

Abstract
White adipose tissue stores energy in the form of lipids, and brown adipose tissue expends energy via uncoupled fatty acid oxidation, which leads to the generation of heat. Obesity reflects an imbalance between energy storage and energy expenditure and is strongly associated with metabolic and cardiovascular disease. Therefore, there are important medical and biological implications for elucidating the mechanisms that promote energy expenditure in humans. Animal models with altered vitamin D receptor (VDR) expression have changes in energy expenditure. However, the specific mechanism for this effect has not been elucidated and the relevance for humans is unclear. Here we show, using human patient samples from individuals with hereditary vitamin D resistant rickets, that the VDR directly inhibits the expression of uncoupling protein-1 (UCP1), the critical protein for uncoupling fatty acid oxidation in brown fat and burning energy. The inhibition is enforced by VDR occupancy of a negative response element in the promoter proximal region of the UCP1 gene. Deletion of VDR increases UCP1 expression and results in a “browning” of adipocytes. Importantly, we found that this process occurs cell autonomously and is independent of the physiologic VDR hormone ligand, 1,25-dihydroxyvitamin D. These results identify a mechanism for modulating energy balance in humans.
The vitamin D receptor (VDR), a member of the steroid/thyroid/retinoid nuclear receptor superfamily, heterodimerizes with retinoid X receptor (RXR) and binds to VDR response elements encoded in DNA (VDREs) to modulate the transcription of target genes (1, 2). Hereditary vitamin D-resistant rickets (HVDRR) is an autosomal recessive disease caused by heterogeneous loss-of-function mutations in the VDR (3). Patients with this disease develop a constellation of clinical manifestations including severe rickets with osteomalacia and hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism. These prominent features are due to loss of ligand-dependent VDR activity (3). VDR-null mice (VDR knockout [KO]) have been developed that recapitulate the HVDRR phenotype (4, 5).
The physiologic ligand for the VDR is 1α,25-dihdroxyvitamin D3 (1,25(OH)2D3 or calcitriol). Calcitriol is synthesized in the body by a series of tissue-specific steps in the skin, liver, and kidney. The final renal 1α-hydroxylation step is regulated by parathyroid hormone in response to hypocalcemia. In the small intestine, VDR is activated by calcitriol to induce genes that promote calcium and phosphate absorption (6). This calcitriol action is a primary regulator of calcium homeostasis in the body and is important in skeletal development and in bone mineralization. Not surprisingly, patients with loss-of-function mutations in the VDR gene develop hypocalcemia and are unable to normally mineralize bone, resulting in rickets (7).
However, it has become clear that, beyond a restricted role in the regulation of calcium, the VDR has a variety of important functions in fundamental biological processes (8). An early indication of the role of the VDR beyond calcium regulation was the observation that patients with HVDRR harboring certain VDR mutations (DNA binding domain, RXR dimerization domain, or premature stop mutations) have alopecia (7). VDRKO mice also have alopecia (4, 5). Several studies have now demonstrated a role for the VDR in hair follicle cycling as well as skin biology that explain the alopecia phenotype (reviewed in Refs. 7 and 9). Importantly, however, mice with a deletion in the calcitriol biosynthetic enzymes have normal hair cycles and only modest skin abnormalities despite severe hypocalcemia and rickets (10). These data, as well as other evidence (11, 12), indicate that VDR has both ligand-dependent and ligand-independent activities.
Careful analysis in cell lines and mouse models has suggested a role for the VDR in several additional important fundamental biological processes including cell proliferation, differentiation, and inflammation (13, 14). For example, previous studies have observed a role for the VDR in adipogenesis. In particular, bone marrow stromal cells from VDRKO mice were found to exhibit increased adipogenesis compared with wild-type (WT) cultures (15). One suggested mechanism for this activity is that VDR inhibits peroxisomal proliferator-activated receptor (PPAR)γ expression (16), a well-established critical regulator of adipogenesis.
The role of VDR in mature adipose tissue physiology has been less extensively studied. However, analysis of genetically modified mice suggests that modulating VDR levels in adipose tissue can impact the systemic metabolism of rodents. For example, VDRKO mice have reduced body weight and hypoleptinemia and are resistant to high-fat diet-induced obesity (17, 18). Interestingly, these studies revealed that white adipose tissue from VDRKO mice have areas of multilocular cell clusters and an increase in the expression of uncoupling protein 1 (UCP1), a central regulator of uncoupled fatty acid oxidation that drives energy expenditure (19). These findings are consistent with a beige phenotype in the white fat, in which increased features of brown fat occur in the white adipose depot (20). Because this study used mice with a global deletion of VDR, it is unclear whether the phenotype results from a tissue-specific or systemic effect. Wong and colleagues (21) generated transgenic mice that overexpresses VDR under an aP2 promoter/enhancer element (which is active in adipocytes, macrophages, endothelial, and other cells) and discovered that these mice have the opposite phenotype of the VDRKO mice, with increased fat mass and decreased levels of UCPs and energy expenditure. These findings suggest that an intrinsic adipose tissue effect, rather than systemic changes, induces the phenotype, but a causative indirect effect is feasible and was not excluded. It was also unknown whether these phenotypes result from a toggle in the ratio of beige to white adipocytes in the WAT or from cell autonomous changes in the adipocytes. Finally, the molecular mechanism and the relevance of these findings to humans remained unresolved.
In humans, obesity, as well as the associated metabolic diseases, are enormous and growing public health problems. Increasing the rate of the body's energy usage is an attractive strategy for developing novel therapies for these disorders. Uncoupling fatty acid oxidation results in enhanced energy metabolism, as demonstrated in brown adipose tissue, and increased brown fat inversely correlates with obesity in humans (22–25). Therefore, we were stimulated to examine how this process is regulated in humans. In this study, we used cells from patients with HVDRR to reveal that regulation of UCP1 by VDR is direct and occurs cell autonomously. In addition, we discovered that this regulation is maintained by a ligand-independent VDR activity. Revealing these mechanisms advances our understanding of how energy metabolism is controlled at the cellular level in humans.
Materials and Methods
Cell culture
Isolation of dermal skin fibroblasts from normal and HVDRR patients has been previously described (Table 1). 3T3-L1 cells were obtained from American Type Culture Collection (ATCC). Cells were grown in DMEM containing 4.5 g glucose, 10 mM sodium pyruvate, and 10% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2/95% air.
Table 1.
Genotype of HVDRR Mutant Cells
| VDR Mutation | Functional Defect | Reference |
|---|---|---|
| V26M | DNA binding | (39) |
| H305Q | Ligand binding | (41) |
| R391C | RXR binding | (36) |
| E420K | Coactivator binding | (38) |
| Y295X | Truncated protein, proximal | (43) |
| Y401X | Truncated protein, distal | (37) |
| H305Q/BSCL2 (DM) | VDR: Ligand binding BSCL2: Truncated protein | (28, 41) |
The V26M mutation is in the VDR DNA-binding domain and affects interactions with DNA and inhibits VDR transactivation. The V26M mutation does not affect 1,25(OH)2D3 binding or interactions with RXR or coactivators. The H305Q mutation is in the VDR ligand-binding domain and alters the contact point for the 25-hydroxyl group of 1,25(OH)2D3 and reduces VDR's affinity for 1,25(OH)2D3. The R391C mutation reduces RXR binding and inhibits VDR transactivation. The E420K mutation abolishes coactivator binding to the activation function-2 domain in the ligand-binding domain and inhibits VDR transactivation. The E420K mutation does not affect 1,25(OH)2D3 binding, DNA binding, or RXR heterodimerization. The Y295X and Y401X mutations introduce a premature stop signals that truncate the VDR. The VDR Y295X truncated protein is not expressed due to nonsense-mediated mRNA decay. The VDR Y401X truncated protein is expressed and is able to heterodimerize with RXR and bind to DNA but lacks 1,25(OH)2D3 binding. The patient with the Y401X mutation does not have alopecia, indicating that this mutant VDR maintains some functional activity. The H305Q/BSCL2 double mutant (DM) has homozygous mutations in the VDR (H305Q) and BSCL2 (1016/−3 C to G) genes.
Chromatin immunoprecipitation (ChIP) assay
Human fibroblasts grown to confluence were fixed in 1% formaldehyde for 10 minutes. Cells were sonicated in a Bioruptor (Diagenode, Inc). Samples were precipitated with anti-VDR C-20 antibody (Santa Cruz Biotechnology) or normal rabbit IgG. Immunoprecipitates were collected using ChIP Grade Protein G Magnetic Beads (Cell Signaling Technology). Real-time PCR was performed using the DyNAmo ColorFlash quantitative PCR (qPCR) kit and primers listed in Supplemental Table 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org.
Differentiation of human fibroblasts
Cells were seeded in 6-well plates in DMEM with 10% FBS and grown to confluence. Cells were then treated with either vehicle (0.1% ethanol-0.1% DMSO), 1 μM dexamethasone, 0.5 mM isobutyl-methylxanthine, 10 μM insulin (DIM), or DIM plus 1 μM rosiglitazone (DIMR). Cells were treated for 10–14 days.
Lipolysis assay
To measure lipolysis activity, control and differentiated fibroblasts were starved in DMEM containing 1% FBS overnight. Cells were then incubated for 24 hours in Hank's balanced salt solution containing 2% fatty-acid-free BSA and 10 μM isoproterenol. The culture medium was collected for glycerol measurement using the free glycerol reagent (Sigma).
Oil red O staining
After differentiation the cells were washed twice with 1×PBS and then fixed in 10% formaldehyde in 1× PBS for 60 min. Cells were washed twice with water and stained with oil red O for 50 minutes at room temperature. The cells were washed 3 times with water.
Real-time RT-PCR
Fibroblasts were treated for 20 hours with vehicle (0.1% ethanol), 1 μM rosiglitazone, or DIM plus 1 μM rosiglitazone in DMEM containing 10% FBS. RNA was isolated using RNeasy Plus Mini Kit (Qiagen). cDNA was prepared using Maxima Universal First Strand cDNA Synthesis kit (Fisher Scientific). Real-time PCR was performed using the DyNAmo ColorFlash qPCR kit (Fisher Scientific). Relative changes in mRNA expression were assessed by the 2-ΔΔC(T) method and normalized to that of the reference gene glyceraldehyde phosphate dehydrogenase. Assays were performed in triplicate. The primers used are listed in Supplemental Table 1.
VDR knockdown
3T3-L1 cells were transfected with VDR short hairpin RNA (shRNA) (Mission shRNA, Sigma) or vector only (pLKO.1-puro) using FuGeneDH according to the manufacturer's protocol (Promega). Transformed cells were selected using puromycin (2.5 μg/mL) and single colonies were isolated. VDR protein levels were determined by immunoblotting using anti-VDR antibody (D-6, Santa Cruz Biotechnology). Transformed cells were treated with DIM for 48 hours and then maintained on media containing insulin (1 μg/mL) to induce adipogenesis. Cells were treated with 1 μM rosiglitazone overnight and then treated with fresh rosiglitazone for an additional 3 hours before RNA was extracted. UCP1 levels were determined by real-time PCR and normalized to the reference gene RPL19.
Reporter constructs and assay
DNA fragments of sequence upstream of the human UCP1 MET translational start site were amplified by PCR from human genomic DNA and cloned into pGL3-basic or pGL4.1 TATA luciferase reporter vectors (Promega). Deletions and point mutations of putative VDREs were generated by PCR using Pfu polymerase (Stratagene). The primers used for cloning and mutagenesis are listed in Supplemental Table 1. The reporter plasmids were transiently cotransfected with VDR, RXRα, and PPARγ expression vectors in COS-7 cells using Polyfect (Qiagen). Cells were then treated with 1 μM rosiglitazone. Forty-eight hours later, cells were collected and reporter gene assays were carried out using the Dual Luciferase System (Promega).
Statistics
All experiments were performed with biological triplicates. Error bars represent standard deviations from the mean. Student's t tests were used for pair-wise comparisons and P < .05 was considered significant.
Study approval
Informed consent was obtained from the patients and parents under a Stanford University IRB approved protocol.
Results
Human HVDRR dermal fibroblasts can be differentiated into adipocyte-like cells
To test the impact of VDR on adipocytes in humans, we used an ex vivo model of adipogenesis by differentiating human skin fibroblasts. We examined the role of VDR using our collection of primary cells obtained from patients with HVDRR with several different loss-of-function mutations in VDR (Table 1). Human dermal fibroblasts have been shown to develop features of adipocytes when induced with an adipogenic cocktail (26). Therefore, we treated the fibroblasts with dexamethasone, insulin, isobutyl-methylxanthine (DIM), alone or in combination with the PPARγ agonist rosiglitazone (DIMR). Microscopic examination revealed many visible fat droplets within 5–6 days in the HVDRR fibroblasts cells treated with DIMR. At day 14, the HVDRR cells treated with DIMR contained many cells with intracellular droplets (Figure 1, A and B). Treatment with DIM alone also induced lipid droplet formation in the HVDRR cells but to a lesser degree. When compared with the HVDRR cells, the normal control cells exhibited fewer oil red O (which binds neutral lipids) staining positive cells after treatment with DIM or DIMR (Figure 1A). We also examined adipogenesis in fibroblasts from a patient with homozygous mutations in both the VDR (H305Q) and BSCL2 genes compared with those of a sibling with only the VDR (H305Q) mutation. The BSCL2 mutation in intron 6 (1016/-3C to G) causes exon 7 to be skipped and results in a frameshift and premature stop codon (27). Mutations in BSCL2 cause Berardinelli-Seip congenital lipodystrophy (28). BSCL2 is a membrane protein located in the endoplasmic reticulum and is thought to be involved in lipid droplet formation (29). The cells with the VDR/BSCL2 mutations had very few lipid droplets after treatment with DIMR, confirming a role for BSCL2 in droplet formation. None of the HVDRR cells or normal control cells treated with vehicle without DIM (Figure 1A, top row) or rosiglitazone alone (data not shown) had visible droplets and were negative for oil red O staining.
Figure 1.

Differentiated human fibroblasts have mutlilocular lipid droplets and exhibit functional properties of adipocytes. A, Human fibroblasts were treated with vehicle, DIM, or DIM plus rosiglitazone for 14 days and examined by light microscopy. B, Oil red O-positive staining was evident in the HVDRR cells (E420K)-treated DIM plus rosiglitazone indicating the presence of lipids. Relative gene expression of PPARγ and FABP4 in HVDRR cells treated with DIM (C and E) or DIMR (D and F) compared with WT control that was set to 1. G, Glycerol release in the supernatant of human skin fibroblasts treated with vehicle (undiff) or differentiated (diff) with DIMR adipogenic media following treatment with isoproterenol. The quantity of glycerol released (in micrograms) was normalized to the total amount of protein (in milligrams) for each sample. Three independent experiments were performed. Error bars represent SD from the mean (s.d.). a.u., arbitrary units; DM, double mutant; *, P < .05; **, P < .01; ***, P < .001.
Having successfully differentiated the fibroblasts into what histologically appeared to be adipocyte-like cells, we next analyzed the expression of the adipocyte markers PPARγ and FABP4. These transcripts are highly expressed in both brown and white adipocytes, and coinduction of expression of these markers indicates an adipocyte-specific expression profile in the cells (30). Gene expression studies showed that both WT control and HVDRR cells expressed the pan-adipocyte markers PPARγ and FABP4 (Figure 1, C–F). PPARγ expression was low in the undifferentiated WT control and HVDRR cells but was up-regulated after treatment with DIM or DIMR (Supplemental Figure 1). The WT control and HVDRR cells expressed similar levels of PPARγ mRNA after DIM treatment (Figure 1C). DIMR treatment further induced PPARγ expression approximately 10-fold in the WT control cells and about 10- to 30-fold in the HVDRR cells compared with undifferentiated cells (Supplemental Figure 1). With the exception of the Y295X cells, HVDRR cells expressed higher levels of PPARγ compared with the DIMR-treated WT control cells (Figure 1D). FABP4 expression was undetectable in the undifferentiated cells but was significantly up-regulated in the WT control and HVDRR cells when differentiated with DIM alone (Figure 1E). FABP4 was highly up-regulated in several HVDRR cells (R391C, E420K, and Y401X) compared with the WT control cells after DIM treatment (Figure 1E). FABP4 expression was highly expressed in both the WT control and HVDRR cells when differentiated with DIMR (Figure 1F). The VDR/BSCL2 double mutant (DM) expressed significantly lower levels of PPARγ and FABP4 even when compared with the sibling with the same VDR mutation but no BSCL2 mutation, confirming earlier reports of a role for BSCL2 in adipogenesis (31) and indicating that this BSCL2 activity is upstream of VDR (Figure 1, C–F). Together, our results demonstrate that the differentiated fibroblasts exhibit an expression profile signature of adipocytes.
To investigate the functional capabilities of the differentiated human fibroblasts, we examined their ability to carry out lipolysis, by measuring the release of glycerol after exposure to the synthetic catecholamine β-adrenergic agonist isoproterenol. The differentiated fibroblasts showed a 1.5- to 4-fold increase in glycerol release compared with the undifferentiated cells (Figure 1G). Of note, the H305Q/BSCL2 double mutant had significantly less induction of lipolysis compared with the H305Q single mutant, indicating that the level of lipolysis reports successful differentiation. Together, these results demonstrate that both the WT control and HVDRR differentiated cells exhibit functional properties of adipocytes.
Cell-autonomous and ligand-independent regulation of UCP1 by VDR
Studies have shown that the white adipose tissue from VDRKO mice has features of brown fat including small lipid droplets and expression of UCP1 (17, 18), a phenotype that has been referred to as “beige” fat (32). However, the mechanism regulating this finding had not been fully elucidated and the relevance for humans was unknown. Therefore, to determine whether HVDRR cells autonomously regulate the beiging process, we examined the expression of UCP1 in differentiated WT control and HVDRR human cells. Neither WT nor HVDRR cells expressed UCP1 prior to differentiation. We discovered that UCP1 was up-regulated 8-fold in H305Q, 32-fold in R391C, and 4-fold in Y295X HVDRR cells after DIM treatment (Figure 2A). On the other hand, UCP1 was undetectable in the WT control cells and the HVDRR cells with the V26M, E420K, and Y401X mutations after DIM treatment. The addition of rosiglitazone to the DIM induction (DIMR) further stimulated UCP1 levels dramatically in specific HVDRR cells (175-fold in H305Q, 68-fold in R391C, and 70-fold in Y295X). Of note, compared with these mutant HVDRR cells, the levels of UCP1 expression after DIMR treatment remained significantly lower in the WT and other HVDRR cells (2-fold in V26M 11-fold in E420K and 0.8-fold in Y401X). In the VDR/BSCL2 double mutant (DM), UCP1 was undetectable after DIM treatment and remained below WT control after DIMR (Figure 2B). Although significantly lower than in the H305Q, R391C, and Y295X HVDRR cells, the detectable levels of UCP1 expression in WT and V26M, E420K, and Y401X HVDRR cells after DIMR treatment may have been induced by rosiglitazone, because chronic exposure to PPARγ agonists induces UCP1 expression in white adipocyte cultures (33). Importantly, the very strong induction in the H305Q, R391C, and Y295X HVDRR cells suggests that VDR regulates a distinct, and cell autonomous, mechanism for modulating UCP1 expression.
Figure 2.
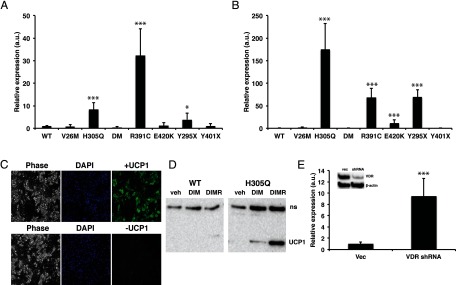
Differentiated human HVDRR fibroblasts express the brown fat marker UCP1. Relative gene expression of UCP1 in HVDRR cells treated with DIM (A) or DIMR (B) compared with WT control that was set to 1. C, HVDRR (H305Q) fibroblasts were differentiated with DIMR for 14 days and labeled with DAPI (blue) or stained with antibodies against UCP1 (green). The left panels show corresponding phase contrast image. D, Immunoblots of UCP1 in WT control cells and in HVDRR cells with H305Q mutation following treatment with vehicle (veh), DIM, or DIMR. Nonspecific band (ns). E, Differentiated 3T3-L1 cells stably transfected with VDR shRNA or empty vector (vec) were treated with rosigliatozone and examined for UCP1 expression by real time RT-PCR. Inset shows immunoblot of VDR levels in transfected cells. β-Actin was used as a loading control. Three independent experiments were performed. Error bars are SDs. a.u., arbitrary units; DM, double mutant; *, P < .05; ***, P < .001.
We also examined the expression of several other genes thought to be selectively expressed in brown fat (33–35, 47). NRBF1 was significantly increased in H305Q and R391C mutant cells and CITED was increased in H305Q. However CPT1b was not increased in the VDR mutant cells (Supplemental Figure 2). These data suggest that the impact of VDR in regulating brown fat identity may extend beyond UCP1. However, more studies defining the human brown fat expression profile are required in order to fully compare against these mutant VDR cells.
The data from the HVDRR cells demonstrate that UCP1 expression is negatively regulated by the VDR in human cells. The high level of UCP1 in the R391C mutant suggests that RXR binding is necessary for this inhibitory action of VDR, because this mutant has otherwise intact VDR protein (36). Of note, the HVDRR cells with the E420K mutation (located in the activation function 2 domain) and Y401X mutation (that deletes the activation function 2 domain) are defective in binding coactivators (eg, VDR-interacting protein 205, steroid recptor coactivator 1) (37, 38). Both of these mutant cells express UCP1 at lower levels, suggesting that coactivator interaction is not required for the inhibitory activity of the VDR on UCP1 expression. In addition, the Y401X mutant does not bind calcitriol (37), supporting a role of an unliganded activity of the VDR in regulating UCP1. In the V26M mutant (with an amino acid change in the first zinc finger of the DNA-binding domain) (39), the levels of UCP1 are also similar to the WT control. This finding supports prior studies suggesting that the first zinc finger in VDR DNA-binding domain is not required for repression activity (40). Unexpectedly, the cells with the H305Q mutation (with lower affinity for calcitriol from disrupting the contact point for the 25-hydroxyl group of calcitriol (41, 42) have significantly elevated UCP1 levels. The findings in the H305Q and Y401X mutants further demonstrate that the repressive action of the WT VDR on UCP1 expression occurs independently from ligand binding. Moreover, these results suggest that H305 is a critical amino acid involved in unliganded VDR actions and reveal a previously unknown function of this amino acid.
To determine whether UCP1 protein is expressed in the mutant cells, we performed immunohistochemistry on the differentiated HVDRR cells using a UCP1 antibody. Strong UCP1 staining was observed in the cytoplasm of the lipid droplet-containing cells, compared with almost undetectable levels in the undifferentiated cells (Figure 2C). No staining was observed in the HVDRR cells without the UCP1 antibody. Immunoblotting confirmed that UCP1 was not expressed in undifferentiated (vehicle treated) WT control or HVDRR cells. HVDRR cells that expressed UCP1 RNA after differentiation also had detectable UCP1 protein following DIM treatment that was further induced with DIMR, whereas in the WT control cells, UCP1 was undetectable in DIM or DIMR samples (Figure 2D).
To further examine the effect of VDR on UCP1 levels, we stably transfected the adipogenic cell line 3T3-L1 with VDR shRNA. Immunoblotting showed that the shRNA transfected cells had approximately 90% reduction in VDR levels compared with cells transfected with control vector. UCP1 levels were significantly elevated in the differentiated VDR knockdown cells (Figure 2E), further supporting the hypothesis that VDR represses UCP1 expression.
It is important to highlight that the studies described above were done without the addition of calcitriol, and there is no induction of 24-hydroxylase (CYP24A1), the classic VDR target gene, unless calcitriol is added to the cultures (Figure 3A). Together our results indicate that regulation of UCP1 expression by VDR occurs in a ligand-independent manner. Because the ligand binding affinity of the H305Q mutant is reduced but adding excess calcitriol (10 nM) rescues the induction of 24-hydroxylase expression by VDR ex vivo (Figure 3A) and can be used to treat the rickets that develops in patients with this mutation (41), cells with this mutation provide an opportunity to further test the role of ligand in the regulation of UCP1. Therefore, we tested whether the addition of calcitriol would rescue the repression of UCP1 expression by VDR. We found that even pharmacologic doses of calcitriol did not significantly alter UCP1 levels in the H305Q or any of the other mutants compared with the absence of ligand (Figure 3B). These results confirm that the regulation of UCP1 can be uncoupled from other VDR activities and is ligand independent.
Figure 3.

HVDRR mutant fibroblasts exhibit resistance to calcitriol. A, Control and HVDRR fibroblasts were treated with vehicle (0.1% ethanol) or 10 nM calcitriol for 6 hours. RT-qPCR assays were carried out for CYP24A1 a marker of calcitriol responsiveness. DM denotes double mutant with H305Q mutation in the VDR and BSCL2 mutation. B, Pharmacologic doses of calcitriol do not alter UCP1 levels in differentiated HVDRR cells. Relative gene expression of UCP1 in WT and HVDRR fibroblasts differentiated with DIMR or DIMR plus vehicle or 10 nM calcitriol for 14 days. a.u., arbitrary units; DM, double mutant; *, P < .05; **, P < .01; ***, P < .001.
VDR directly regulates UCP1 expression in human cells
Using a combination of in silico analyses and ChIP scanning, we identified 4 potential VDREs between −3698 bp to −984 bp in the sequence upstream of the translational start site of the human UCP1 gene. To explore the mechanisms underlying VDR-mediated repression of UCP1, we generated 2 constructs containing a PPARγ enhancer and all 4 putative VDREs (−4218 to −984 bp; VDRE1–4) or 2 VDREs (−4218 to −2545 bp; VDRE3–4) upstream of a luciferase reporter gene (Figure 4A). We then generated deletions of each VDRE and tested their effects on rosiglitazone-induced transactivation by PPARγ. These experiments were performed in the absence of calcitriol. As shown in Figure 4B, rosiglitazone induced luciferase activity of the −4218 to −984 construct containing all 4 VDREs. Deletion of VDRE1 and VDRE2 had no effect on this activity. Deletion of VDRE3 resulted in a modest increase in activity whereas deletion of VDRE4 resulted in a doubling of the rosiglitazone-induced reporter activity compared with the parent construct containing all 4 VDREs. The −4218 to −2545 bp construct had ∼2-fold higher levels of rosiglitazone induced activity compared with the longer −4218 to −984 bp construct. Deletion of VDRE3 had no effect on this activity. Deletion of VDRE4, however, resulted in a strong stimulation by rosiglitazone that was approximately 4-fold greater than the intact −4218 to −2545 bp construct and about 10-fold higher than the intact −4218 to −984 bp construct (Figure 4B). Furthermore, when a single guanine residue that is highly conserved in VDREs was mutated to a cytosine in the 3-prime hexamer of VDRE4 (AGGCGA to ACGCGA) a significant induction in basal activity as well as rosiglitazone-stimulated activity was observed (Figure 4C). These results demonstrate that VDRE4 is a critical negative regulatory element controlling UCP1 expression.
Figure 4.

A VDRE in the human UCP1 upstream sequence negatively regulates UCP1. A, In silico and ChIP scanning analyses identified 4 potential VDR binding sites in the region from +1 to −4218 of the UCP1 upstream sequence (VDREs 1–4). Deletions of the VDREs were generated by PCR and analyzed by reporter assays. B, COS-7 cells were transiently cotransfected with the different deletion constructs with VDR, RXR, and PPARγ expression plasmids. Cells were treated with DMSO (veh) or 1 μM rosiglitazone (Rosi) for 48 hours and then assayed for luciferase activity. C, A single G to C mutation created in the VDRE4 sequence was analyzed by reporter assays. *, P < .05; **, P < .01; ***, P < .001.
We then used ChIP assays and qPCR to analyze endogenous occupancy of VDRE4 by VDR in intact human WT and HVDRR cells. We found that the WT VDR and the E420K mutant VDR were competent to occupy VDRE4 (Figure 5A). As expected, the Y295X mutant that does not express VDR protein, due to nonsense-mediated decay of the VDR mRNA (43), showed no binding to the VDRE4. Importantly, neither the H305Q nor the R391C mutants occupied VDRE4 in the UCP1 gene (Figure 5A). Interestingly, the addition of 1,25(OH)2D3 did not significantly increase the levels of VDR occupancy of VDRE4 (Figure 5B). When analyzed with our UCP1 expression data (Figure 3), these results support the model that occupancy of VDRE4 enforces repression of UCP1 expression. This paradigm contrasts with the classic induction of the expression of CYP24A1 via ligand-induced recruitment of VDR to a positive VDRE in the CYP24A1 gene (Figure 5, B and C). Interestingly, only the WT VDR was found on the CYP24A1 VDRE in the absence of ligand. Addition of calcitriol increased WT VDR occupancy and stimulated occupancy by the H305Q mutant VDR (Figure 5C), results that are consistent with activation of CYP24A1 expression by the WT and H305Q mutant (Figure 3A). The E420K mutant VDR was not present on the CYP24A1 VDRE, suggesting that there is differential binding to ligand-dependent and ligand-independent VDREs. Because VDR repression is thought to involve recruitment of corepressors, we examined whether the VDR corepressor SMRT (silencing mediator of retinoid and thyroid hormone receptor) was enriched at VDRE4 using ChIP assays. However, no enrichment of SMRT was observed at VDRE4 whereas we did detect SMRT recruitment to the osteocalcin VDRE as previously reported (44, 45) (Supplemental Figure 3). Together, our results reveal a scenario in which ligand-independent VDR-mediated repression of UCP1 is achieved by WT and mutant VDRs capable of occupancy of the VDRE4-regulatory element. The fact that the H305Q mutant was unable to repress UCP1 and failed to occupy VDRE4 suggests that H305 is a critical amino acid involved in the unliganded actions of the VDR in addition to its known function as a contact point for the 25-hydroxyl group in 1,25(OH)2D3.
Figure 5.

VDR occupancy at the UCP1 VDRE4. A, ChIP analyses of VDR binding to the endogenous UCP1 VDRE4 in WT and HVDRR mutants. B, ChIP analyses of VDR binding to the CYP24A1 VDRE and UCP1 VDRE4 in WT control cells treated with vehicle (0.1% ethanol) or 10 nM 1,25(OH)2D3 for 6 hours. C, ChIP analyses of VDR occupancy of the CYP24A1 VDRE in WT control and HVDRR mutants fibroblasts treated with vehicle (veh) or 10 nM 1,25(OH)2D3 for 6 hours. *, P < .05; **, P < .01; ***, P < .001.
Discussion
The control of energy metabolism in humans is a complex and carefully regulated process. Understanding how energy utilization is regulated at the molecular level has broad medical implications. In particular, the rise in prevalence of obesity and metabolic disease in the population underscores the need to advance our understanding of this process in order to develop better therapeutic options for these patients.
Brown fat, once thought to fully regress in humans after the neonatal period, has now been convincingly identified throughout adult life (23, 46). As brown fat is especially successful at burning energy, increasing the levels or the features of brown fat in cells has emerged as an attractive potential strategy for treating patients with excess energy stores. As opposed to rodents, it appears that most brown fat cells in adult humans have a molecular signature of beige cells (47, 48). It is therefore reasonable to speculate that most physiologically relevant human brown fat in adults is analogous to the beige fat identified in rodent models.
Inspired by studies of mouse models that revealed that altering VDR levels modulates the levels of brown fat features in white adipose depots (17, 18), we speculated that VDR could regulate features of beiging in human cells. Further, we hypothesized that studies utilizing our collection of cells isolated from patients with HVDRR, that contain various naturally occurring mutations in VDR, would help us to resolve whether the connection between VDR and beiging was cell autonomous as well as define the specific mechanism. Our results indicate that unliganded VDR can directly regulate the critical functional mediator of energy consumption, UCP1, by binding to a VDRE in the promoter-proximal region of the gene and inhibiting expression. We show that changes in UCP1 levels can occur cell autonomously, rather than requiring the recruitment of a distinct cell lineage. When VDR is deleted or mutated in a way that disrupts occupancy of a specific VDRE, UCP1 is derepressed and expressed. Importantly, our data suggest a paradigm in which VDR modulates beige vs white adipocyte identity as opposed to a toggle in the recruitment of distinct progenitor cell populations. We believe these results will have important implications for human health. For example, these data support pursuing the therapeutic strategy of developing factors that specifically target releasing the beige fat expression profile of human cells.
Supplementary Material
Acknowledgments
We thank Joseph Zerwekh (University of Texas Southwestern), G. Kerr Whitfield (University of Arizona), and Mark Haussler for the HVDRR cells with the R391C mutation. We thank Maria Costa (Stanford University), David Feldman (Stanford University), Mihir Gupta (Stanford University), and Katie Krueger (Stanford University) for helpful discussions.
This work was supported by The Children's Health Research Institute at Stanford and NIH Director's New Innovator Award (DP2 OD006740) (to B.J.F.). B.J.F. is a Bechtel Endowed Faculty Scholar.
Author Contributions: B.J.F. and P.J.M. developed the hypothesis, experimental plan, interpreted the data, and wrote the manuscript. P.J.M. conducted the experiments.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- chromatin immunoprecipitation
- DIM
- 1 μM dexamethasone, 0.5 mM isobutyl-methylxanthine, 10 μM insulin
- DIMR
- rosiglitazone
- FBS
- fetal bovine serum
- HVDRR
- hereditary vitamin D-resistant rickets
- KO
- knockout
- PPAR
- proliferator-activated receptor
- qPCR
- quantitative PCR
- RXR
- retinoid X receptor
- shRNA
- short hairpin RNA
- SMRT
- silencing mediator of retinoid and thyroid hormone receptor
- UCP1
- uncoupling protein 1
- VDR
- vitamin D receptor
- VDRE
- vitamin D response element
- WT
- wild-type.
References
- 1. Haussler MR, Haussler CA, Whitfield GK, et al. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful aging. J Steroid Biochem Mol Biol. 2010;121:88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pike JW. Genome-wide principles of gene regulation by the vitamin D receptor and its activating ligand. Mol Cell Endocrinol. 2011;347:3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malloy PJ, Feldman D. Genetic disorders and defects in vitamin D action. Endocrinol Metab Clin N Am. 2010;39:333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci USA. 1997;94:9831–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–396 [DOI] [PubMed] [Google Scholar]
- 6. Bouillon R, Van Cromphaut S, Carmeliet G. Intestinal calcium absorption: molecular vitamin D mediated mechanisms. J Cell Biochem. 2003;88:332–339 [DOI] [PubMed] [Google Scholar]
- 7. Malloy PJ, Tiosano D, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets. In: Feldman D, Pike JW, Adams JS, eds. Vitamin D. 3rd ed San Diego: Elsevier;2011:1197–1232 [Google Scholar]
- 8. Rosen CJ, Adams JS, Bikle DD, et al. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev. 2012;33:456–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demay MB. The hair cycle and Vitamin D receptor. Arch Biochem Biophys. 2012;523:19–21 [DOI] [PubMed] [Google Scholar]
- 10. Panda DK, Miao D, Tremblay ML, et al. Targeted ablation of the 25-hydroxyvitamin D α-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci USA. 2001;98:7498–7503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dowd DR, MacDonald PN. The 1,25-dihydroxyvitamin D3-independent actions of the vitamin D receptor in skin. J Steroid Biochem Mol Biol. 2010;121:317–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skorija K, Cox M, Sisk JM, et al. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol Endocrinol. 2005;19:855–862 [DOI] [PubMed] [Google Scholar]
- 13. Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700 [DOI] [PubMed] [Google Scholar]
- 14. Krishnan AV, Moreno J, Nonn L, et al. Novel pathways that contribute to the anti-proliferative and chemopreventive activities of calcitriol in prostate cancer. J Steroid Biochem Mol Biol. 2007;103:694–702 [DOI] [PubMed] [Google Scholar]
- 15. Cianferotti L, Demay MB. VDR-mediated inhibition of DKK1 and SFRP2 suppresses adipogenic differentiation of murine bone marrow stromal cells. J Cell Biochem. 2007;101:80–88 [DOI] [PubMed] [Google Scholar]
- 16. Blumberg JM, Tzameli I, Astapova I, Lam FS, Flier JS, Hollenberg AN. Complex role of the vitamin D receptor and its ligand in adipogenesis in 3T3–L1 cells. J Biol Chem. 2006;281:11205–11213 [DOI] [PubMed] [Google Scholar]
- 17. Narvaez CJ, Matthews D, Broun E, Chan M, Welsh J. Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology. 2009;150:651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wong KE, Szeto FL, Zhang W, et al. Involvement of the vitamin D receptor in energy metabolism: regulation of uncoupling proteins. Am J Physiol Endocrinol Metab. 2009;296:E820–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richard D, Picard F. Brown fat biology and thermogenesis. Front Biosci (Landmark Ed). 2011;16:1233–1260 [DOI] [PubMed] [Google Scholar]
- 20. Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998;102:412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong KE, Kong J, Zhang W, et al. Targeted expression of human vitamin D receptor in adipocytes decreases energy expenditure and induces obesity in mice. J Biol Chem. 2011;286:33804–33810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Celi FS. Brown adipose tissue–when it pays to be inefficient. N Engl J Med. 2009;360:1553–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508 [DOI] [PubMed] [Google Scholar]
- 25. Saito M, Okamatsu-Ogura Y, Matsushita M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Junker JP, Sommar P, Skog M, Johnson H, Kratz G. Adipogenic, chondrogenic and osteogenic differentiation of clonally derived human dermal fibroblasts. Cells Tissues Organs. 2010;191:105–118 [DOI] [PubMed] [Google Scholar]
- 27. Van Maldergem L, Bachy A, Feldman D, et al. Syndrome of lipoatrophic diabetes, vitamin D resistant rickets, and persistent Müllerian ducts in a Turkish boy born to consanguineous parents. Am J Med Genet. 1996;64:506–513 [DOI] [PubMed] [Google Scholar]
- 28. Magré J, Delépine M, Khallouf E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370 [DOI] [PubMed] [Google Scholar]
- 29. Chen W, Chang B, Saha P, et al. Berardinelli-Seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32:1099–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12:722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Payne VA, Grimsey N, Tuthill A, et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008;57:2055–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishibashi J, Seale P. Medicine. Beige can be slimming. Science. 2010;328:1113–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ahfeldt T, Schinzel RT, Lee YK, et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol. 2012;14:209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hansen JB, Jørgensen C, Petersen RK, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci USA. 2004;101:4112–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whitfield GK, Selznick SH, Haussler CA, et al. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol. 1996;10:1617–1631 [DOI] [PubMed] [Google Scholar]
- 37. Malloy PJ, Wang J, Peng L, et al. A unique insertion/duplication in the VDR gene that truncates the VDR causing hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Arch Biochem Biophys. 2007;460:285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol. 2002;16:2538–2546 [DOI] [PubMed] [Google Scholar]
- 39. Malloy PJ, Wang J, Srivastava T, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from a novel missense mutation in the DNA-binding domain of the vitamin D receptor. Mol Genet Metab. 2010;99:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim MS, Fujiki R, Murayama A, et al. 1α,25(OH)2D3-induced transrepression by vitamin D receptor through E-box-type elements in the human parathyroid hormone gene promoter. Mol Endocrinol. 2007;21:334–342 [DOI] [PubMed] [Google Scholar]
- 41. Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rochel N, Hourai S, Moras D. Crystal structure of hereditary vitamin D-resistant rickets–associated mutant H305Q of vitamin D nuclear receptor bound to its natural ligand. J Steroid Biochem Mol Biol. 2010;121:84–87 [DOI] [PubMed] [Google Scholar]
- 43. Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest. 1990;86:2071–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim JY, Son YL, Lee YC. Involvement of SMRT corepressor in transcriptional repression by the vitamin D receptor. Mol Endocrinol. 2009;23:251–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meyer MB, Pike JW. Corepressors (NCoR and SMRT) as well as coactivators are recruited to positively regulated 1α,25-dihydroxyvitamin D3-responsive genes. J Steroid Biochem Mol Biol. 2013;136:120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ouellet V, Labbé SM, Blondin DP, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. 2012;122:545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sharp LZ, Shinoda K, Ohno H, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PLoS One. 2012;7:e49452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu J, Boström P, Sparks LM, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.