their corresponding androgens, catalyzing a unique reaction among CYPs that consists of a three-step process that ultimately results in aromatization of the steroid A-ring2 Due to its role in estrogen biosynthesis, aromatase plays an integral role in the propagation of estrogen-dependent breast cancer cells. Targeted inhibition of aromatase is therefore a major frontline therapy in the treatment of estrogen-dependent breast cancer in post-menopausal women.
Several inhibitors, such as anastrozole,3 letrozole,4,5 and exemestane,6 are currently used clinically to treat hormone-dependent breast cancer in post-menopausal patients. Some (anastrozole, letrozole) are competitive inhibitors of aromatase activity, while others (exemestane) act as non-competitive irreversible suicide substrates, potently inactivating the enzyme until new enzymes can be expressed. Other aromatase inhibitors are also currently in development for clinical application or for use as chemical probes in research, utilizing novel scaffolds and mechanisms of action.7–13
The resolved crystal structure for human placental aromatase comprises of residues 45 through 496 and contains an androstenedione ligand bound to the active site.14, 15 The crystal structure has led to discovery of a putative active site access/egress channel and the development of membrane integration models.16 Aromatase, like all mammalian CYPs, associates with the endoplasmic reticulum membrane and integrates with the membrane through a single N-terminal transmembrane helix, with the bulk of the protein exposed to the cytosolic region. Other residues, such as those in the F-G helix loop and in the A helix have also been shown experimentally to contribute to membrane integration. Despite the availability of the crystal structure, much remains to be discovered about the molecular characteristics of aromatase and its interactions with the lipid bilayer, with itself, and with other proteins such as cytochrome P450 reductase (CPR).
This crystal structure has enabled researchers to apply molecular docking and high-throughput screening to develop new inhibitors of aromatase. Furthermore, molecular dynamics (MD) simulations of aromatase in solution have yielded further insight into protein motion and receptor flexibility that may be useful in molecular docking applications.17 Normal mode analysis of the crystal structure18 has revealed possible internal motions of the protein and has suggested the possibility of higher-ordered organization of the protein in the membrane. Knowledge of the crystal structure has also enabled the structure-based design of novel aromatase inhibitors that exploit the unique structural features of the protein in its active site cavity and active site channels.19 Additional MD of membrane-bound aromatase16 and analysis on the active site channels suggest that the membrane has a significant influence on the presence of certain access and egress channels connecting the active site to the exterior protein environment.
The extensive sets of structural data for solubilized CYPs obtained by X-ray crystallography give little direct insight into the association with the membrane, and this lack of detail may prevent the effective and efficient computational discovery of inhibitors. The relaxed complex scheme (RCS)20 is a computer-aided drug discovery approach that accounts for protein flexibility. The RCS has impacted the development of HIV integrase drugs,21 novel trypanocidal compounds,22 novel antibacterials23–25 and p53 reactivation compounds.26 The development of MD simulations of proteins such as CYPs in model membrane environments may lead to greater understanding and insight into the molecular characteristics of these systems in their natural environment. The work presented here addresses the current lack of such models and presents the predictions of the dynamics of aromatase in a model lipid bilayer from all-atom, explicitly-solvated molecular dynamics simulations. This work may assist in the development of next-generation aromatase inhibitors with novel mechanisms of action. Furthermore, it has recently been reported that the aromatase catalytic activity is strongly pH-dependent, with enhancement of the activity in terms of dissociation constant and Michaelis-Menten constant by subtle decreases of pH from 7.4 to 6.5, which however is not observed in Asp309Asn aromatase mutants.27 We investigate the possible effects of proton titration by directly simulating both protonation states of this Asp309 residue in separate simulations, each with a starting state resembling the native X-ray structure (PDB accession code 3EQM14) but in the presence or absence of the ASP309 side-chain proton. From these simulations, conformational dynamics of the bound androstenedione ligand and the aromatase active site and the corresponding role of active site titration were observed over the course of molecular dynamics trajectories. Our work provides insight into the conformational dynamics of aromatase in the native, endoplasmic reticulum membrane-bound environment and suggests novel avenues for drug design for this important anticancer target.
METHODS
Model Construction
The starting coordinates for model construction were obtained from the coordinates of human placental aromatase in 3EQM.14, 15 Phosphate ions present in the crystallization conditions and observed in the resolved structure were removed prior to model construction. Missing hydrogen atoms for all residues in the crystal structure were then added with Maestro.28 An initial peptide linker, connecting the transmembrane domain to the catalytic domain and comprising of residues 39 to 44, was modeled as an extended structure. Transmembrane residues 18 to 38 were modeled as an alpha helix, as defined previously,29 although terminal residues 16–17 were not incorporated, due to difficulties associated with accommodating multiple proline residues in our alpha-helical model. Using the membrane orientation of the crystal structure as computed by the OPM database,30 the transmembrane helix was rotated until it was approximately perpendicular to the membrane plane and was spanning the height of the bilayer. The membrane orientation and transfer free energy (from solvent to lipid bilayer) of our models were calculated using the implicit solvent model of the lipid bilayer as described by Lomize31 using the PPM server32 and appear to improve the thermodynamics of membrane association according to the calculations of the PPM server.
Because of the placement of both the F-G loop and entire transmembrane helix inside the lipid bilayer, as well as energetically favorable placement of tryptophans33 and basic residues34 near the protein-lipid interface, we considered our model to be reasonable for use in molecular dynamics simulations. Our model is in good agreement with epitope analysis of aromatase,35 as all epitopes are reasonably far away from the membrane as to allow full access by site-directed antibodies.
System Construction
The methods provided by the CHARMM-GUI Membrane Builder application36 were used to insert the protein into a constructed heterogeneous lipid bilayer membrane and assemble the final system. The androstenedione and heme were added to the active site, with the Cys-437 residue deprotonated to a cysteinate residue in order to coordinate to the heme iron. The composition of the constructed membrane was determined using a previously determined lipid content of the rat liver endoplasmic reticulum.37 Due to limitations in the availability of certain lipids in the available CHARMM36 lipid force field, the final membrane lipid content was slightly modified to consist of 10.0% cholesterol, 9.0% DPPE, 11.0% DOPE, 8.4% SDPC, 8.1% DAPC, 13.2% SOPC, 6.3% DPPC, 16.4% DOPC, and 17.6% POPS. The resulting bilayer consisted of 266 lipids, with slightly more lipids in the upper leaflet due to integration of protein residues into the lower leaflet. This model is not expected to be an exact depiction of bilayer conditions near aromatase, but it is constructed to include the biologically relevant lipids in the cellular compartment.
The system was then solvated in a TIP338 water box, along with 46 K+ ions to neutralize the protein-membrane system and an additional 34 K+ (80 total K+ ions) and 34 Cl- ions representing approximately 100 mM KCl salt. For the system topology, the CHARMM27 protein39, 40 and CHARMM36 lipid41 force fields were utilized, along with separately derived parameters for androstenedione from SwissParam.42 With the exception of the active site Asp-309 residue, amino acid side chain protonation states were assigned from default CHARMM protonation states, which were determined to be consistent with predictions from the PROPKA 3.0 web server.43 According to the PROPKA 3.0 web server, the predicted pKa for this Asp-309 residue in the presence of the ligand was 7.69, close to commonly-observed values of physiological pH. In the protonated Asp-309 state, a hydrogen bond between this delta proton of the side chain and the O9 oxygen of androstenedione can be inferred by the crystal structure. The final system contained 98,165 atoms for the deprotonated Asp-309 trajectory, and 98,157 atoms for the protonated Asp-309 trajectory. Figure 1 shows the model and the assembled system for molecular dynamics.
Figure 1.

Depiction of the simulated systems. A) The protein, heme, and ligand assembly. B) Illustration of the orientation of aromatase with respect to the bilayer, with the explicit lipid bilayer model shown, constructed as described in the methods. The depicted system in the illustration is from simulations of aromatase with a protonated Asp-309 residue, and the modeled transmembrane domain is colored in magenta.
Molecular Dynamics Simulations
NAMD 2.844 was used for the minimization, equilibration, and free dynamics, using parameters derived from the CHARMM27 protein39, 40 and CHARMM36 lipid41 force fields, with separately derived parameters for androstenedione molecule from the SwissParam42 web server. The simulations were performed in parallel on 320 CPU cores of either the Teragrid XSEDE Texas Advanced Computing Center (TACC) Ranger cluster or the San Diego Supercomputing Center (SDSC) Trestles cluster, with benchmark times of 0.16 days/nanosecond and 0.09 days/nanosecond, respectively.
The entire system was initially minimized for 10,000 steps to remove any unfavorable contacts and interactions. During the minimization, k = 5 kcal mol−1 Å−2 harmonic restraints were applied to the protein backbone, and k = 2.5 kcal mol−1 Å−2 harmonic restraints were applied to the protein side chains, heme, and the ligand atoms. The system was subsequently equilibrated for 25 ps with the same applied harmonic restraints from minimization. A longer 5 ns equilibration followed in which k = 1 kcal mol−1 Å−2 harmonic restraints were applied to the protein backbone, and k = 0.5 kcal mol−1 Å−2 harmonic restraints were applied to the side chains, heme, and ligand atoms. A final equilibration phase, with no restraints, was then run for 2 ns.
The equilibration steps were run at 310 K temperature and 1 atmosphere pressure with a 1 fs time step in the NPT ensemble. The temperature was maintained at 310 K using Langevin dynamics with a 5 ps−1 damping coefficient, and pressure was maintained at 1 atm using the hybrid Nose Hoover-Langevin piston method45, 46 with a 100 fs period time and a 50 fs decay time. A multi-step algorithm computed bonded forces every 1 fs, non-bonded van-der-Waals forces every 2 fs, and full electrostatic interactions every 4 fs. The SHAKEH47 algorithm was employed to constrain hydrogen bond lengths, and the Particle Mesh Ewald48 method was used with a grid spacing of 1.0 Å to compute long-range electrostatic interactions. Harmonic restraints as specified above were applied to the first two equilibrations. In the final equilibration, the surface tension of the membrane was kept constant to ensure an almost tensionless membrane.
Free dynamics of the system, with constant bilayer surface area determined by the final state of equilibration, were subsequently run for a total of 250 ns with identical parameters as the equilibration, except that the 1 fs time step was increased to 2 fs. Snapshots of the system configuration were recorded every 25 ps, resulting in a trajectory of 10,000 individual conformations.
Analysis
Following free dynamics, both trajectory ensembles of 10,000 individual, non-redundant conformations extracted at 25 ps intervals from the MD simulations were clustered using the GROMOS++ algorithm20, 49, 50 as implemented in GROMACS g_cluster.20 Clustering was performed on the subset of active site residues surrounding the androstenedione substrate molecule, defined as all residues containing at least one atom within 5.0 Å of the androstenedione molecule (comprising of residues 115, 133, 134, 221, 224, 228, 305, 306, 309, 310, 370, 372, 373, 374, 395, 398, 477, and 478). Trajectory frames were aligned, by minimizing the RMSD between the subset of protein atoms of each frame and the corresponding atoms of the first frame. This least-squares alignment removed external translational and rotational motion of the protein during dynamics so that subsequent RMSD calculations could focus on the internal conformational variance of the selection. After varying the RMSD cutoff values to optimize the number of clusters without compromising the conformational variance of the trajectory, a cutoff value of 1.5 Å was chosen for the deprotonated trajectory, producing 27 clusters of distinct protein conformations. The first five most prevalent clusters represented about 91.5% of the trajectory. For the protonated trajectory, a cutoff value of 1.65 Å was chosen, producing 18 clusters of distinct protein conformations. The first five most prevalent clusters represented about 95.8% of the trajectory. From these five clusters each in both trajectories, the centroid frame of each respective cluster was chosen for analysis as the representative structure for all frames in the cluster.
Active site tunnels were identified for the representative structures of the five most prevalent clusters in both trajectories using MOLE 2.0 and MOLEonline 2.0.51 The MOLE 2.0 and MOLEonline 2.0 algorithm involves (1) the calculation of Delaunay triangulation and Voronoi diagrams, (2) refinement of Voronoi diagrams into smaller cavity diagrams, (3) identification of starting points and endpoints in the cavity diagrams, and (4) use of Dijkstra shortest path algorithm to find tunnels between pairs of starting points and endpoints. The starting point was defined as the centroid of the androstenedione ligand, and heteroatoms were excluded from the calculation to correctly identify tunnels leading out of the active site.
Computational solvent mapping was performed using the FTMap server21, 22 to identify favorable ligand-binding “hot spots”, a.k.a. druggable hot spots, on the surface and interior cavities of aromatase. The centroid frames of the five most prevalent clusters of both trajectories were used for individual computation solvent mapping calculations. In addition, the four currently available crystal structures of aromatase (3EQM, 3S7S, 4GL5, 4GL7) were additionally used for computational solvent mapping calculations to observe whether molecular dynamics simulations samples new conformations that might potentially reveal novel druggable hot spots. The FTMap algorithm determines the energetically favorable binding regions of sixteen solvent-like fragments along a protein surface via the following steps: (1) rigid body docking of fragments using a fast Fourier transform approach, (2) minimization and rescoring of fragment-protein complexes, (3) clustering and ranking of low-energy fragment-protein complexes, and (4) determination of consensus sites. Consensus sites are regions of the protein surface where low-energy fragment clusters of multiple fragment types co-localize. Following the calculations of each individual centroid frame, the results were further clustered using FTprod,52 which identified “hot spots” that common to all protein clusters which represent potential druggable sites. These “hot spots” were further visually inspected to identify druggable regions on the surface and interior of aromatase.
Visual inspection of the trajectories and individual frames extracted from the trajectories were done with both VMD53 and UCSF Chimera.54
Water occupancy analysis was performed using the VolMap tool in VMD, while RMSD analysis was computed using the RMSD Trajectory Tool in VMD. All other analyses, such as the calculation of root mean square fluctuations (RMSF) and the membrane position and orientation of the protein, were performed using custom written Tcl scripts and programs used under the Tk Console in VMD. All illustrations were prepared and rendered with VMD and UCSF Chimera.
RESULTS AND DISCUSSION
Model Validation
Epitope mapping studies of aromatase indicate that residues 250–262, 376–390, and 474–485 are solvent-exposed.35 Furthermore, these residues must be placed reasonably away from the lipid membrane to allow unhindered access to site-directed antibodies. In our model, all of these residues were solvent-exposed throughout the simulation, and consistently located a reasonable distance away from the membrane to allow full antibody access (Supporting Information Figure S1). Similarly, all residues known to interact with Cytochrome P450 reductase (CPR), such as Lys-108 and Lys-420, were solvent-exposed throughout the dynamics.
Protein rotation studies of various cytochrome P450 proteins additionally indicate that the angle between the heme plane and the membrane plane varies between 38° to 78°.55 In our simulation, the angle was observed to be 34.3±5.3° throughout the deprotonated trajectory and 31.4±5.2° throughout the protonated trajectory (Supporting Information Figure S2). Our observation is further consistent with values from molecular dynamics of membrane-bound CYP2C9,56 where the angle was found to be 35±5° throughout the simulation.
We computed a vertical height for aromatase of 4.5±0.2 nm above the membrane throughout the deprotonated trajectory and 4.4±0.1 nm throughout the protonated trajectory (Supporting Information Figure S3). These values are consistent with Sgrignani and Magistrato's16 height measurement of approximately 4.5 nm in a molecular dynamics simulation of membrane-bound aromatase. Atomic force microscopy (AFM) studies of aromatase by Praporski et al.57 measured the vertical height of membrane-bound wild-type aromatase aggregates at around 2 nm. There is a good possibility that the contact tip could have underestimated the height of the protein in these experiments, as seen in the case of AFM studies of CYP2B4 where membrane height was initially determined to be 1.2±0.3 nm before force-dependent height analysis corrected the height to a more reasonable 3.5±0.9 nm.58 Likewise, Praporski et al. measured the vertical height of membrane-bound cytochrome P450 reductase (CPR) at 1–2 nm, similar to initial measurements of CPR by Bayburt et al.59 However, later force-dependent height analysis corrected the height of CPR to 5.6±2.2 nm.60
Computational modeling of the transmembrane helix of aromatase using the PPM anisotropic solvent model of the lipid bilayer31 further determined that the ideal tilt angle of the transmembrane helix relative to the membrane was 21±8°. This is consistent with the observations where the tilt angle was 24.0±9.0° in the deprotonated trajectory, and 15.0±6.7° in the protonated trajectory.
We observed no major changes in the secondary structure during the simulations with the exception of the partial unfolding of the A' helix in the protonated simulation. Interestingly, Sgrignani and Magistrato observed a similar unfolding in their simulations of membrane-bound aromatase. We observed that the minor unfolding of residues in the A' helix was mainly caused by interactions between the charged lipid head groups of the membrane and the backbone of the protein helix.
Calculated root mean square fluctuation (RMSF) values for each residue of the model throughout both simulations are also in good agreement with B-factors derived from the crystal structure (Figure 2). When the RMSF values are converted into crystallographic B-factors using the equation Bfactor = 8/3 π2 (RMSF)2 and compared with the B-factors of the crystal structure, both follow similar trends and patterns. Furthermore the RMSF values that we have computed are in general agreement with the RMSF values computed by Suvannang et al.17 as well as Sgrignani and Magistrato in their MD simulations of aromatase.
Figure 2.

Root-mean-squared fluctuations (RMSF) in aromatase simulations. A) The RMSF over the course of 250 ns simulation for the MD trajectory with deprotonated Asp-309 (blue) and protonated Asp-309 (red) compared to estimates of the RMSF from crystallographic data for the aromatase-androstenedione complex 3EQM (black) using the reported B-factors, approximated with the formula Bfactor = 8/3 π2 (RMSF)2. B) Color-coded representation of the fluctuations in the deprotonated Asp-309 trajectory.
Calculated root mean square deviation (RMSD) values of the model throughout both simulations further indicate the stability of the protein structure during the simulations (Supporting information Figure S4). The calculated RMSD of the non-hydrogen atoms of the residues present in the crystal structure, comprising of residues 45 to 496, was 1.83±0.16 Å in the deprotonated trajectory and 1.74±0.12 Å in the protonated trajectory, both computed relative to the crystal structure. The calculated RMSD of the non-hydrogen atoms of all residues present (including the modeled transmembrane helix) was 2.90±0.56 Å in the protonated trajectory and 2.83±0.51 Å in the deprotonated trajectory, a slightly larger value with greater fluctuations due to the flexibility of the modeled transmembrane helix linker residues and the position of the helix through the simulations.
Identification and Analysis of Active Site Tunnels
Using MOLE 2.0 to detect active site tunnels in the most prevalent cluster representative frames, we observed three main channels in the trajectories – the S channel, the 2d channel, and Channel 5 (Figure 3). These channel identifications are adopted from Wade's nomenclature.61
Figure 3.

Illustration of the main active site channels of aromatase found by MOLE, with the transmembrane helix hidden for clarity, from a top-down view of the protein looking through the membrane. The S channel is in cyan, the 2d channel is in green, and Channel 5 is in purple. A) Channels in the second cluster representative of the trajectory with a deprotonated Asp-309 residue. B) Channels in the first cluster representative of the trajectory with a protonated Asp-309 residue..
The S channel is formed by residues Ile-133, Phe-134, Arg-192, Phe-221, Trp-224, Gln-225, Asp-309, Thr-310, Val-313, Val-370, Leu-477, and Ser-478 (seen in Figure 3). The channel opening was located between the F and I helices, the ß9 sheet, and the ß8–ß9 loop, and was formed by residues Phe-221, Gln-225, Asp-309, and His-480. This channel approximately corresponds to the pathway identified with Caver,50 the second most prevalent expulsion route in RAMD simulations of the androstenedione ligand, and the most prevalent expulsion route in RAMD simulations of molecular oxygen by Sgrignani and Magistrato16. Furthermore, the channel approximately corresponded to the second and third most favorable pathways for androstenedione dissociation from the active site in SMD simulations in this work of Sgrignani and Magistrato. We observed numerous instances of solvent exchange through the S channel between the interior of the protein and the exterior environment during the trajectory, confirming its importance in the regulation of water access to the active site. According to our analysis, the channel appears to be more accessible in the deprotonated model of Asp-309 (Table 1) suggesting that substrate exit and entry is facilitated in the deprotonated state and the channel may be locked closed by the protonation of this catalytic residue.
Table 1.
Analysis of the channel state in the deprotonated and protonated trajectories. Data was obtained and analyzed in MOLE 2.0 and assigned to specific channels using Wade's nomenclature. The centroid structures of the top five clusters of both trajectories from GROMOS++ clustering (see methods) are reported along with similar analysis of the 3EQM crystal structure in the Protein Databank. The channel widths are reported for each channel in the result (some channels have multiple openings detected by the algorithm).
| S Channel | Channel 5 | 2d Channel | Channel 1 | |
|---|---|---|---|---|
| Crystal Structure | 1.57 Å, 0.37 Å | 0.87 Å | Closed | Closed |
| Deprotonated Cluster 1 (67%) | 1.94 Å, 0.89 Å | 0.46 Å, 1.10 Å | 0.86 Å, 0.86 Å | Closed |
| Deprotonated Cluster 2 (11%) | 1.94 Å, 0.89 Å | Closed | 0.89 Å, 0.89 Å | Closed |
| Deprotonated Cluster 3 (6%) | 1.36 Å | Closed | 0.97 Å, 1.28 Å | Closed |
| Deprotonated Cluster 4 (4%) | 1.03 Å | 0.97 Å, 0.95 Å, 0.95 Å | Closed | Closed |
| Deprotonated Cluster 5 (4%) | 1.22 Å, 1.40 Å | 1.04 Å | Closed | Closed |
| Protonated Cluster 1 (61%) | Closed | 1.48 Å | Closed | Closed |
| Protonated Cluster 2 (14%) | Closed | 1.37 Å | Closed | Closed |
| Protonated Cluster 3 (10%) | 0.57 Å, 0.57 Å | 0.84 Å, 0.84 Å, 0.84 Å | Closed | Closed |
| Protonated Cluster 4 (6%) | Closed | 1.19 Å | 1.00 Å | Closed |
| Protonated Cluster 5 (4%) | Closed | 0.83 Å, 0.78 Å | 0.90 Å | 0.70 Å |
The 2d channel was formed by residues Gly-71, Asn-75, Ile-133, Phe-134, Trp-224, Val-370, Asp-371, Leu-372, Met-374, Leu-477, Ser-478, and Leu-479. The channel opening was located between the A' and K' helices, ß8–9 sheets, and the K-ß3 loop, and was formed by residues Asp-371, Leu-372, and His-475. We also observed solvent exchange through the 2d channel, although in a significantly fewer number of instances compared to the S channel.
Channel 5 was formed by residues Ile-133, Phe-134, Trp-224, Thr-310, Arg-365, Pro-368, Val-369, Val-370, Ile-398, Gly-399, Arg-403, Phe-427, Pro-429, Phe-430, and Cys-437. The channel opening was located between the K and K' helices, as well as the extended K' loop, and was formed by residues Arg-365, Pro-368, Gly-399, His-402, and Pro-410. We did not observe any instances of solvent exchange through Channel 5. Unlike the S channel and the 2d channel, channel 5 did not lead directly to the membrane surface. It appears that the channel is more accessible in the protonated Asp-309 model of aromatase relative to the deprotonated model in the analysis with MOLE 2.0 (Table 1).
We further evaluated the influence of the protonation state of Asp-309 on the opening and closing of active site tunnels, which may serve as the access and egress routes of important substrates such as androstenedione, water, and oxygen. Remarkable differences in the presence and structure of these channels were observed between the two different trajectories (Table 1). The S channel, for example, was open in all five of the most prevalent cluster representatives of the deprotonated trajectory, but nearly absent in the same representatives of the protonated trajectory. The 2d channel furthermore is open in the three most prevalent cluster representative frames of the deprotonated trajectory, while closed in the same representative frames of the protonated trajectory. Channel 5, on the other hand, was open in all five of the most prevalent cluster representative frames of the protonated trajectory but open in only some of the same representative frames of the deprotonated trajectory. Accordingly, solvent exchange in the protonated trajectory occurred mainly through the 2d channel, while solvent exchange in the deprotonated trajectory occurred mainly through the S channel. Therefore, the protonation state of the Asp-309 residue may help in stabilizing certain channels by inducing local structural changes in the protein, especially near the active site region.
We have further deduced an explanation for the differences observed in the case of the S channel and further a possible mechanism for the opening/closing of the S channel following the deprotonation/protonation of Asp-309 (Figure 4). By observing the relative orientations of Phe-221 and Asp-309, the two putative “gatekeeper” residues of the S channel, we observed that the sidechains of both residues tend to be stacked (the planes of each sidechain are roughly parallel to the other) throughout both trajectories. Furthermore, we observed that movements in the sidechain of one of the residues induced the movement of the other sidechain to maintain this stacking, in part due to steric forces. In the deprotonated trajectory, the Asp-309 side chain points outward away from the ligand to form a hydrogen bond with either a nearby water molecule or the sidechain of Arg-192. With this orientation, Phe-221 is positioned to open the S channel entrance formed by Gln-225, Asp-309, Ser-478, and His-480. In the protonated trajectory, however, the Asp-309 sidechain points inward towards the ligand in order to form a hydrogen bond with the keto group of the ligand due to its new capacity to function as a hydrogen bond donor. This new position brings Phe-221 into steric contact with Gln-225, effectively closing the S channel entrance. Protonation of Asp-309 therefore stabilizes the closed conformation of the S channel, while deprotonation stabilizes the open conformation of the S channel.
Figure 4.
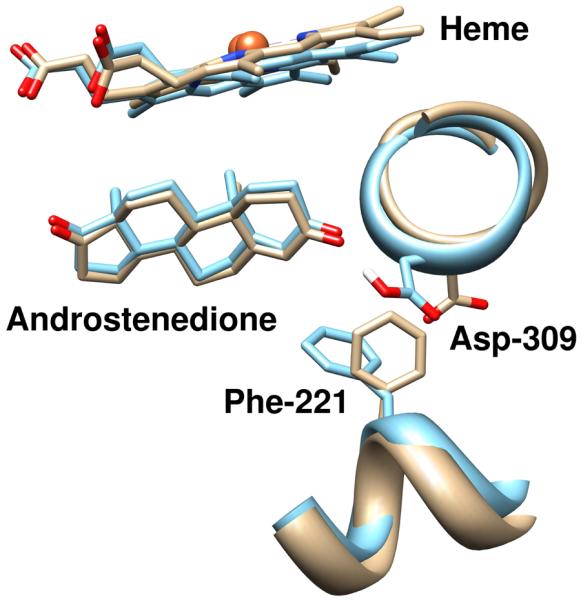
Representation of the effect of protonation of Asp-309 on the conformation of androstenedione binding and in the locking of the channel conformation. The trajectory with a deprotonated Asp-309 (tan) exhibits significant deviations around the androstenedione O3 oxygen atom, and the conformations of Asp-309 and Phe-221 are altered relative to the crystal structure. The simulated trajectory with a protonated Asp-309 (blue) demonstrates a conformation in this region more consistent with the crystal structure. The figures are rendered from the centroid frame of the most prevalent clusters from GROMOS++ clustering using the protocol described in the text.
This mechanism may help to account for observations on the effect of pH of the local environment on the catalytic activity of aromatase27. Increases in the pH of the local environment that increase the dissociation of the androstenedione substrate and reduce its catalytic activity are correlated with the protonation/deprotonation of Asp-309. This increase in pH would favor the deprotonated state of the catalytic Asp-309 residue, reducing substrate affinity due to the breakage of a hydrogen bond between the residue and the ligand.
Based on our observations of the channels during the simulation, water mainly enters and exits the interior protein cavity through the S channel and the 2d channel, depending on the protonation state of Asp-309. Our simulations also indicate the effect of pH on substrate binding and catalytic activity properties of aromatase, wherein the S channel appears to be a likely candidate for the access/egress route for the substrate. For oxygen, however, we did not any observe any possible access/egress channels with MOLE 2.0. Since Sgrignani and Magistrato observed their proposed channels for oxygen using Implicit Ligand Sampling16, it is possible that these channels were too small or narrow for MOLE 2.0 to detect.
Effect of Protonation State on Ligand Binding and the Active Site
The protonation of Asp-309 appears to stabilize the position of the androstenedione ligand in the active site, due to formation of a hydrogen bond with the O3 keto oxygen atom of androstenedione. This interaction creates a stable void between the ligand and the heme molecule that allows for the positioning of the dioxygen moiety during catalysis of the aromatization reaction. Furthermore, the protonation state of Asp-309 maintains a significant influence on the structure and presence of water networks within the active site that may serve a catalytic role during the conversion of androgens into estrogens.
In the protonated trajectory, the closure of the S channel appears to exclude most water molecules into the active site, leading to the absence of a stable water network. The few water molecules that do enter the active site mainly associate with the iron atom of the heme ligand, as well as Thr-310. Furthermore, despite the open conformation of the 2d channel throughout most of the protonated trajectory, few water molecules are able to enter the active site, and instead remain between Val-370, Leu-372, and Leu-477. These water molecules, however, are on the O17 keto oxygen side of the ligand, away from the catalytic side of the ligand, and likely serve no significant catalytic role. The relative scarcity of water molecules in the active site also appears to stabilize the ligand, as indicated by the ligand RMSF of 1.016 Å in the deprotonated trajectory, and 0.885 Å in the protonated trajectory.
On the other hand, the deprotonated trajectory revealed extensive water networks within the active site, facilitated by the open conformation of the S channel throughout the simulation. The network consisted of water molecules that were (1) located between the androstenedione ligand the heme molecule, (2) located near Asp-309, Thr-310, Ala-306, and Ser-478, and (3) located within the S channel. We observed two localized positions for water molecules located between the androstenedione ligand and the heme molecule that could be stabilized positions for the dioxygen moiety and the presume catalytic water molecule during catalysis.
Dynamical Ligand-binding Hot Spots Indicate New Areas for Substrate Binding and Inhibitor Design
We discovered nine distinct consensus sites (Supporting Information Figure S5), or “hot spots” on the surfaces and cavities of the combined protonated and deprotonated trajectory using our customized clustering algorithm to determine unique non-overlapping consensus sites of interest. Of these consensus sites, two consensus sites provided detailed insight onto the molecular characterization of novel druggable hot spots, which build the framework for future novel inhibitors with novel mechanisms of action.
The first consensus site corresponded to the active site cavity, containing the heme and androstenedione molecules, as well as the S active site channel (Figure 5). Residues forming this consensus site included Met-107, Arg-115, Ile-132, Ile-133, Phe-134, Trp-141, Arg-145, Phe-148, Leu-152, Leu-188, Arg-192, Val-214, Ile-217, Gln-218, Tyr-220, Phe-221, Asp-222, Trp-224, Gln-225, Leu-228, Glu-302, Met-303, Ile-305, Ala-306, Ala-307, Pro-308, Asp-309, Thr-310, Met-311, Ser-312, Val-313, Ser-314, Val-369, Val-370, Asp-371, Leu-372, Val-373, Met-374, Arg-375, Ile-398, Pro-429, Phe-430, Phe-432, Arg-435, Glu-436, Phe-437, Ala-438, Ala-443, Leu-477, Ser-478, Leu-479, and His-480.
Figure 5.
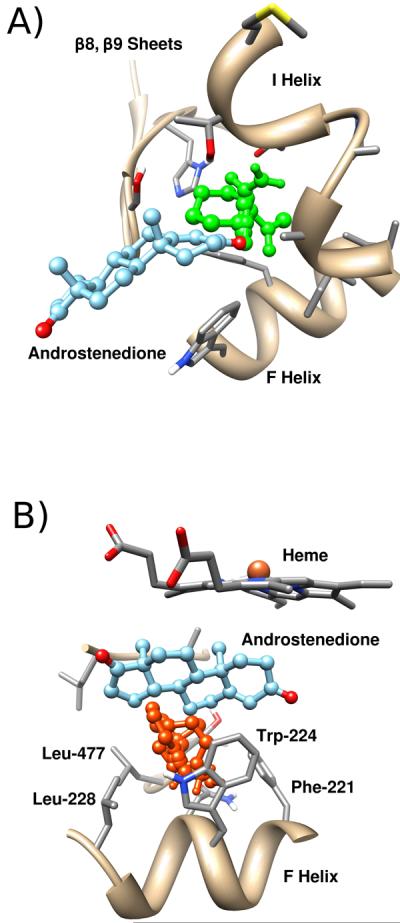
Dynamic druggable hot spots in the active site region of aromatase. A) Mainly polar probes (green) bind and extend out to the S channel in one of the trajectory clusters from simulations of aromatase with a deprotonated Asp-309 residue. B) Mainly aromatic probes (orange) bind into a crevice into the active site in one of the aromatase trajectory clusters with a protonated Asp-309 residue.
The position of most solvent probes within the first consensus site generally followed the position of the ligand within the active site. Other solvent probes were additionally located within the active site tunnel and a secondary crevice formed by residues near the O17 side of the ligand. While the active site tunnel corresponded with the S channel, the secondary crevice generally corresponded with the 2d channel. In the relatively hydrophobic first consensus site, hydrophobic van der Waals interactions predominate interactions between the solvent probes and active site residues such as Phe-134, Phe-221, Trp-224, Ile-305, Ala-306, Val-370, Val-373, Met-374, and Leu-477. However, significant hydrogen bonding between solvent probes and active site residues were also observed. These hydrogen bonds included those with the backbone amide with Met-374, backbone carbonyl of Leu-372, guanidium side chain of Arg-115, hydroxyl side chain of Thr-310, backbone carbonyl of Ala-306, backbone carbonyl of Leu-477, carboxylate side chain of Asp-309, backbone carbonyl of Leu-479, and hydroxyl side chain of Ser-478. Additional ring-stacking interactions with Phe-221 and Trp-224 were also observed in solvent probes that were bound to the active site channel and the secondary crevice, respectively.
In contrast to computational solvent mapping calculations of both trajectories, calculations for the four crystal structures reveal that solvent probes are absent in the active site channel portion of the first consensus site. Additionally, fewer solvent probes are observed in the secondary crevice of the first consensus site. MD simulations predict the flexibility and motions of the protein in the modeled environment, and, according to our model, the opening of the active site channels and secondary crevices promote new areas for substrate and inhibitor binding. These new druggable sites, which are absent in the four crystal structures, have great potential to influence the design and development of the next-generation of aromatase inhibitors.
Of additional interest was the second consensus site, a hydrophobic heme-proximal surface cavity, and the putative binding site of the integral association between CPR and aromatase during electron transfer for catalysis of the hydroxylation and the aromatization reactions. Residues forming the second consensus site included Phe-104, Tyr-361, Phe-418, Lys-420, Asn-421, Val-422, Pro-423, Tyr-424, Phe-427, Gln-428, Pro-429, Phe-430, Gly-431, Phe-432, Gly-433, Gly-436, Cys-437, and Lys-440.
The computational solvent mapping revealed that most probe molecules that bound to the cavity were hydrophobic in nature, complementing the hydrophobic nature of the heme proximal cavity. Probe molecules, however, were positioned to form hydrogen bonds with the backbone amide of Phe-430, backbone carbonyl of Phe-427, amine sidechain of Lys-440, backbone amide of Val-422, backbone carbonyl of Gly-431, and amide side chain of Gln-428. The computational solvent mapping of this hydrogen-bonding region suggests that this location is an optimal site for an alcohol substituent, as it allows for the oxygen of the alcohol moiety to hydrogen bond with the backbone amide atoms of Phe-430, and allows the hydrogen of the alcohol moiety to hydrogen bond with the backbone carbonyl of Gly-431. The hydrophobic residues Phe-104, Tyr-361, Tyr-424, Phe-427, Phe-430, and Phe-432 placed on the surface of the cavity further suggest further that this location is an optimal site for an aromatic substituent on the inhibitor in order to engage in stabilizing pi stacking.
Additional conserved consensus sites between clusters of both trajectories, whose pharmaceutical potential and significance remain unknown, were exhibited. An example of such a consensus site is a charged hydrophilic pocket that contains the openings of the 2d channel and Channel 5, formed by the A, K, and K' helices, ß8–9 sheets, and the K-ß3 loop. If the 2d channel or Channel 5 plays a significant role in the access and egress of substrates such as water molecules or oxygen molecules, inhibitors targeting this consensus site may potentially interfere with catalysis by blocking movement of key substrates and products. Another conserved consensus site is formed by the A' and G' helices, B'-B and F-G loops, and the ß1–4 sheets and consists of a hydrophobic pocket near the opening of the S channel. Inhibitors binding to this pocket may potentially induce changes in the nearby active site or the active site channel and interfere with catalysis.
CONCLUSION
Our results indicate the value in sampling various conformational states of aromatase in its native, membrane-bound cellular environment. Our work here suggests that the protonation state of the active site catalytic Asp-309 residue plays a significant role in reshaping the architecture of the active site and active site access channels. Furthermore, the effect of the protonation state of this residue on water networks and ligand binding suggests that the mechanism of action of aromatase involves dynamic protonation/deprotonation of Asp-309.
Conformational sampling in the membrane environment suggests possible new druggable hot spots for inhibitor binding, which are not currently utilized by ligands in the available crystal structures. Current aromatase inhibitors may act through the following mechanisms of action: (1) as a competitive inhibitor to the enzyme by mimicking the natural substrates of the enzyme (a Type I ligand), (2) as a competitive inhibitor to the enzyme by direct coordination with the heme iron while maintaining similarity to the substrate (a Type II ligand), (3) as an irreversible suicide inhibitor to the enzyme by chemically attacking key residues to permanently inactivate the enzyme by formation of a covalent substrate-enzyme complex. The next generation of aromatase inhibitors may build upon current inhibitors by exploiting the novel, dynamic druggable sites presented here.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Prof. Tom Poulos for inspiring discussions. This work was funded in part by through the NIH Director's New Innovator Award Program DP2-OD007237 and the National Science Foundation's XSEDE Supercomputer resources grant LRAC CHE060073N to R.E.A. Support from the National Biomedical Computation Resource, the Center for Theoretical Biophysics, and UCSD Drug Discovery Institute is gratefully acknowledged.
Footnotes
SUPPORTING INFORMATION Illustration of the solvent-exposure of epitope sites and cytochrome P450 reductase binding interface. Angle between the normal to the plane of the heme and the normal to the plane of the membrane (the z-axis) in both trajectories. Vertical distance between aromatase and the center of the membrane in both trajectories. RMSD measurements for both trajectories. Results of the GROMOS++ clustering of trajectory ensembles in the deprotonated and protonated trajectories. Depiction of the nine consensus sites identified by FTprod after computational solvent mapping calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Guengerich P. Human Cytochrome P450 Enzymes. In: de Montellano PRO, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd ed. Springer; New York: 2005. pp. 450–452. [Google Scholar]
- 2.Hong Y, Li H, Yuan YC, Chen S. Molecular Characterization of Aromatase. Ann. N.Y. Acad. Sci. 2009;1155:112–120. doi: 10.1111/j.1749-6632.2009.03703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plourde PV, Dyroff M, Dowsett M, Demers L, Yates R, Webster A. ARIMIDEX: A new oral, once-a-day aromatase inhibitor. J. Steroid Biochem. Mol. Biol. 1995;53:175–179. doi: 10.1016/0960-0760(95)00045-2. [DOI] [PubMed] [Google Scholar]
- 4.Trunet PF, Bhatnagar AS, Chaudri HA, Hornberger U. Letrozole (CGS 20267), A New Oral Aromatase Inhibitor for the Treatment of Advanced Breast Cancer in Postmenopausal Patients. Acta Oncol. 1996;35:15–8. doi: 10.3109/02841869609083962. [DOI] [PubMed] [Google Scholar]
- 5.Lipton A, Demers LM, Harvey HA, Kambic KB, Grossberg H, Brady C, Adlercruetz H, Trunet PF, Santen RJ. Letrozole (CGS 20267). A phase I study of a new potent oral aromatase inhibitor of breast cancer. Cancer. 1995;75:2132–2138. doi: 10.1002/1097-0142(19950415)75:8<2132::aid-cncr2820750816>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 6.Evans TR, Di Salle E, Ornati G, Lassus M, Benedetti MS, Pianezzola E, Coombes RC. Phase I and Endocrine Study of Exemestane (FCE 24304), a New Aromatase Inhibitor, in Postmenopausal Women. Cancer Res. 1992;52:5933–5939. [PubMed] [Google Scholar]
- 7.Gobbi S, Cavalli A, Rampa A, Belluti F, Piazzi L, Paluszcak A, Hartmann RW, Recanatini M, Bisi A. Lead Optimization Providing a Series of Flavone Derivatives as Potent Nonsteroidal Inhibitors of the Cytochrome P450 Aromatase Enzyme. J. Med. Chem. 2006;49:4777–4780. doi: 10.1021/jm060186y. [DOI] [PubMed] [Google Scholar]
- 8.Cepa MMDS, Tavares da Silva EJ, Correia-da-Silva G, Roleira FMF, Teixeira NAA. Structure–Activity Relationships of New A,D-Ring Modified Steroids as Aromatase Inhibitors: Design, Synthesis, and Biological Activity Evaluation. J. Med. Chem. 2005;48:6379–6385. doi: 10.1021/jm050129p. [DOI] [PubMed] [Google Scholar]
- 9.Woo LWL, Sutcliffe OB, Bubert C, Grasso A, Chander SK, Purohit A, Reed MJ, Potter BVL. First Dual Aromatase-Steroid Sulfatase Inhibitors. J. Med. Chem. 2003;46:3193–3196. doi: 10.1021/jm034033b. [DOI] [PubMed] [Google Scholar]
- 10.Su B, Diaz-Cruz ES, Landini S, Brueggemeier RW. Novel Sulfonanilide Analogues Suppress Aromatase Expression and Activity in Breast Cancer Cells Independent of COX-2 Inhibition. J. Med. Chem. 2006;49:1413–1419. doi: 10.1021/jm051126f. [DOI] [PubMed] [Google Scholar]
- 11.Neves MAC, Dinis TCP, Colombo G, Sá e Melo ML. Fast Three Dimensional Pharmacophore Virtual Screening of New Potent Non-Steroid Aromatase Inhibitors. J. Med Chem. 2009;52:143–150. doi: 10.1021/jm800945c. [DOI] [PubMed] [Google Scholar]
- 12.Woo LWL, Bubert C, Sutcliffe OB, Smith A, Chander SK, Mahon MF, Purohit A, Reed MJ, Potter BVL. Dual Aromatase–Steroid Sulfatase Inhibitors. J. Med. Chem. 2007;50:3540–3560. doi: 10.1021/jm061462b. [DOI] [PubMed] [Google Scholar]
- 13.Woo LWL, Bubert C, Purohit A, Potter BVL. Hybrid Dual Aromatase-Steroid Sulfatase Inhibitors with Exquisite Picomolar Inhibitory Activity. ACS Med. Chem. Lett. 2011;2:243–247. doi: 10.1021/ml100273k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh D, Griswold J, Erman M, Pangborn W. Structural Basis for Androgen Specificity and Oestrogen Synthesis in Human Aromatase. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh D, Griswold J, Erman M, Pangborn W. X-ray structure of human aromatase reveals an androgen-specific active site. J. Steroid Biochem. Mol. Biol. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sgrignani J, Magistrato A. Influence of the Membrane Lipophilic Environment on the Structure and on the Substrate Access/Egress Routes of the Human Aromatase Enzyme. A Computational Study. J. Chem. Inf. Model. 2012;52:1595–1606. doi: 10.1021/ci300151h. [DOI] [PubMed] [Google Scholar]
- 17.Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular Docking of Aromatase Inhibitors. Molecules. 2011;16:3597–3617. [Google Scholar]
- 18.Jiang W, Ghosh D. Motion and Flexibility in Human Cytochrome P450 Aromatase. PLoS One. 2012;7:e32565. doi: 10.1371/journal.pone.0032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh D, Lo J, Morton D, Valette D, Xi J, Griswold J, Hubbell S, Egbuta C, Jiang W, An J, Davies HM. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amaro RE, Baron R, McCammon JA. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided Mol. Des. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. Discovery of a Novel Binding Trench in HIV Integrase. J. Med. Chem. 2004;47:1879–1881. doi: 10.1021/jm0341913. [DOI] [PubMed] [Google Scholar]
- 22.Durrant JD, Hall L, Swift RV, Landon M, Schnaufer A, Amaro RE. Novel Naphthalene-Based Inhibitors of Trypanosoma brucei RNA Editing Ligase 1. PLoS Negl Trop Dis. 2010;4:e803. doi: 10.1371/journal.pntd.0000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durrant JD, Cao R, Gorfe AA, Zhu W, Li J, Sankovsky A, Oldfield E, McCammon JA. Non-Bisphosphonate Inhibitors of Isoprenoid Biosynthesis Identified via Computer-Aided Drug Design. Chem. Biol. Drug. Des. 2011;78:323–332. doi: 10.1111/j.1747-0285.2011.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinko W, de Oliveira C, Williams S, Van Wynsberghe A, Durrant JD, Cao R, Oldfield E, McCammon JA. Applying Molecular Dynamics Simulations to Identify Rarely Sampled Ligand-Bound Conformational States of Undecaprenyl Pyrophosphate Synthase, an Antibacterial Target. Chem. Biol. Drug. Des. 2011;77:412–420. doi: 10.1111/j.1747-0285.2011.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Lindert S, Cao R, Li K, Wang K, Wang Y, Liu YL, Sankovsky A, de Oliveira CA, Mitchell DA, Nizet V, McCammon JA, Oldfield E. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2013;110:123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wassman CD, Baronio R, Demir O, Wallentine BD, Chen CK, Hall LV, Salehi F, Lin DW, Chung BP, Hatfield GW, Chamberlin AR, Luecke H, Lathrop RH, Kaiser P, Amaro RE. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. [(accessed Jan 30, 2013)];Nat. Commun. 2013 4 doi: 10.1038/ncomms2361. http://www.nature.com/ncomms/journal/v4/n1/full/ncomms2361.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Nardo G, Breitner M, Sadeghi J, Nicolai E, Mei G, Bandino A, Ghosh D, Panzica G, Gilardi G. A pH-Dependent Structural Rearrangement of the Human Aromatase Active Site: Implications in Catalysis. FEBS J. 2010;277:175. [Google Scholar]
- 28.Maestro. version 9.2 Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 29.Ghosh D, Jiang W, Lo J, Egbuta C. Higher order organization of human placental aromatase. Steroids. 2011;76:753–758. doi: 10.1016/j.steroids.2011.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of Proteins in Membranes Database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 31.Lomize AL, Pogozheva ID, Mosberg HI. Anisotropic Solvent Model of the Lipid Bilayer. 2. Energetics of Insertion of Small Molecules, Peptides, and Proteins in Membranes. J. Chem. Inf. Model. 2011;51:930–946. doi: 10.1021/ci200020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. Nucleic Acids Res. 2011;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yau WM, Wimley WC, Gawrisch K, White SH. The Preference of Tryptophan for Membrane Interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 34.Boyd D, Beckwith J. Positively charged amino acid residues can act as topogenic determinants in membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1989;86:9446–9450. doi: 10.1073/pnas.86.23.9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong Y, Li H, Ye J, Miki Y, Yuan YC, Sasano H, Evans DB, Chen S. Epitope Characterization of an Aromatase Monoclonal Antibody Suitable for the Assessment of Intratumoral Aromatase Activity. PLoS One. 2009;4:e8050. doi: 10.1371/journal.pone.0008050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 37.Davison SC, Wills ED. Studies on the Lipid Composition of the Rat Liver Endoplasmic Reticulum After Induction with Phenobarbitone and 20-Methylcholanthrene. Biochem. J. 1974;140:461–468. doi: 10.1042/bj1400461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 39.Mackerell AD, Jr., Feig M, Brooks CL., III Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 40.MacKerell AD, Bashford D, Bellott, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 41.Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr., Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 43.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKaValues. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 44.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 46.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 47.Ryckaert J, Ciccotti G, Berendsen H. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 48.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089. [Google Scholar]
- 49.Daura X, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Reversible Peptide Folding in Solution by Molecular Dynamics Simulation. J. Mol. Biol. 1998;280:925–932. doi: 10.1006/jmbi.1998.1885. [DOI] [PubMed] [Google Scholar]
- 50.Baron R, McCammon JA. Dynamics, Hydration, and Motional Averaging of a Loop-Gated Artificial Protein Cavity: The W191G Mutant of Cytochrome c Peroxidase in Water as Revealed by Molecular Dynamics Simulations. Biochemistry. 2007;46:10629–10642. doi: 10.1021/bi700866x. [DOI] [PubMed] [Google Scholar]
- 51.Berka K, Hanak O, Sehnal D, Banas P, Navratilova V, Jaiswal D, Ionescu CM, Svobodova Varekova R, Koca J, Otyepka M. MOLEonline 2.0: interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 2012;40:W222–W227. doi: 10.1093/nar/gks363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Votapka L, Amaro RE. Multistructural hot spot characterization with FTProd. Bioinformatics. 2012;29:393–394. doi: 10.1093/bioinformatics/bts689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 54.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 55.Ohta Y, Kawato S, Tagashira H, Takemori S, Kominami S. Dynamic Structures of Adrenocortical Cytochrome P-450 in Proteoliposomes and Microsomes: Protein Rotation Study. Biochemistry. 1992;31:12680–12687. doi: 10.1021/bi00165a019. [DOI] [PubMed] [Google Scholar]
- 56.Berka K, Hendrychova T, Anzenbacher P, Otyepka M. Membrane Position of Ibuprofen Agrees with Suggested Access Path Entrance to Cytochrome P450 2C9 Active Site. J. Phys. Chem. A. 2011;115:11248–11255. doi: 10.1021/jp204488j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Praporski S, Ng SM, Nguyen AD, Corbin CJ, Mechler A, Zheng J, Conley AJ, Martin LL. Organization of Cytochrome P450 Enzymes Involved in Sex Steroid Synthesis: PROTEIN-PROTEIN INTERACTIONS IN LIPID MEMBRANES. J. Biol. Chem. 2009;284:33224–33232. doi: 10.1074/jbc.M109.006064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayburt TH, Sligar SG. Single-molecule height measurements on microsomal cytochrome P450 in nanometer-scale phospholipid bilayer disks. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6725–6730. doi: 10.1073/pnas.062565599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bayburt TH, Carlson JW, Sligar SG. Reconstitution and Imaging of a Membrane Protein in a Nanometer-Size Phospholipid Bilayer. J. Struct. Biol. 1998;123:37–44. doi: 10.1006/jsbi.1998.4007. [DOI] [PubMed] [Google Scholar]
- 60.Bayburt TH, Carlson JW, Sligar SG. Single Molecule Height Measurements on a Membrane Protein in Nanometer-Scale Phospholipid Bilayer Disks. Langmuir. 2000;16:5993–5997. [Google Scholar]
- 61.Cojocaru V, Winn PJ, Wade RC. The Ins and Outs of Cytochrome P450s. Biochim. Biophys. Acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.