

Abstract
The TAR DNA-binding protein 43 (TDP-43) has been identified as a critical player in a range of neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). Recent discoveries demonstrate the important role of carboxyl-terminal fragments of TDP-43 in its proteinopathy. Herein, we report the characterization of β-domains in the C-terminal fragments of TDP-43 using scanning tunneling microscopy (STM). Careful comparison of the wild-type TDP-43 (Wt) and the three mutant TDP-43 peptides: an ALS-related mutant peptide: phosphorylated A315T mutant TDP-43 (A315T(p)) and two model peptides: A315T mutant TDP-43 (A315T), A315E mutant TDP-43 (A315E) reveals that A315T(p) has a longer core region of the β-domain than Wt. A315E possesses the longest core region of the β-domain and A315T(p) mutant TDP-43 has the second longest core region of the β-domain. The core regions of the β-domains for A315T and Wt TDP-43 have the same length. This observation provides a supportive evidence of a higher tendency in beta-sheet formation of A315T(p) containing TDP-43 fragment, and structural mechanism for the higher cytotoxicity and accelerated fibril formation of the A315T(p) mutation-containing TDP-43 peptide as compared with Wt TDP-43.
Keywords: TAR DNA-binding protein 43, β-Domain, STM, Amyloid, Fibril formation
1. Introduction
Neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS), are characterized by the progressive loss of structure or function of neurons, including degeneration or death of neurons. ALS and FTLD are neuronal diseases caused by the degeneration of neurons located in the ventral horn of the spinal cord, or frontal and temporal lobes of the brain, respectively. Pathology with ubiquitin-positive, tau-negative, and TDP-43 positive neuronal inclusions have been recognized in FTLD, ALS and other, neuronal diseases, collectively classified as TDP-43 proteinopathy (Lagier-Tourenne et al., 2010; Mackenzie et al., 2010; Cohen et al., 2011). TAR DNA-binding protein 43 (TDP-43) is a multifunctional DNA/RNA binding protein that plays important roles in a number of cellular processes: transcription regulation, splicing and other steps of post-transcriptional gene regulation (Neumann et al., 2006; Kuo et al., 2009; Wang et al., 2008). Since the first discovery by Neumann et al. that TDP-43 was the main protein constituent of the ubiquitinated inclusions in ALS patients (Neumann et al., 2006), intensive interest has been drawn to the TDP-43 related research. It has been reported that nearly half of the FTLD cases and, most of the ALS cases and some other neurodegenaration diseases exhibit TDP-43 pathology, and the disease-related TDP-43 proteins are commonly characteristic of hyperphosphorylation, cleavage, ubiquitination, mislocalization and aggregation into insoluble aggregates (Neumann et al., 2006; Chen-Plotkin et al., 2010; Piao et al., 2003; Arai et al., 2009; Kadokura et al., 2009). However, molecular mechanisms underlying TDP-43 neurotoxicity remain to be elucidated.
More than thirty different mutations have been identified in the TDP-43 gene among ALS patients with most of them clustered in the carboxyl terminal region of the TDP-43 protein (Lagier-Tourenne et al., 2010; Neumann et al., 2006). Both in vitro and animal model studies suggest that C-terminal domains may play a significant role in TDP-43 proteinopathy (Lagier-Tourenne et al., 2010; Neumann et al., 2006; Arai et al., 2009; Hasegawa et al., 2008; Igaz et al., 2008, 2009). Several mutations in the C-terminal domain of TDP-43 have been shown to increase its propensity to form aggregation (Sreedharan et al., 2008; Guo et al., 2011; Zhang et al., 2009). Previous studies demonstrate that an ALS phosphorylated mutant, A315T(p) has increased propensity to form amyloid fibrils in solution and protein aggregates in cultured neurons than Wt TDP-43 (Guo et al., 2011; Barmada et al., 2010), and leads to accelerated neurodegenration in transgenic animals (Guo et al., 2011; Wegorzewska et al., 2009).
A previous study using small angle X-ray scattering (SAXS) and GST pull-down assay suggested the dimer formation in TDP-43 aggregates (Kuo et al., 2009). Time-lapse atomic force microscopy (AFM) studies on aggregation of Wt and A315T(p)-containing TDP-43 peptides demonstrate that the A315T(p) phosphorylated mutant exhibits a higher tendency to form fibrils than Wt TDP-43 (Guo et al., 2011). However, the structural basis for these observations remains to be defined. So far, structural analyses of TDP-43 carboxyl terminal domain using X-ray diffraction (XRD) or nuclear magnetic resonance (NMR) techniques are largely unsuccessful due to the heterogeneous amyloidal aggregation of TDP-43 C-terminal domain (Guo et al., 2011). The direct observation of the assembly and aggregation structures of the TDP-43 proteins without the requirement of the crystallization and complex computations at single molecule level would be highly rewarding.
Our previous reports have demonstrated that scanning tunneling microscopy (STM) could be applied for amyloid structure analysis due to its high structural resolution and adaptability to various environments (Ma et al., 2009; Mao et al., 2009, 2011; Liu et al., 2009, 2011; Wang et al., 2011). We have reported previously the high resolution structures of β-amyloid peptide1-42 relating to Alzheimer’s disease (Ma et al., 2009) and amylin peptides relating to type II diabetes (Mao et al., 2009, 2011). The modulation of amyloid assembly (Liu et al., 2009, 2011) and amyloid-dye molecule interactions (Mao et al., 2011; Wang et al., 2011) were also investigated by STM in our previous reports, which is beneficial for the detection and treatment of amyloidosis. Due to the high structural resolution of STM, it is feasible to directly visualize the peptide strand with beta-sheet or beta-like structures and compare the structural difference between Wt and mutant TDP-43 peptides. The studies on intrinsic or modulated peptide structures at single-molecule level would give more insight into the structure–activity relationship.
Herein we report characterization of an amyloidogenic region in TDP-43 carboxyl terminal domain using STM and comparison of the Wt TDP-43 with A315T mutant, phosphorylated mutant A315T(p) and A315E mutant TDP-43 peptides. Synthetic peptides were prepared corresponding to the carboxyl terminal region of TDP-43 peptide that has been shown to form amyloid fibrils and cause neurotoxicity (Guo et al., 2011), Wt and A315T(p) peptides. All peptides contain residue Gln286 to Gln331 in the full length TDP-43 protein, and A315T(p) phosphorylated mutant peptide contains phosphorylated threonine at the residue 315. A315T mutant peptide is considered and compared with A315T(p) for understanding the impact of the phosphorylation, and A315E mutant peptide was introduced for understanding the effect of negative charges in A315T(p) on the amyloid aggregation and related neurotoxicity. The sequences of Wt, A315T, A315T(p) and A315E peptides are shown in Scheme 1a. The molecular structures and assembly structures of Wt, A315T, A315T(p) and A315E peptides are investigated by using STM, and the aggregation kinetics of these peptides are monitored by thioflavin T (ThT) fluorescence.
Scheme 1.
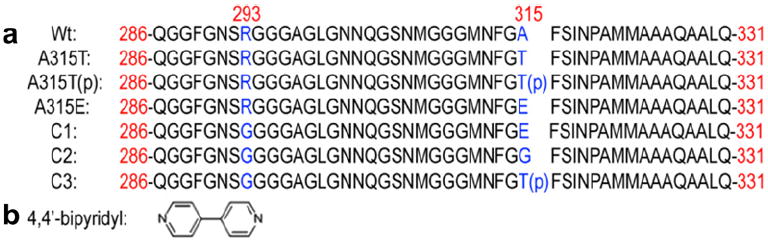
The sequences of Wt, A315T, A315T(p), A315E, A293G/A315E (C1), A293G/A315G (C2) and A293G/A315T(p) (C3) TDP-43 peptides (a) and molecular structure of 4,4′-bipyridyl. In Scheme 1a, the red numbers represent the residues in the Wt, A315T, A315T(p), A315E, C1, C2 and C3 peptides in the 414-mer TDP-43. The blue capital letters “A”, “T”, “E”, “G” and “R” highlight the mutation site, and “p” in the bracket points out the phosphorylated residue Thr315. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
In order to identify the carboxyl terminal of the peptides, small molecule 4,4′-bipyridyl (4Bpy, Scheme 1b) were introduced to interact with the C terminal of the peptides, which could facilitate the length measurement of the peptide strands with beta-like structures (Mao et al., 2011). The peptides are observed to assemble homogeneously on graphite surface as a reflection of tendency of β-sheet formation. By determining the lengths of extended peptide fragments of both C-terminus and N-terminus, we are able to identify the β-domains and also the associated mutation effect. Light scattering experiments were also conducted to reveal the aggregation propensity of the C-terminal fragment of TDP-43 and its ALS mutants. The longer β-domains from the STM results are consistent with the stronger aggregation propensity of A315E than the other three peptides from ThT fluorescence results.
2. Materials and methods
2.1. Sample preparation
All the Wt, A315T, A315T(p), A315E, and their counter parts with reversed sequence, reverse Wt (re-Wt), reverse A315T (re-A315T), reverse A315T(p) (re-A315T(p)) and reverse A315E (re-A315E) TDP-43 peptides (corresponding to amino acid residues 286–331 of human TDP-43 protein) (Guo et al., 2011) and three comparison peptides A293G/A315E (C1), A293G/A315G (C2) and A293G/A315T(p) (C3) were purchased from Shanghai Science Peptide Biological Technology Co., Ltd. with purity of >98%. 4Bpy and ThT molecules are obtained from Sigma–Aldrich and used without further purification. The TDP-43 peptides were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Sigma–Aldrich) initially in sterile microcentrifuge tubes, stored at 4 °C for extended time, followed by evaporation of the HFIP solvent under vacuum. The obtained peptide deposits were dissolved in MilliQ water. 4Bpy was also dissolved in MilliQ water and then mixed with the peptide solution to reach the final concentration ca. 0.05 mM. The mixed solutions were kept standstill for 2 h, and then 2 μL of the mixed solutions were dropped onto freshly cleaved highly oriented pyrolytic graphite (HOPG) surface for STM observations after the solvent was evaporated.
2.2. STM experiment
All the STM experiments were carried out with a Nanoscope IIID system (Bruker Nano, USA) under ambient conditions. Mechanically formed Pt/Ir (80/20) tips were used in the STM experiments. All STM images were obtained in constant current mode, and the tunneling conditions are shown in the corresponding figure captions.
2.3. ThT fluorescence experiment
The aqueous solutions of Wt, A315T, A315T(p) and A315E TDP-43 synthetic peptides with forward and reversed sequence were prepared at the concentration of 125 μM in 0.2 mM phosphate buffer (5 mM sodium dihydrogen phosphate and sodium phosphate, pH = 7.4) in the presence of 1% of DMSO, but the concentrations of the A315T(p), A315E and C1, C2, C3 are 250 μM in 0.2 mM phosphate buffer in Fig. 4. Stock solutions of the peptides were incubated at 37 °C and transferred into the quartz cells immediately before ThT fluorescence detection. The peptide solutions were incubated again immediately between neighboring ThT fluorescence detections. The ThT fluorescence experiments were performed on a Hitachi F4600 fluorescence spectrophotometer at room temperature using quartz cells with ThT fluorescence intensity obtained at emission wavelength of 480 nm and excitation wavelength of 450 nm with a spectrum bandwidth of 1 nm.
Fig. 4.
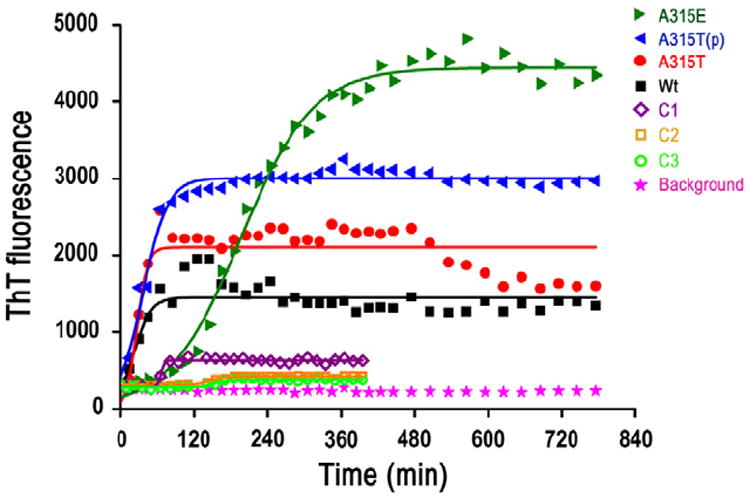
The time course of ThT fluorescence experiments of TDP-43 peptides A315E (green solid triangles), A315T(p) (blue solid triangles), A315T (red solid circles), Wt TDP-43 peptide (black solid squares) and C1 (purple open diamonds), C2 (yellow open squares), C3 (light green open circles). The background phosphate buffer control is shown as pink solid stars. The time course of ThT fluorescence experiments of TDP-43 peptides shows an increased aggregation propensity of A315E than the other TDP-43 peptides, and aggregation propensity of A315T(p) TDP-43 peptide ranks second. The ThT fluorescence intensities of the Wt, A315T, A315T(p), A315E (125 μM in 0.2 mM phosphate buffer) TDP-43 peptides and C1, C2, C3 (250 μM in 0.2 mM phosphate buffer) are determined in aqueous solutions. The aggregation propensity of A315E and A315T(p) is stronger than C1, C2 and C3, which confirmed the significance of the electrostatic interaction between the positive charge on Arg293 and negative charges at 315 sites on A315T(p) or A315E, although the concentrations of A315E and A315T(p) are half of the concentrations of C1, C2 and C3. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3. Results and discussion
To investigate the structural features of TDP-43 carboxyl-terminal fragments, the synthetic peptides that were shown to form amyloid fibrils (Guo et al., 2011) were labeled with 4Bpy for STM imaging (Liu et al., 2009, 2011; Mao et al., 2011; Wang et al., 2011). The pyridyl species are introduced targeting the carboxyl terminus of peptides via hydrogen bond interaction between the nitrogen atoms of 4Bpy and the carboxyl groups of peptide C-termini. Because the higher electron density of the labeling molecule 4Bpy gives rise to the higher contrast in STM imaging, the C-terminus of the TDP-43 peptides can be clearly identified.
The STM images for 4Bpy-labeled Wt or A315T (Fig. 1a and c) and A315T(p) or A315E (Fig. 1e and g) peptide co-assembly structures on HOPG surface reveal the lamella characteristics with 4Bpy molecular arrays bearing higher contrast between peptide stripes. The high-resolution STM images show the molecularly resolved 4Bpy moiety and the peptide backbones. The lamella structure of the peptide assembly could be attributed to the formation of β-like structures (β-domain). With the labeling of the 4Bpy molecules, one can directly measure the peptide strand length from C-terminus, corresponding to the length of the β-domain (Mao et al., 2011). The corresponding statistical histograms of the length distributions of the Wt, A315T, A315T(p) and A315E TDP-43 peptide stands are shown in Fig. 1b, d, f and h respectively. The green line segments in each STM images represent the measured length and the green Arabic numerals “eight” features highlight the 4Bpy molecules. The measured lengths for Wt TDP-43 peptide are within the range from 2.60 to 4.550 nm, corresponding to a β-domain consisting of 9–14 amino acids. From the Gaussian fitting of the length distribution, the most probable β-domain consists of 11 amino acids at the carboxyl terminus. The measured length for A315T, A315T(p) and A315E TDP-43 peptide range from 2.60 to 4.875 nm, 3.575–5.525 nm and 5.850–7.80 nm respectively, corresponding to a β-strand with 8–15, 12–17 and 18–24 amino acids. The Gaussian fitting gives the most probable β-domain consisting of 11, 16 and 22 amino acids. The longer core fragments of β-domain for A315E and A315T(p) that those of A315T and Wt TDP-43 peptides is indicative of higher tendency of the β-sheet formation of the A315E and A315T(p).
Fig. 1.

STM experiments show that the Wt, A315T, A315T(p) and A315E TDP-43 peptide form the different length beta-sheet at the carboxyl terminus. STM images of two-dimensional assemblies of 4Bpy-labeled Wt, A315T, A315T(p) and A315E peptides are shown in panels (a, c, e and g) respectively. The tunneling conditions are: 600 mV and 360 pA, TDP-43 peptides on the HOPG surface. Histograms with Gaussian fitting of the length distribution of core β-domain for Wt (b), A315T (d), A315T(p) (f) and A315E (h) peptides. The numbers shown in the histograms mark the number of the amino acids in the core regions. The green lines and green Arabic numerals “eight” figures highlight the length of beta-sheet we measured and the 4Bpy molecules. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The key residues of the β-domains for Wt, A315T, A315T(p) and A315E TDP-43 peptides, Ala321, Ala321, Phe316 and Gly310, were marked by thin red arrows in Fig. 2a–d respectively. The gray arrows with superimposed peptide main-chains and side-chains stand for the β-domains identified in the STM images (Fig. 2a–d). The flexible strings consisting of schematically colored spheres represent the undiscernible regions of the peptide chains of TDP-43 peptides in STM images. The side-chain conformations in the discernible parts are shown schematically. The similar key sites of C-terminal β-domains at Ala321 for Wt and A315T peptides could be ascribed to the possible β-turn structure due to the neighboring proline residue Pro320 (Hutchinson and Thornton, 1994; Rose et al., 1985). This phenomenon gives a hint that the only mutation from Ala315 to Thr315 has negligible effect on the β-structure formation and the sequential amyloid aggregation. The key site for the C-terminal β-domains of A315T(p) is located at Phe316, which is the neighboring residue to the phospholated Thr315 residue in A315T(p) peptide. The expanded β-domain compared to A315T peptide indicates that the impact of the phosphorylation on the β-sheet structures for C-terminal of TDP-43 could arise from the negative charges introduced by phosphorylation. The even more prolonged C-terminal β-domains of A315E to Gly310 confirm the hypothesis that the increased propensity of beta-structure formation could be ascribed to the mutation from Ala315 to Glu315 with negative charges. These results indicate that the introduced negative charges in TDP-43 peptides (A315T(p) and A315E) could bring extra electrostatic interactions, most likely with the only positive charged Arg293 residue.
Fig. 2.
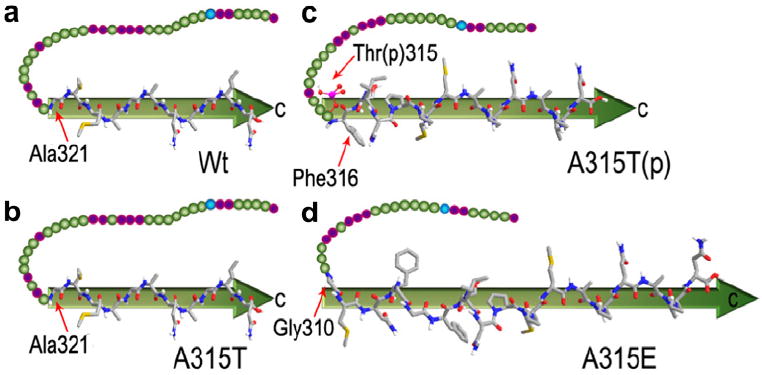
Schematic illustration of the observable core β-domain with highlighted folding sites at key residues (highlighted by red arrows) for Wt (a), A315T (b), A315T(p) (c) and A315E (d), and the residues of Arg293, Thr315 and Glu315 are pointed out.
Because the beta-sheet structures are dominated by the hydrogen bonding between backbones and jointly by the diverse interactions between side groups, most of the interactions for peptide with forward and reverse sequences could be expectedly unchanged. The same lengths for the β-structures can also be expected for the reverse and forward sequences in the peptides. Because the 4Bpy can only label the C-termini of peptides on the surface, we synthesized the re-Wt, re-A315T, re-A315T(p) and re-A315E TDP-43 peptides from native l-amino acids with reverse sequences corresponding to Wt, A315T, A315T(p) and A315E, in which C-terminus of the re-peptides corresponding to the N-terminus of normal sequences. In this way, we can get both the N-terminal and C-terminal β-domains on the HOPG substrate. With the same approach, the β-domains of re-Wt, re-A315T, re-A315T(p) and re-A315E were determined by STM images to obtain the N-terminus fragments of normal sequences Wt, A315T, A315T(p) and A315E as shown in Fig. 3a, c, e and g. The histograms of the β-domain length distributions with Gaussian fitting for re-Wt, re-A315T, re-A315T(p) and re-A315E are shown in Fig. 3b, d, f and h respectively. In contrast to the Wt, A315T, A315T(p) and A315E TDP-43 peptides, all of the four reverse TDP-43 peptides show bimodal distribution and the predominant lengths of β-domain containing 14 amino acids. The minor peaks for β-domain lengths are centered at 20 and 21 amino acids. The key site at Leu299 residue and the minor sites at Ser305, Asn306 residues for re-Wt, re-A315T, re-A315T(p) and re-A315E peptides could be ascribe to the favorable flexible turn at the neighboring Gly298, Gly300 and Gly304. These results show that A315E TDP-43 contains a longest β-domain in the C-terminal part than the other three counterpart TDP-43 peptides, and A315T(p) TDP-43 peptide has the second longest β-domain, and Wt, A315T TDP-43 peptides share nearly the same β-domain length in the C-terminal part. It should be noted that the 4Bpy molecules can form hydrogen bond with carboxyl groups at C-terminal, and it is also possible that 4Bpy molecules bind onto side groups, such as Asp or Glu. The STM images featuring of the peptide strands are convolution of geometrical topography and electronic states, and the side groups out of the surface should have inevitable conformational variations and fluctuations. Such fluctuations could blur the STM imaging resolution of the side groups, and the recognition of the side chains is still a challenging task.
Fig. 3.
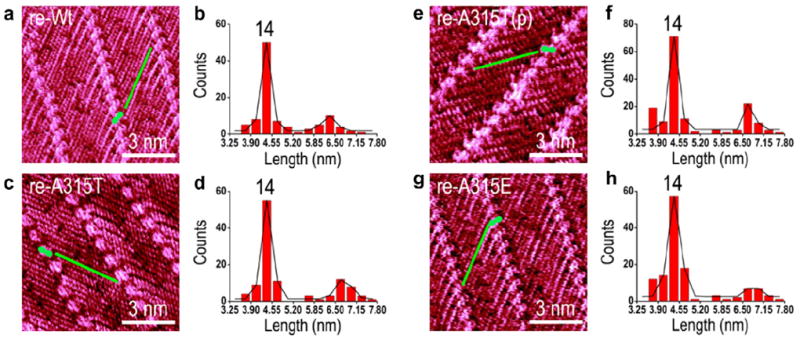
STM experiments show that the re-Wt, re-A315T, re-A315T(p) and re-A315E TDP-43 peptide form the same length beta-sheet at the carboxyl terminus. STM images of two-dimensional assemblies of 4Bpy-labeled re-Wt, re-A315T, re-A315T(p) and re-A315E peptides are shown in panels (a, c, e and g) respectively. The tunneling conditions are: 600 mV and 360 pA, TDP-43 peptides on the HOPG surface. Histograms with Gaussian fitting of the length distribution of core β-domain for re-Wt (b), re-A315T (d), re-A315T(p) (f) and re-A315E (h) peptides. The numbers shown in the histograms mark the number of the amino acids in the core regions. The green lines and green Arabic numerals “eight” figures highlight the length of beta-sheet we measured and the 4Bpy molecules. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
From the energetic point of view, the aggregation propensity should be dependent on the total lengths of the core β-domains if there are multiple β-strands within the whole sequence. The reverse sequences all show major peaks at 14 residues, which means the contribution from the N-terminal β-domains could be plausibly considered the same. Thus the aggregation propensity could be mainly dependent on the length of the C-terminal β-stand where the main mutation site is located. To further examine the aggregation behaviors of Wt, A315T, A315T(p) and A315E TDP-43 peptides in PBS solution, ThT fluorescence experiments were performed following incubation at 37 °C. It should be noted that the synthetic TDP-43 peptides with forward and reverse sequences show nearly the same aggregation trends from the ThT fluorescence experiments (Fig. S1). In Fig. 4, the ThT fluorescence results revealed that all of the Wt A315T, A315T(p) and A315E TDP-43 peptides showed significant aggregation propensity. In addition, the A315E peptide seems to form aggregates more rapidly than the other three peptides in the aqueous solution. The aggregation processes of Wt, A315T, A315T(p) and A315E peptides are fitted with logistic curves () as shown in black, red, blue and green solid lines in Fig. 4, in which a, b and k are three constants. According to the logistic function, the equilibrium intensities (a) are 1450, 2106, 3008 and 4440 respectively. Much stronger aggregation propensity of the A315E and A315T(p) than the Wt and A315T TDP-43 peptide are observed in ThT fluorescence experiments, which is consistent with the longer C-terminal β-domain in Figs. 1 and 2. The stronger aggregation of A315E and A315T(p) could be attributed to the possible electrostatic interactions with the positive charged Arg293 residue. The increased equilibrium intensities from Wt to A315T to A315T(p) also demonstrate the higher and higher propensity of amyloid aggregation by the mutation and the followed phosphorylation. The intensity difference of A315T(p) and Wt are also consistent with the previously reported time-lapse AFM data of these TDP-43 peptide aggregates formed on mica surface (Guo et al., 2011). Aggregation of the Wt peptide followed the process from small granular aggregates to very thin fibrils after 17 h incubation at 37 °C, whereas the A315T(p) peptide showed an accelerated aggregate formation from short protofibrils to thick fibrils (Sreedharan et al., 2008). The increased equilibrium intensity of A315E compared to the ALS related A315T(p) could be attributed to the longer core β-domain, as well as the strong hydrogen bond formation between Glu315 and Arg293 besides the electrostatic interactions. The stronger interaction facilitated the longer C-terminal β-domain, which results in kinetically slower assembly rate at the initial stage while higher equilibrium intensities. The longer core region of β-domain of A315T(p) peptide detected by STM provides a possible mechanistic explanation for the increased propensity of fibril formation and higher neurotoxicity of the ALS mutant, A315T(p). In Fig. 4 we also can tell that there is a obvious lag time of the aggregation process for A315E, but there is hardly a lag time for Wt, A315T or A315T(p). The reason for A315E having a large nucleation delay than A315T(p) could be associated with the length difference of core β-domains at the C-terminals for A315E and A315T(p). The length difference in C-terminal core β-domains could influence the relaxation time and the electrostatic interaction between N and C-terminal β-domains could help to enhance the inter-sheet interaction leading to higher equilibrium intensity (as shown in Fig. 4). The decreased fluorescence intensity after the aggregation reaches the equilibrium can also be observed, which could be attributed to the possible decrease of the effective quantity of peptide aggregates. When the large size aggregates precipitate at the bottom of the quartz cells, the reduction of the effective amount of fluorescence molecules within the optical path will lead to the decrease of the fluorescence intensity.
In patients affected by TDP-43 proteinopathy, TDP-43 has been identified in skein-like cytoplasmic inclusions. The STM results indicate that the C-terminal β-domain of Wt and A315T is ended at the preferential sites Ala36 residue, whereas the core β-domain of A315T(p) is located between C-terminus and Phe316 residue, and that of A315E is between C-terminus and Gly310 residue. The observed longer β-domains of A315E and A315T(p) in comparison with those of the Wt and A315T may be attributed to the stronger aggregation propensity of A315E, A315T(p) than those of Wt and A315T, as supported by the AFM and ThT fluorescence results. This observation is also consistent with the reported molecular dynamics simulation results of Wt, A315T(p) and A315E TDP-43 peptides (Guo et al., 2011), in which the C-terminal part of wild-type and A315T TDP peptides are suggested to adopt an extended β-sheet conformation and more liable to formed β-turns than the C-terminal part of A315T(p) and A315E TDP-43 peptides. The proline may act as a β-sheet disruptor to give rise to the ending of beta-like structure at Pro320 primarily for Wt TDP-43 peptides (Hutchinson and Thornton, 1994; Rose et al., 1985). In A315T(p) TDP-43 peptides, the phosphate group may alter the interaction between peptide strands within the amyloid fibrils and result in more extended β-domain in A315T(p) peptide. The synthetic peptide A315E as a model further confirm the impact of electrostatic interaction on the amyloid aggregation due to the introduced negative charge of the phosphorylated A315T. For testing the electrostatic attraction in the aggregation process of TDP-43, we use three comparison peptides: A293G/A315E (C1), A293G/A315G (C2) and A293G/A315T(p) (C3) TDP-43 peptides (the sequences are shown in Scheme 1). The result (Fig. 4) indicates that the aggregation propensity of A315T(p) and A315E are much stronger than C1 (purple open diamonds), C2 (yellow open squares), and C3 (light green open circles), which confirmed the significance of the electrostatic interaction between the positive charge on Arg293 and negative charges at 315 sites on A315T(p) or A315E. Carefully comparing the aggregation propensity of C1, C2 and C3, we can clearly observe the strongest aggregation propensity of C1 than the other two peptides. This could be attributed to the possible enhanced inter-peptide interactions associated with the capacity of hydrogen bond formation for Glu315. The slightly higher aggregation propensity for C1 compared to C2 and C3 is also consistent with the longer core C-terminal β-domain in A315E.
4. Summary
We have observed longer β-domain of A315E and A315T(p) in comparison with that of the Wt and A315T through surface mediated peptide assemblies by using STM. Such difference may be attributed to the stronger aggregation capacities of A315E and A315T(p) than those of Wt and A315T, as supported by the ThT fluorescence results. Substantially stronger aggregation propensity of the A315E and A315T(p) could be attributed to the possible electrostatic interactions with the positive charged Arg293 residue. The model synthetic peptide, A315E, further confirms the impact of electrostatic interaction on the amyloid aggregation due to the introduced negative charge to ALS mutation, the phosphorylated A315T. The impact of β-domain length on the aggregation propensity and finally the cytotoxicity of the amyloid peptides indicates the feasibility of analyzing biological effects of amyloid peptides both in vitro and in vivo by studying the structural characteristics of beta-motifs at molecular level. These results could help gain insight of the correlation between the length of β-domain and the aggregation propensity of amyloid peptides and also the associated mutation effects.
Supplementary Material
Acknowledgments
This work was supported by the National Basic Research Program of China (2011CB932800, 2009CB930100, 2009CB825402) and Chinese Academy of Sciences (KJCX2-YW-M15, CASNN-GWPPS-2008). JYW is supported by National Institutes of Health (RO1AG033004; R56NS074763) and ALS Therapy Alliance. Financial supports from National Natural Science Foundation of China (20911130229, 20973043) are also gratefully acknowledged.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jsb.2012.10.011.
References
- Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 Is toxic to neurons & enhanced by a mutation associated with familial ALS. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM-Y, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17:659–667. doi: 10.1016/j.molmed.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WR, Chen YB, Zhou XH, Kar A, Ray P, Chen XP, et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol. 2011;18:822–830. doi: 10.1038/nsmb.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson EG, Thornton JM. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- gaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, et al. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009;284:8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura A, Yamazaki T, Lemere CA, Takatama M, Okamoto K. Regional distribution of TDP-43 inclusions in Alzheimer’s disease (AD) brains: their relation to AD common pathology. Neuropathology. 2009;29:566–573. doi: 10.1111/j.1440-1789.2009.01017.x. [DOI] [PubMed] [Google Scholar]
- Kuo PH, Doudeva LG, Wang YT, Shen CKJ, Yuan HS. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37:1799–1808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang L, Mao XB, Niu L, Yang YL, Wang C. Chaperon-mediated single molecular approach toward modulating Aβ peptide aggregation. Nano Lett. 2009;9:4066–4072. doi: 10.1021/nl902256b. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang L, Niu L, Xu M, Mao XB, Yang YL, et al. Observation of reduced cytotoxicity of aggregated amyloidogenic peptides with chaperone-like molecules. ACS Nano. 2011;5:6001–6007. doi: 10.1021/nn201773x. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Liu L, Mao XB, Niu L, Deng K, Wu WH, et al. Amyloid-β (1–42) folding multiplicity and single molecule binding behavior studied by STM. J Mol Biol. 2009;388:894–901. doi: 10.1016/j.jmb.2009.03.054. [DOI] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao XB, Ma XJ, Liu L, Niu L, Yang YL, Wang C. Structural characteristics of the beta-sheet-like human and rat islet amyloid polypeptides as determined by scanning tunneling microscopy. J Struct Biol. 2009;167:209–215. doi: 10.1016/j.jsb.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Mao XB, Wang CX, Wu XK, Ma XJ, Liu L, Zhang L, et al. Beta structure motifs of islet amyloid polypeptides identified through surface-mediated assemblies. Proc Natl Acad Sci USA. 2011;108:19605–19610. doi: 10.1073/pnas.1102971108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao XB, Guo YY, Wang CX, Zhang M, Ma XJ, Liu L, et al. Binding modes of thioflavin t molecules to prion peptide assemblies identified by using scanning tunneling microscopy. ACS Chem Neurosci. 2011;2:281–287. doi: 10.1021/cn200006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Piao YS, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, et al. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003;13:10–22. doi: 10.1111/j.1750-3639.2003.tb00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CKJ. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang CX, Mao XB, Yang AH, Niu L, Wang SN, Li DH, et al. Determination of relative binding affinities of labeling molecules with amino acids by using scanning tunneling microscopy. Chem Commun. 2011;47:10638–10640. doi: 10.1039/c1cc12380g. [DOI] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2009;106:18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.