

Abstract
Second malignant neoplasms (SMNs) are therapy-induced malignancies and a growing problem in cancer survivors, particularly survivors of childhood cancers. The lack of experimental models of SMNs has limited understanding of their pathogenesis. It is currently not possible to predict or prevent this devastating late complication. Individuals with Neurofibromatosis I (NF1) are at increased risk of developing therapy-induced cancers for unclear reasons. To model SMNs, we replicated clinical radiotherapy and delivered fractionated abdominal irradiation to Nf1+/− and wildtype mice. Similar to irradiated cancer survivors, irradiated wildtype and Nf1+/− mice developed diverse in-field malignancies. In Nf1+/− mice, fractionated irradiation promoted both classical NF1-associated malignancies and malignancies unassociated with the NF1 syndrome but typical of SMNs. Nf1 heterozygosity potentiated the mutagenic effects of irradiation, as evidenced by the significantly reduced survival after irradiation and tumor development that was often characterized by synchronous primary tumors. Interestingly, diverse radiation-induced tumors arising in wildtype and Nf1+/− mice shared a genetic signature characterized by monoallelic loss of Nf1 and the adjacent Trp53 allele. These findings implicate Nf1 loss as mediating tumorigenesis in a broad range of cell types and organs extending beyond the classical NF1 tumor histologies. Examining clinical SMN samples, we found LOH of NF1 in SMNs from non-NF1 patients. Nf1 heterozygosity confers broad susceptibility to genotoxin-induced tumorigenesis and this paradigm serves as an experimental platform for future studies of SMNs.
Keywords: Neurofibromatosis I, radiation, abdominal, sarcomas, carcinomas, genetic
Introduction
Secondary malignant neoplasms (SMNs) are late complications arising after exposure to genotoxins. These cancers can arise from a variety of tissues within an irradiated anatomic compartment (1–3), reflecting the wide range of cell types sharing susceptibility to mutagenesis. It is currently not possible to predict which patients will develop an SMN nor are there known shared mechanisms linking the pathogenesis of diverse SMN histologies. The incidence of SMNs is growing primarily because the number of pediatric and other at-risk cancer survivors has increased over the last few decades (4–6).
The mechanisms underlying SMN development are very poorly understood, with radiation exposure and young age at the time of treatment being the strongest risk factors (2, 3, 7, 8). Defining the mechanisms responsible for SMN pathogenesis is critical to developing strategies to accurately predict and mitigate this risk in current and future cancer survivors. Mechanistic analyses of both patient and treatment factors are difficult to perform through retrospective clinical analyses, and thus experimental models of SMNs are needed.
Genetic background contributes to tumor susceptibility in humans and mice, for example, the high frequency of SMNs arising in the setting of Li-Fraumeni syndrome and other genetic disorders of DNA repair (9, 10). Furthermore, experimental models of radiation-induced tumorigenesis do not replicate radiotherapy delivery and often employ genetically unstable mouse strains. The resultant tumors do not reconstitute the full spectrum of histologies comprising SMNs, and the latency to tumor development is often extremely abbreviated (within a few months) in contrast to the latency of SMNs in humans, which is over several years and sometimes decades. The inability to broadly model SMN risk in relation to relevant radiotherapy variables of dose, fractionation and targeting has limited our understanding of how clinical treatment, patient variables and environmental exposures influence SMN risk. As a result, there are no validated approaches to predicting or preventing SMNs in at-risk individuals, such as survivors of childhood cancers.
Heterozygosity for the NF1 gene causes the Neurofibromatosis I Syndrome (NF1), and individuals with NF1 are at increased risk of developing SMNs after radiotherapy (11). The NF1 gene, and its conserved murine homologue Nf1, encode the protein neurofibromin, which is ubiquitously expressed in mammalian cells and necessary for development (12). Neurofibromin possesses a GTPase activating protein (GAP) domain, a region that negatively regulates Ras signaling (13–16).
We hypothesized that the clinical susceptibility of NF1 patients to SMN development could be modeled in Nf1 mutant mice to study SMN pathogenesis. In earlier work, we showed that focal, fractionated cranial irradiation of Nf1+/− mice produced both hematologic and in-field solid malignancies (17). To develop a more robust model that 1) tested whether focal fractionated irradiation potentiates tumorigenesis among diverse tissues and 2) efficiently generated large numbers of tumors in both genetically resistant and susceptible backgrounds for analysis we delivered abdominal irradiation to wildtype and Nf1 mutant mice. We replicated a clinical paradigm using customized techniques to deliver focal, fractionated radiation to the abdomen (AI) (0 Gy, 3 Gy, 15 Gy or 30 Gy) (Figure 1 and Supplementary Figure 1). This procedure concentrates radiation exposure to the superficial tissues and generates a dose gradient to deeper internal organs, permitting the safe delivery of radiation doses that would be lethal as whole body exposures. This application of fractionation mimics clinical radiotherapy practice and was an approach we used in our cranial irradiation model (17).
Figure 1. Overall survival in Nf1 mutant mice decreases after abdominal irradiation in a dose-dependent manner.
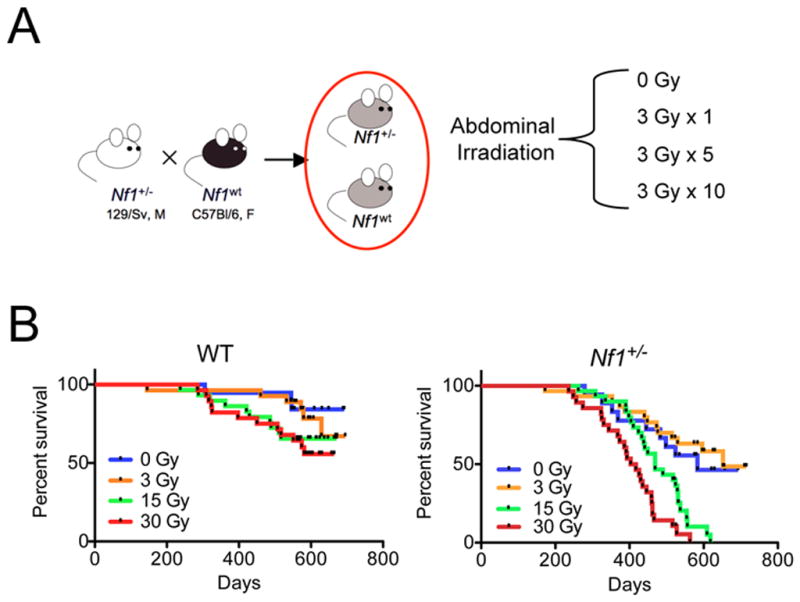
A. Experimental schema B. Kaplan-Meier survival analysis of WT and Nf1 mutant mice shows no statistically significant decrease in overall survival after irradiation in WT mice. In contrast, overall survival in Nf1 mutant mice is significantly reduced after 15 and 30 Gy AI (p<0.0001, log-rank).
Both irradiated wildtype and Nf1+/− mice developed diverse in-field malignancies in a dose-dependent manner, with Nf1+/− mice developing greater numbers of malignancies than matched wildtype mice at each radiation dose level. Diverse tumor histologies shared a signature loss of heterozygosity (LOH) of wildtype Nf1 and Trp53 alleles, rendering tumors from Nf1+/− mice null for Nf1 and consistent with a common pathogenetic mechanism of Nf1- dependent tumorigenesis active in multiple cell types. Interestingly, this signature loss was also present in a subset of radiation-induced tumors from wildtype mice, which lost either parental allele at similar frequencies. This suggests that Nf1 haploinsufficiency can drive radiation-induced tumor formation in the wildtype background as well, and may be a genetic mechanism promoting radiation-induced tumors in between different genetic backgrounds. We extended our genetic analysis to human radiation-induced breast cancers and identified loss of heterozygosity of NF1 in a subset of these SMNs from unrelated individuals without NF1. These findings suggest that Nf1 loss in our mouse model mirrors NF1 loss in human SMNs and suggest that targeting the biochemical consequences of NF1 loss may be a useful strategy against SMN development.
Materials and Methods
Mouse Strains, Breeding and Treatment
Nf1+/− and wildtype mice were generated as previously described (18). In brief, Nf1+/− mice maintained in the 129/Sv background (19) were crossed with wild-type C57Bl/6 mice. Mice were placed in a cesium-137 source (J.L. Shepherd & Associates, San Fernando, CA) animal irradiator and shielded with an iron collimator that focused the beam width. Five to eight week old mice were given abdominal irradiation at a fractionation of either one fraction of 3 Gy, five fractions of 3 Gy or ten fractions of 3 Gy, delivered at a rate of five fractions per week, one fraction per day. The UCSF IACUC approved all animal procedures.
Pathologic Analysis
Animals were followed for a minimum of 18 months post-irradiation. Animals with signs of systemic illness were euthanized and visible masses/growths, peripheral blood, and bone marrow were collected immediately. The mice were then perfused with 4% paraformaldehyde and the following organs were collected: brain, skin in irradiated region, skin outside irradiated region, skull, heart, lungs, spleen, liver, kidneys, segment of small intestine. Pathologic review was performed on hematoxylin and eosin-stained sections by A.H. and S.K. Photographs of histology were taken with an Olympus BX41 microscope, using Olympus UplanFl 10X/0.3 and 20X/0.5 objectives. An attached Olympus DP72 camera and Adobe Photoshop CS2 were used to capture the images. Complete blood counts were performed on blood samples obtained at the time of sacrifice by cardiac puncture and analyzed immediately on a Hemavet provided by the UCSF Mouse Pathology Core.
Mouse tumor Genotyping and Mutation Analysis
Trp53 loss of heterozygosity was analyzed at the D11Mit29 and D11Mit31 loci by amplifying tumor DNA with the following primers: forward 5′ TTGAGGCATGAGGGGATTAG 3′, reverse 5′ TTTCCGTCATTGCTAAAGGG 3′, 5′ TTTCCCAGTCACGACGTTGGCCTGAATTCACATGGTGG 3′, reverse 5′ AGAATAAGTAAACCCAGCTGCG 3′. D11Mit219 was amplified with the following primers: forward 5′ TTTCCCAGTCACGACGTTGTTGTATGTATAGATGCATTTGAATGG -3′, reverse 5′ GGTTTGTATAAATTCTCACCTGTGC 3′. The UCSF Genome Core using Peak Scanner software from Applied Biosystems performed PCR fragment analyses. The SNP rs13481119 was sequenced using the following primers: forward 5′ GCCCGCTACATGCTGATGCTG 3′ reverse 5′ GCTTGTAGGCCTGGTGAGTC 3′. SNP products were sequenced using a commercial sequencing service.
Clinical FFPE Tissue DNA Isolation and Amplification
Formalin-fixed, paraffin-embedded (FFPE) sections of 8 radiation induced breast cancers were obtained from City of Hope Medical Center (Duarte, California, USA). H & E slides of FFPE sections were reviewed by a pathologist (AH), who delineated tumor and normal areas for dissection. Tumor and normal tissues were then dissected off of unstained slides and genomic DNA was isolated and subjected to whole genome amplification according to manufacturers instructions using the REPLI-g FFPE kit (Qiagen). DNA was purified using sodium acetate, and quantified using Picogreen (Qiagen).
Taqman SNP Genotyping
Analysis of clinical samples was performed with approval from the UCSF Committee on Human Research. Clinical DNA samples were genotyped at four selected SNPs in the human NF1 gene (NCBI ref SNPs rs2952994, rs2953014, rs9902893, and rs2107359 using C1547650_10, C2557613_10, C2533273_10 and C16032374_10, all from Applied Biosytems, respectively). Primers flanking selected SNPs were designed using Integrated DNA Technologies primer design program. Assay C2557613_10 was used to genotype using the Taq amplification method in a 7900 HT Fast Real-Time PCR system (Applied Biosystems, USA). Five μl PCR reactions were performed using 2.42 μl DNA in H20 (4 ng total) or H20 for negative controls, 0.08 μl probe (20 μmol) and 2.5 μl Master Mix (20 μmol; Applied Biosystems). Reactions underwent heat activation at 95 °C for 10 minutes, then cycled 40 times with a denaturation step of 92 °C for 15 seconds, and an annealing and elongation step of 60 °C for 90 seconds. All samples were run in replicates of 6. Allelic discrimination plots generated by SDS3.0, an Applied Biosystems program used to make genotyping calls.
Trp53 sequencing
Whole exomic sequencing was performed to assess for the presence of mutations in the Trp53 gene. Two kb fragments of the Trp53 gene spanning exons 2–4, 5–7, and 8–10 were PCR-amplified from all primary tumor and select matching tail DNA samples. Similarly, 1 kb fragments were PCR amplified from exons 1 and 11. PCR amplification was performed with Qiagen Taq DNA polymerase. PCR cleanup was done with ExoSAP-IT (Affymetrix). The PCR fragments were then sequenced (Quintara Biosciences) with sequencing primers (listed in Supplementary Table 2). The Catalogue of Somatic Mutations in Cancer Database (COSMIC, www.sanger.ac.uk/genetics/CGP/cosmic, Wellcome Trust Sanger Institute, United Kingdom) was used to search for references to human tumors also possessing the identified mutations.
Statistical Analysis
Survival curves are calculated from Kaplan-Meier product limit estimators to determine the association of CI on mortality. Log-rank tests are used to test for differences in survival curves between groups. All analyses were performed using Prizm v.4 (GraphPad).
Results
High dose AI reduces survival in Nf1 mutant mice but not wildtype mice
In irradiated cancer survivors, the risk of SMNs is well recognized although the precise SMN histology can be difficult to predict. Furthermore, cancer survivors may develop multiple subsequent malignancies and diverse SMNs over time (1). Multi-organ SMN development is therefore not well-modeled using tissue-restricted conditional mutations. To replicate whole organismal tumor susceptibility we sought to model the germline Nf1 loss responsible for the NF1 syndrome.
To establish a homogeneous genetic background, we intercrossed wildtype (WT) C57BL6 and heterozygous Nf1 mutant (Nf1+/−) Sv129 mice to generate F1 Nf1+/− and control cohorts. Between 5 – 8 weeks of age, mice were assigned to one of four AI treatment regimens: 0 Gy, 3 Gy (single dose), 15 Gy (5 daily doses of 3 Gy), or 30 Gy (10 daily doses of 3 Gy) (Figure 1A, Supplementary Figure 1). To accurately reflect current clinical practice, the mice were irradiated five times per week, one fraction per day. They were then observed until they developed signs of illness necessitating euthanasia or until 18 months post-irradiation.
The survival of WT mice was not significantly reduced after any AI dose, and median survival was not reached by any experimental group (Figure 1B). In contrast, radiation-exposure reduced the overall survival of Nf1+/− mice in a dose-dependent manner (log-rank test p<0.0001, Figure 1B) at higher dose levels (15 and 30 Gy). The median survivals of Nf1+/− mice receiving 0, 3, 15, and 30 Gy were 583, 652, 469 and 410 days, respectively.
AI drives multi-organ tumorigenesis in WT and Nf1+/− mice
Necropsy revealed multi-organ abnormalities after irradiation. Hematologic malignancies such as myeloproliferative neoplasms and leukemias can arise as secondary malignancies after radiation exposure (20). To screen for this we assessed spleen and liver weight and performed a complete blood count (CBC) at the time of euthanasia and found significantly increased mean terminal splenic weight in unirradiated Nf1+/− mice as compared to WT mice (Supplementary Figure 2A). However, splenic weight was not significantly affected by radiation dose in either the WT or Nf1+/− cohorts. Similarly, terminal hemoglobin levels did not significantly differ between any genotype/radiation dose combination, except for Nf1+/−/3 Gy and Nf1+/−/15 Gy (Supplementary Figure 2B). Examination of H&E-stained spleen, liver and bone marrow sections from animals demonstrating splenomegaly and/or peripheral anemia or leukocytosis revealed changes consistent with varied hematologic diseases such as histiocytic sarcoma, myeloproliferative neoplasms, and lymphoma.
Radiation-induced SMNs in humans consist of varied malignant histologies, predominantly solid tumors, reflecting a breadth of cell types and tissues that are susceptible to genotoxin-induced mutagenesis (6). Capturing this diversity is a critical requirement for pre-clinical platforms in which to test SMN-modifying approaches for multiple at-risk tissues. Comprehensive necropsy of experimental animals revealed solid tumor development as the cause of death in most animals (Figure 2, Supplementary Table 1). Similar to irradiated patients, irradiated mice developed diverse solid tumors originating from organs in the irradiated field (in-field tumors). Tumors were more common in Nf1+/− mice than WT mice, and for Nf1+/− mice tumor incidence increased with increasing radiation dose (p < 0.0001 by χ2 analysis, Figure 2), while for WT mice, a relationship between tumor incidence and radiation dose failed to reach significance (p= 0.0511 by χ2 analysis). Solid tumors in both WT and Nf1+/− mice included epithelial tumors such as mammary carcinoma, adenocarcinoma, as well as soft tissue sarcomas (Supplementary Table 1). Importantly, patients with NF1 are not predisposed to these cancers, and nor do these tumor histologies spontaneously develop in Nf1+/− mice. Thus, many of these SMN-replicating histologies are not simply amplifications of the known NF1 phenotype (tumors arising from neural crest-derived tissues) but are malignancies that suggest a broader role for neurofibromin as a tumor suppressor. Among the solid tumors arising in our model, sarcomas are well recognized SMNs after high-dose radiation therapy (21–23), and were significantly more common in irradiated Nf1+/− mice than irradiated WT mice (p = 0.0175, log-rank). Mirroring overall survival, solid tumor free survival and hematologic disease free survival were both reduced in a dose-dependent manner in Nf1+/− mice but not WT mice (Figure 3, log rank test, p<0.0001 for solid tumor free survival in Nf1+/− mice and p=0.0001 for hematologic disease free survival in Nf1+/− mice).
Figure 2. Tumor incidence in experimental groups.
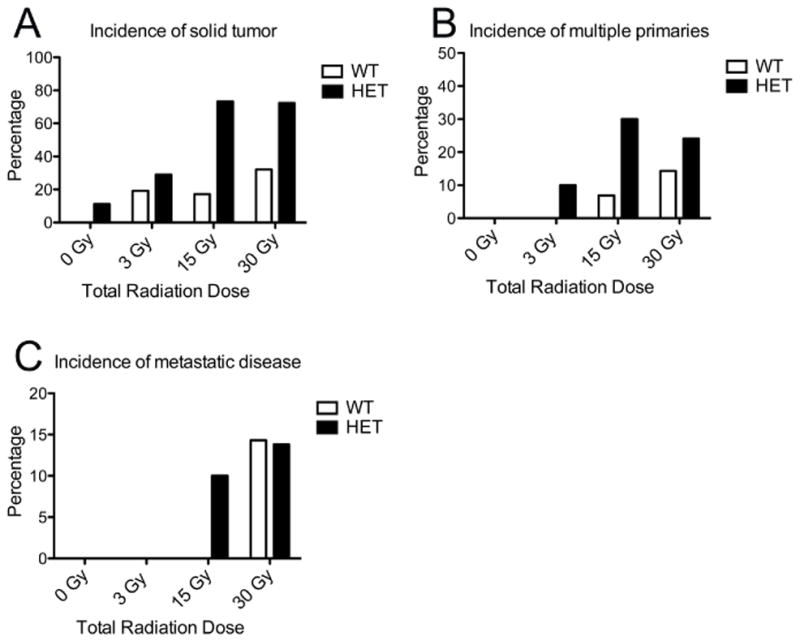
WT – wildtype, HET – Nf1 mutant. Percentages of mice developing tumors (A), multiple (synchronous) primary tumors (B), and metastases (C) are shown for each experimental group. Tumors were more common in Nf1+/− mice than WT mice, and for Nf1+/− mice tumor incidence increased with increasing radiation dose (p < 0.0001 by χ2analysis), while for WT mice, a relationship between tumor incidence and radiation dose failed to reach significance (p= 0.0511 by χ2analysis). Synchronous tumorigenesis occurred in both WT and Nf1+/− mice, with synchronous malignancies in the Nf1+/− background increasing with increasing radiation doses (p= 0.0264 by χ2analysis).
Figure 3. Solid tumor and hematologic disease free survival after AI.

Kaplan-Meier survival analysis of WT mice shows no significant dose response for death due to either solid tumor or hematologic disease (log rank test, p=0.13 for solid tumor free survival, p=0.68 for hematologic disease free survival). In contrast, Nf1 mutant mice demonstrate a dose response for both solid tumor and hematologic disease-related death (log rank test, p<0.0001 for solid tumor free survival and p=0.0001 for hematologic disease free survival).
Multi-organ examination revealed that radiation-induced tumors displayed important features of aggressive malignancies, including local invasiveness and distant metastases (Figure 2C). Appropriate site-specific SMN presentations were observed in irradiated Nf1+/− mice, for example a locally invasive chest wall sarcoma causing a pathologic fracture of an adjacent rib (Figure 4). Metastatic disease in the lungs and liver, common clinical manifestations of advanced and aggressive cancers, were the sites of metastatic disease in this mouse model (Figure 2C). The most common metastatic histology was pheochromocytoma, followed by sarcoma and carcinoma.
Figure 4. Radiation-induced tumors in mice replicate clinical SMN presentations.
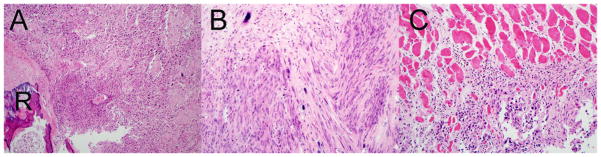
A. 100X magnification B. 200X magnification images of hematoxylin and eosin stained radiation-induced chest wall sarcoma arising in an Nf1 mutant mouse. The soft tissue mass surrounds and invades an adjacent rib (R) with tumor cells present in the marrow. C. Local invasiveness of the sarcoma evidenced by invasion of the adjacent skeletal muscle by tumor cells.
Individual patients can develop multiple independent SMNs in different organs within an irradiated region (1), and this aspect of SMN risk has not been modeled to date. The irradiation of the large anatomic compartment enclosed by the peritoneum concurrently mutagenizes multiple diverse tissues (for example hematopoietic, gastrointestinal, endocrine, and connective). Further, irradiating Nf1+/− mice capitalizes on the large volume mutagenesis of AI and allows tests of multi-organ Nf1+/− dependent susceptibility to radiation-induced tumors. Synchronous tumorigenesis occurred in both WT and Nf1+/− mice, with synchronous malignancies in the Nf1+/− background increasing with increasing radiation doses (p= 0.0264 by χ2 analysis). Consistent with the enhanced and broad susceptibility conferred by Nf1 heterozygosity, synchronous malignancies were more common in the irradiated Nf1+/− background (Figure 2B). Synchronously occurring malignancies in both genotypic backgrounds included pheochromocytomas, sarcomas, and carcinomas.
Concurrent loss of Nf1 and Trp53 is a signature genetic lesion in radiation-induced mouse tumors
The genetic mechanisms driving SMN development are poorly understood. While genetic background clearly modulates tumorigenesis risk, a common signature of genetic alterations has not been defined for SMNs. The human NF1 and TP53 tumor suppressor genes are located 21.8 Mb apart on chromosome 17. Similarly, the homologous murine genes Nf1 and Trp53 are both on chromosome 11, and the intervening sequences are conserved in both species. Previous mouse models have shown that engineered Nf1 and Trp53 loss results in tumor development (24, 25), but spontaneous cooperativity between these two tumor suppressor genes is less well established. We performed microsatellite analysis of four markers spanning these two genes (D11Mit31, D11Mit4, D11Mit219, D11Mit38) and also sequenced the single nucleotide polymorphism (SNP) rs13481119, located in exon 41 of Nf1, to determine whether loss of heterozygosity (LOH) at these loci occurred and which parental allele was lost (Figure 5). Microsatellite and SNP sequencing analysis revealed that loss of heterozygosity was a common event in tumors isolated from Nf1 mutant mice (Figure 5), and the frequency of LOH at rs13481119 was similar across the three major classes of solid malignancies (pheochromocytoma, carcinoma and sarcoma).
Figure 5.
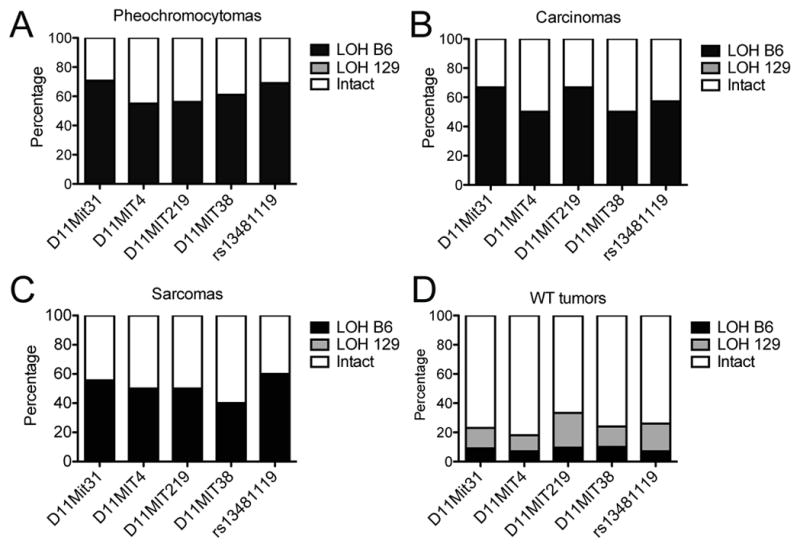
Frequency of LOH in SNP and Microsatellite sites flanking Trp53 and Nf1 genes (on Chromosome 11) from solid tumors arising in WT and Nf1+/− mice. The percentage of tumors demonstrating intact heterozygosity (Intact), LOH of the B6-derived allele (LOH B6), or LOH of the Sv129-derived allele (LOH 129), are shown for A. pheochromocytomas, B. carcinomas, C. sarcomas, and D. solid tumors arising in wildtype mice (mixed histologies).
LOH of Nf1 was accompanied by LOH of at least one of the microsatellites in 87% percent of all tumors, and in fact LOH commonly involved all four tested microsatellites and intragenic SNP from the same parental strand, consistent with a loss of a chromosomal segment spanning Nf1 and Trp53. Comparison to parental control DNA revealed that LOH in tumors arising in Nf1+/− mice commonly involved the C57Bl/6-derived allele at all positions (Figure 5A–C). This led to loss of the wildtype Nf1 allele and adjacent Trp53 allele (Figure 5D). LOH of the wildtype Nf1 allele and the adjacent Trp53 allele occurred at similar frequencies for the major tumor classes (carcinomas, sarcomas and pheochromocytomas), indicating that Nf1 nullizygosity is a shared genetic mechanism of tumorigenesis in different tissue lineages. Because our mouse model employs a genetic background in which mice possess two wildtype copies of Trp53, loss of a chromosomal segment spanning Nf1 and Trp53 would render tumors heterozygous for Trp53. To determine whether tumors in this model are driven by nullizygosity for Trp53 in addition to nullizygosity for Nf1, we assessed the remaining Trp53 allele by exon sequencing. Of 123 primary and metastatic tumors we isolated from this mouse model Trp53 exon sequencing was obtained on 98 (79%). Interestingly, most tumors retained a copy of wildtype Trp53 allele, and we identified only five mutations in the remaining Trp53 allele (Figure 6). These included point mutations and small deletions localizing to the DNA-binding domain, a known hotspot for Trp53 mutations, and occurring in conjunction with LOH of NF1 (Figure 6A). Examination of these mutations in the Catalogue of Somatic Mutations in Cancer (COSMIC, Wellcome Trust Sanger Institute, United Kingdom) revealed that each of these mutations occur in diverse human cancers (Figure 6A).
Figure 6. Somatic Trp53 mutations in radiation-induced tumors.

A. Table summarizing mutational spectrum in Trp53. Mouse genotype refers to the genotype of the animal from which the tumor was isolated. WT – wildtype, HET – Nf1 mutant. Using the COSMIC database we found that these mutations also are found in human tumors (COSMIC ID number/mutation). LOH of the B6-derived Trp53 allele (B6), or LOH of the Sv129-derived Trp53 allele (129) is noted for each mutation. B. Sequencing chromatogram from a tail control and radiation-induced tumor showing somatic deletion(Δ) in exon 7. C. Schematic of p53 exons showing localization of identified mutations.
In contrast to tumors arising in Nf1 mutant mice, most tumors from WT mice demonstrated intact heterozygosity at the tested markers (Figure 5D). However, surprisingly in five WT tumors we identified segmental loss of Nf1 and Trp53, similar to tumors arising in Nf1+/− mice. This suggests that concurrent loss of these genes is also an important mechanism for radiation-induced tumorigenesis in a WT background. Among these WT tumors, segmental LOH involved either parental allele at similar frequency (Figure 5D), consistent with allelic loss that is unbiased by an inactivated allele on either parental strand. This pattern stands in contrast to segmental LOH in tumors derived from the Nf1+/− mice, in which the mutant Nf1 allele is maintained on the Sv129-derived allele and preferential loss of the C57Bl/6-derived allele occurred.
From 11 mice identified to have developed metastatic disease, we isolated sufficient quantities of metastatic tumor DNA from 7 (6 primary tumors, each with associated metastasis, and one pair of metastases developing from an unknown primary), permitting comparisons of genetic loss between primary and metastatic disease. Assessing for LOH at Nf1 and Trp53 in metastatic disease revealed partial concordance with the primary, as three of seven metastases shared Nf1 LOH status with their associated primary tumor, while three metastases did not. In one animal bearing both lung and liver metastases, both metastases demonstrated loss of the C57Bl/6-derived wildtype Nf1 allele, suggesting that Nf1 nullizygosity preceded metastases development. Only one tumor demonstrated LOH at two tested loci but involving opposite strands; an osteosarcoma metastasis arising in an irradiated WT animal demonstrated loss of the C57Bl/6-derived allele at the D11Mit31 locus and loss of the Sv129-derived allele at rs13481119, located in exon 41 of Nf1. As a consequence, this metastasis lost Trp53 and Nf1 on opposite parental strands, while interestingly its matched primary osteosarcoma maintained intact heterozygosity at these loci.
Concurrent distinct primary malignancies (synchronous primaries) are occasionally diagnosed in patients, with synchronous primaries being more common in individuals with tumor predisposition syndromes. Synchronous primary tumors were more common in Nf1+/− mice and, for both genotypes, high radiation doses (Figure 2). Synchronously developing tumors are unique reagents for analysis as these tumors arise in the same individual and therefore share the same organismal history genetically and environmentally. We analyzed the synchronously arising tumors arising in our model to determine whether tumors from a given individual, initiated by the same mutagenic exposure, share genetic alterations at Nf1 and Trp53. From 25 total mice bearing synchronous primary malignancies we analyzed the previously named microsatellites and SNP in 47 primaries derived from 20 mice. Malignancies in 13 of these 20 mice demonstrated discordance between tumors with regard to LOH at the Nf1 locus at rs13481119. Interestingly, segmental loss of Nf1 and Trp53 was not always shared even among synchronous tumors that were of the same histology, for example, among 7 Nf1+/− mice that developed bilateral pheochromocytomas, three pairs demonstrated genetic discordance characterized by LOH of C57Bl/6-derived Nf1 and Trp53 associated markers in one tumor and intact heterozygosity in the other.
Somatic loss of NF1 occurs in human radiation-induced breast cancers
The genetic basis of human SMNs is poorly understood, most likely due to the scarcity of high quality SMN tissue samples. Our mouse model implicates Nf1 loss as an important driver of SMN development, but the incidence of and significance of NF1 loss in human SMNs are not known. We assessed NF1 status in human radiation-induced breast cancers, which are well recognized SMNs developing in women who receive chest irradiation as children (7, 26). Radiation-induced breast cancers and matched normal tissue from patients (8 total patients, none known to have the NF1 syndrome) were analyzed using Taqman-based SNP genotyping at multiple loci across the NF1 gene. Allelic discrimination plots demonstrate two radiation-induced breast cancers (Figure 7, two pairs of matched control normal tissue and tumor, designated E and H shown, Supplementary Figure 3) whose normal tissues cluster with heterozygous controls (green, A/T genotype) but whose tumors demonstrate LOH at SNP rs9902893 in NF1 (clustering with either T/T or A/A genotype controls). To our knowledge this is the first evidence of somatic NF1 gene alterations in radiation-induced SMNs. Further analyses of larger numbers of SMN samples are necessary to more accurately estimate the incidence of NF1 gene alterations. However, these data suggest concordance between clinical SMNs and our mouse model and justify further comparative studies.
Figure 7. Loss of Heterozygosity of NF1 in Radiation-Induced Breast Cancers.
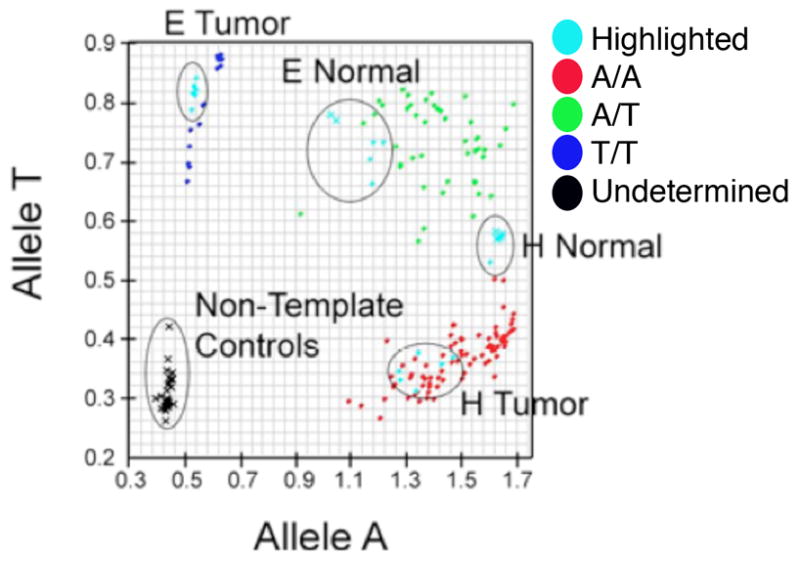
Taqman SNP Genotyping of rs9902893. Allelic Discrimination plot was generated by Applied Biosystems software SDS 3.0, which assigns genotypes A/A (blue), A/G (green), or G/G (red). VIC and FAM fluorescence in each well was recorded and plotted (VIC on y-axis, FAM on x-axis), corresponding to Allele A and Allele G, respectively. Samples were run in replicates of 6, and each replicate is plotted. Black indicates that genotype cannot be determined. Controls were circled, and samples showing LOH are highlighted in blue, circled, and labeled.
Discussion
The pathogenesis of SMNs is not well understood, but clinical analyses indicate that the process is influenced by both cancer therapies and features of cancer patients themselves. In addition to radiotherapy, chemotherapeutic agents such as alkylating agents and topoisomerase II inhibitors such as epipodophyllotoxin have been associated with SMN development (23, 27). In particular, leukemias as therapy-induced malignancies have been associated with chemotherapies (28, 29). Genetic tumor predisposition syndromes are associated with SMN risk (7), and individuals with strong family histories of cancer also have enhanced risk. SMNs are substantially more likely to arise in patients treated as young children (6). Post-treatment endocrinopathies and obesity are common in survivors of pediatric cancers, and may also influence risk of SMNs known to be hormone-sensitive such as breast cancer (30).
The AI model replicates the central elements of clinical radiotherapy, namely anatomic targeting and dose fractionation, in resistant (WT) and susceptible (Nf1+/−) genetic backgrounds. The resulting radiation-induced diseases reproduce the wide range of SMN histologies developing in irradiated patients. In contrast to an earlier mouse model we developed that employed fractionated focal cranial irradiation (17), the AI model revealed a radiation dose-dependent reduction in survival in the Nf1-mutant background only. Overall, large numbers of radiation-induced tumors were generated in the AI model, likely explained by the relatively large volume of tissue irradiated with AI. Further, the increased tumor incidence associated with higher doses was driven by the development of heterogeneous tumor types, revealing the general susceptibility of the Nf1 mutant background to mutagen-induced tumorigenesis.
Segmental chromosomal loss identified in radiation-induced tumors from our model appears consistent with a mutagenesis mechanism driven by double strand breaks, as are classically induced by ionizing radiation. Germline point mutations and microdeletions are well-described genetic mechanisms for NF1 loss in individuals with the NF1 Syndrome (31, 32) rather than large chromosomal losses and thus the mechanism of somatic loss of Nf1 in tumors from wildtype and Nf1 mutant mice may be unique to radiation-induced mutagenesis.
Wildtype Nf1 loss occurred jointly with Trp53 in tumors from both wildtype and Nf1 mutant backgrounds, suggesting that concurrent loss of these genes can serve as a central event in radiation-induced tumorigenesis regardless of genetic background. In wildtype tumors, LOH involving Nf1 may prime affected cells for tumorigenesis similar to Nf1+/− mice. LOH of Trp53 was also accompanied by somatic mutations of the remaining allele in a minority of tumors analyzed by Trp53 exon sequencing. The several mutations identified included substitutions and deletions in conserved regions in several tumors that serve as examples of bi-allelic inactivation of Trp53. p53 is mutated in 50% of human cancers, with mutations in regulators in p53 occurring in many tumors lacking Trp53 mutations (33). It is possible that in this model dysregulation of p53 function may occur through alternative mutations. Comprehensive genomic analyses of our mouse tumors lacking bi-allelic Trp53 loss might reveal these alternative mechanisms of Trp53 inactivation, and are planned for future studies.
An important strength of the AI model is the robust cancer phenotype that includes metastatic disease as well as multiple synchronous tumors. These disease features lend the AI model to comparative studies between 1) primary and metastatic disease and 2) primary tumors arising from different organs. In the case of synchronously arising primary tumors, the genetic background and tumor-initiating mutagen are identical, and thus comparisons may identify shared as well as organ-specific mechanisms of pathogenesis. Comparisons of matched primary and metastatic disease may identify common and required mechanisms of tumor growth, which similarly holds translational potential for advanced cancers.
Comparative oncogenomics utilizes experimental mouse and human cancer genetics to gain insights into conserved and robust mechanisms of disease. The genetic and histopathologic analysis of human SMNs has been limited by the relative scarcity of well-archived samples, and thus these tumors are poorly described compared to their non-therapy associated counterparts. Harnessing robust animal models to precious human SMN samples may be a particularly productive approach towards gaining insights into SMN pathophysiology. To our knowledge, comparative genetic analyses between a mouse model and clinical SMNs have not been performed. The presence of LOH of NF1 in radiation-induced human breast cancers suggests a role for NF1 in SMN pathogenesis. Somatic NF1 loss in irradiated tissues may promote SMN development. Further analyses of both mouse and human SMN samples are needed to determine the degree of concordance between these systems and to identify potential pathways that may be modified for chemoprevention.
Taken together, our data in wildtype and Nf1+/− mice suggest that genomic injury after radiation exposure resulting in monoallelic somatic loss of NF1 and Trp53 is a common genetic mechanism that promotes radiation-induced tumors. Loss or inactivation of the remaining NF1 allele may be a critical secondary event that accelerates tumorigenesis in multiple tissue types, as evidenced by the marked susceptibility of Nf1+/− mice, which already bear the first of presumably two required losses. However, since we detected LOH of NF1 in human samples another possibility is that the upregulation of Ras signaling apparent with monoalleleic NF1 loss is a primed signaling backdrop that increases the efficiency with which additional alterations, for example in growth-factor mediated signaling, promote cell proliferation and transformation. Genomic and proteomic analyses of these tumors may be particularly productive for identifying these cooperating pathways.
Genome-wide association studies (GWAS) have identified potential predictors of cancer susceptibility in cancer survivors (34, 35). Experimental validation is needed to justify and optimize testing chemoprevention strategies for patients. As a result, our model fills a vital role in studies of SMN susceptibility and the pre-clinical validation of chemopreventive strategies against SMNs.
Supplementary Material
Acknowledgments
We thank Kevin Shannon, Benjamin Braun and their labs for helpful discussions. The UCSF Mouse Pathology CORE assisted with mouse tissue processing.
Grant Support
JLN was supported by grants from the US National Institutes of Health, National Cancer Institute, the Irene Perstein Fund, the UCSF Breast SPORE Program, and Alex’s Lemonade Stand Foundation. SK is a Leukemia and Lymphoma Society Scholar and also supported by R01CA095274 NCI. SB is supported by R01 CA139633 NCI.
Footnotes
Potential conflicts of interest: None for any of the authors
References
- 1.Armstrong GT, Liu W, Leisenring W, Yasui Y, Hammond S, Bhatia S, et al. Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol. 2011;29(22):3056–64. doi: 10.1200/JCO.2011.34.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong GT, Liu Q, Yasui Y, Neglia JP, Leisenring W, Robison LL, et al. Late mortality among 5-year survivors of childhood cancer: a summary from the Childhood Cancer Survivor Study. J Clin Oncol. 2009;27(14):2328–38. doi: 10.1200/JCO.2008.21.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman DL, Whitton J, Leisenring W, Mertens AC, Hammond S, Stovall M, et al. Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2010;102(14):1083–95. doi: 10.1093/jnci/djq238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawkins MM, Wilson LM, Burton HS, Potok MH, Winter DL, Marsden HB, et al. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996;88(5):270–8. doi: 10.1093/jnci/88.5.270. [DOI] [PubMed] [Google Scholar]
- 5.Garwicz S, Anderson H, Olsen JH, Dollner H, Hertz H, Jonmundsson G, et al. Second malignant neoplasms after cancer in childhood and adolescence: a population-based case-control study in the 5 Nordic countries. The Nordic Society for Pediatric Hematology and Oncology. The Association of the Nordic Cancer Registries. Int J Cancer. 2000;88(4):672–8. doi: 10.1002/1097-0215(20001115)88:4<672::aid-ijc24>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 6.Neglia JP, Meadows AT, Robison LL, Kim TH, Newton WA, Ruymann FB, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325(19):1330–6. doi: 10.1056/NEJM199111073251902. [DOI] [PubMed] [Google Scholar]
- 7.Bhatia S, Sklar C. Second cancers in survivors of childhood cancer. Nat Rev Cancer. 2002;2(2):124–32. doi: 10.1038/nrc722. [DOI] [PubMed] [Google Scholar]
- 8.Constine LS, Tarbell N, Hudson MM, Schwartz C, Fisher SG, Muhs AG, et al. Subsequent malignancies in children treated for Hodgkin’s disease: associations with gender and radiation dose. Int J Radiat Oncol Biol Phys. 2008;72(1):24–33. doi: 10.1016/j.ijrobp.2008.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–52. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 10.Hisada M, Garber JE, Fung CY, Fraumeni JF, Jr, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–11. doi: 10.1093/jnci/90.8.606. [DOI] [PubMed] [Google Scholar]
- 11.Sharif S, Ferner R, Birch JM, Gillespie JE, Gattamaneni HR, Baser ME, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24(16):2570–5. doi: 10.1200/JCO.2005.03.8349. [DOI] [PubMed] [Google Scholar]
- 12.Gutmann DH, Collins FS. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron. 1993;10(3):335–43. doi: 10.1016/0896-6273(93)90324-k. [DOI] [PubMed] [Google Scholar]
- 13.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356(6371):713–5. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 14.Andersen LB, Ballester R, Marchuk DA, Chang E, Gutmann DH, Saulino AM, et al. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol Cell Biol. 1993;13(1):487–95. doi: 10.1128/mcb.13.1.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12(2):144–8. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 16.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104(4):593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura JL, Phong C, Pinarbasi E, Kogan SC, Vandenberg S, Horvai AE, et al. Dose-dependent effects of focal fractionated irradiation on secondary malignant neoplasms in nf1 mutant mice. Cancer Res. 2011;71(1):106–15. doi: 10.1158/0008-5472.CAN-10-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao RC, Pyzel U, Fridlyand J, Kuo YM, Teel L, Haaga J, et al. Therapy-induced malignant neoplasms in Nf1 mutant mice. Cancer Cell. 2005;8(4):337–48. doi: 10.1016/j.ccr.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7(3):353–61. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 20.Iwanaga M, Hsu WL, Soda M, Takasaki Y, Tawara M, Joh T, et al. Risk of myelodysplastic syndromes in people exposed to ionizing radiation: a retrospective cohort study of Nagasaki atomic bomb survivors. J Clin Oncol. 2011;29(4):428–34. doi: 10.1200/JCO.2010.31.3080. [DOI] [PubMed] [Google Scholar]
- 21.Henderson TO, Whitton J, Stovall M, Mertens AC, Mitby P, Friedman D, et al. Secondary sarcomas in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2007;99(4):300–8. doi: 10.1093/jnci/djk052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahan WG, Woodard HQ, et al. Sarcoma arising in irradiated bone; report of 11 cases. Cancer. 1948;1(1):3–29. doi: 10.1002/1097-0142(194805)1:1<3::aid-cncr2820010103>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 23.Tucker MA, D’Angio GJ, Boice JD, Jr, Strong LC, Li FP, Stovall M, et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med. 1987;317(10):588–93. doi: 10.1056/NEJM198709033171002. [DOI] [PubMed] [Google Scholar]
- 24.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26(1):109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 25.Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crump M, Hodgson D. Secondary breast cancer in Hodgkin’s lymphoma survivors. J Clin Oncol. 2009;27(26):4229–31. doi: 10.1200/JCO.2009.22.9120. [DOI] [PubMed] [Google Scholar]
- 27.Pui CH. Epipodophyllotoxin-related acute myeloid leukaemia. Lancet. 1991;338(8780):1468. doi: 10.1016/0140-6736(91)92779-2. [DOI] [PubMed] [Google Scholar]
- 28.Schellong G, Riepenhausen M, Creutzig U, Ritter J, Harbott J, Mann G, et al. Low risk of secondary leukemias after chemotherapy without mechlorethamine in childhood Hodgkin’s disease. German-Austrian Pediatric Hodgkin’s Disease Group. J Clin Oncol. 1997;15(6):2247–53. doi: 10.1200/JCO.1997.15.6.2247. [DOI] [PubMed] [Google Scholar]
- 29.Tucker MA, Meadows AT, Boice JD, Jr, Stovall M, Oberlin O, Stone BJ, et al. Leukemia after therapy with alkylating agents for childhood cancer. J Natl Cancer Inst. 1987;78(3):459–64. [PubMed] [Google Scholar]
- 30.Tavani A, Gallus S, La Vecchia C, Negri E, Montella M, Dal Maso L, et al. Risk factors for breast cancer in women under 40 years. Eur J Cancer. 1999;35(9):1361–7. doi: 10.1016/s0959-8049(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 31.Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 32.Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, et al. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32(2):213–9. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- 33.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 34.Best T, Li D, Skol AD, Kirchhoff T, Jackson SA, Yasui Y, et al. Variants at 6q21 implicate PRDM1 in the etiology of therapy-induced second malignancies after Hodgkin’s lymphoma. Nat Med. 2011 doi: 10.1038/nm.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mertens AC, Mitby PA, Radloff G, Jones IM, Perentesis J, Kiffmeyer WR, et al. XRCC1 and glutathione-S-transferase gene polymorphisms and susceptibility to radiotherapy-related malignancies in survivors of Hodgkin disease. Cancer. 2004;101(6):1463–72. doi: 10.1002/cncr.20520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.