

Abstract
Hippocampal cultures infected with the ΔRR vector for the HSV-2 anti-apoptotic gene ICP10PK survive cell death triggered by a wide variety of insults. Survival includes robust protection of uninfected neurons, but the mechanism of this bystander activity is still unclear. Here we report that ICP10PK+ neurons release soluble factors that protect uninfected neurons from NMDA and MPP+-induced apoptosis. Release depends on ICP10PK-mediated activation of the Ras signaling pathways MEK/ERK and PI3-K/Akt, and it was not seen for cultures infected with the ICP10PK negative vector ΔPK. The released neuroprotective factors include vascular endothelial growth factor (VEGF) and fractalkine, the levels of which were significantly higher in conditioned media from hippocampal cultures infected with ΔRR (NCMΔRR) than ΔPK or phosphate-buffered saline (mock infection). VEGF neutralization inhibited the neuroprotective activity of NCMΔRR, indicating that the VEGF protective function is through neuron-neuron cross-talk. NCMΔRR also stimulated microglia to release increased levels of IL-10 and decreased levels of TNF-α that were protective for uninfected neurons. These release patterns were not seen for microglia given NCMΔRR in which fractalkine was neutralized, indicating that the fractalkine protective function is through bidirectional neuron-microglia communication. Collectively, the data indicate that ΔRR is a multiple target strategy to rescue neurons from excitotoxic injury.
Keywords: chemokines, cytokines, excitotoxicity, growth factors, microglia, SOD1
Ischemic brain injury, or stroke, is the third most common killer of Americans and the leading cause of severe, long-term disability. Neuronal death is because of excessive activation of glutamate receptors, a process known as excitotoxicity that is accompanied by the induction of apoptosis. Effective treatments remain elusive, due at least in part to the failure to override apoptosis. Clinical challenges include the identification of relevant target genes and delivery platforms and the contribution of neuroactive compounds released by neurons and glial cells towards neuronal cell life and death decisions.
Neuroactive compounds determine the composition of the neuronal milieu and define all aspects of the CNS function. They include growth factors, cytokines and chemokines, a family of small proteins that are secreted by neurons and microglia (Adler et al. 2006). Neuronal and glial cell dysfunction can drastically alter the neuronal milieu from one that supports the life and function of neurons to one that initiates or exacerbates their death (He and Sun 2007; Salmina 2009). It is becoming increasingly evident that the paracrine activity of the active compounds in the neuronal milieu is subject to constraints imposed by the milieu composition and defines cell-to-cell interactions. For example, neurons release growth factors that modulate neuron-neuron communication, but their activity can be amplified through bidirectional neuron-microglia cross-talk that involves microglial secretion of neurotrophic factors (Nakajima and Kohsaka 1998). One factor in the neuronal milieu is the neuronally-derived vascular endothelial growth factor (VEGF) (Schiera et al. 2007) that was shown to protect hippocampal neurons from death caused by ischemia, glucose deprivation or other insults (Jin et al. 2000; Svensson et al. 2002) and inhibit motor neuron degeneration (Gomes et al. 2007; Nicoletti et al. 2008). Neuroprotective activity is believed to involve parallel/concurrent modulation of cells in the vascular and nervous systems (Sköld and Kanje 2008) as well as microglia (Forstreuter et al. 2002), but it is still controversial (Ferrari et al. 2006; Nicoletti et al. 2008; Benton et al. 2009). In fact, exogenously delivered VEGF caused a potent and therapeutically undesirable immune response, was trapped by circulating receptor, did not cross the blood-brain barrier and was associated with neurotoxicity (Storkebaum et al. 2005).
Neurons also release chemokines. One of these, fractalkine (FKN, also known as CX3CL1), is constitutively expressed in neurons where it is tethered to the cell membrane by a mucin-like stalk. Upon cell activation, this stalk is cleaved by metalloproteinases and the chemokine domain is released as a soluble factor with neuroprotective (Mizuno et al. 2003; Limatola et al. 2005) or neurotoxic (Chapman et al. 2000) activity. Because its receptor (CX3R1) is primarily expressed on microglia, FKN released into the neuronal milieu is believed to induce microglial activation (Tarozzo et al. 2003) that may contribute to neurodegeneration. FKN was associated with inflammation-related neuropathic pain (Milligan et al. 2008), development of Parkinson’s disease (Shan et al. 2009) through overproduction of inflammatory cytokines [viz. tumor necrosis factor-α (TNF-α)] (Minghetti et al. 2005; Clausen et al. 2008; Whitney et al. 2009) and cerebral ischemia (Dénes et al. 2008). The microglia-derived anti-inflammatory cytokine IL-10 was also associated both with neuroprotection (Milligan et al. 2008) and neurotoxicity (Rentzos et al. 2009). It is becoming increasingly evident that although individual soluble factors may have neuroprotective potential, their use as independent therapies is limited by the finding that they can also contribute to neurotoxicity. Therefore, effective therapeutic strategies for neurodegenerative diseases must have multiple target activities the development of which depends on a better understanding of the complex cell-to-cell interactions that define the composition of the neuronal milieu and regulate neuronal cell life/death decisions.
We have previously shown that neurons expressing the HSV-2 protein ICP10PK are protected from death caused by various signals, including virus infection, treatment with a protein kinase C inhibitor, disruption of osmolar environment, growth factor withdrawal, toxin injury and excitotoxicity through activation of survival pathways that inhibit caspase-dependent and independent apoptosis/programmed cell death (Smith et al. 1994, 1998, 2000; Perkins et al. 2002a,b, 2003; Gober et al. 2006; Laing et al. 2006, 2008; Golembewski et al. 2007; Wales et al. 2007, 2008). ICP10PK is only expressed in neurons, but its protective activity appears to involve a multiple target strategy that includes glial cell modulation and the inhibition of inflammatory processes (Laing et al. 2006; Golembewski et al. 2007; Laing and Aurelian 2008). This is likely because of paracrine effects mediated by neuroactive compounds released by the ICP10PK+ neurons, because neurons transfected with ICP10PK protected non-expressing neurons from HSV-1 induced apoptosis (Perkins et al. 2003). However, the mechanism responsible for the ability of ICP10PK to modulate cell-to-cell interactions in the CNS and protect non-expressing neurons from death stimuli (bystander activity), are still unknown. The studies described in this report were designed to address these questions.
Materials and methods
Viruses
ICP10 is encoded by the HSV-2 gene UL39 and has kinase (PK) and ribonucleotide reductase (RR) (large subunit) activities, which function independently of each other. The PK activity is located within sequences encoded by the 5′-end while the RR activity is located within sequences encoded by the 3′-end of UL39. Both functions are required for virus growth in neurons (Smith et al. 1998, 2000). The generation and properties of the ΔRR and ΔPK viruses were previously described (Smith et al. 1998, 2000; Perkins et al. 2002a; Gober et al. 2006; Laing et al. 2006). Briefly, to generate ΔRR, the 3′-end RR-encoding UL39 sequence was replaced with LacZ DNA fused in frame with ICP10PK, giving rise to a 175 kDa (p175) mutant protein. ΔPK is deleted in the UL39 5′-end PK-encoding sequence but it retains the RR encoding sequence, giving rise to a 95 kDa protein (p95) (Smith et al. 1998). This is schematically represented in Fig. S1. The mutant proteins are driven by the authentic ICP10 promoter which is regulated with immediate early kinetics and responds to activator protein-1 transcription factors that are up-regulated/activated by neurotoxic stress stimuli (Gober et al. 2005; Laing et al. 2006).
Cell culture
Vero (African green monkey kidney) were grown in minimal essential medium, supplemented with 1 mM sodium pyruvate, 2 mM L-glutamine, 100 μM non-essential amino acids (Gibco-BRL, Gaithersburg, MD, USA) and 10% fetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA, USA). PC12 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Mediatech, Manassas, VA, USA) supplemented with 10% FBS and 7.75 g/L sterile glucose and plated on rat tail collagen-coated glass slides or polystyrene flasks (Corning Life Sciences, Corning, NY, USA). For neuronal differentiation they were cultured (4 days) in Neurobasal medium with 2 mM L-glutamine and B27 supplement (Gibco-BRL) and 100 ng/mL nerve growth factor (NGF) (2.5S; Roche Molecular Biochemicals, Indianapolis, IN, USA). Fresh NGF was supplied every other day (Wales et al. 2007). E18 rat primary hippocampal tissues were purchased from Neuromics (Edina, MN, USA), dissociated with papain [2 mg/mL (Sigma, St. Louis, MO, USA)] as per manufacturer’s instruction and plated at a density of 5 × 105 cells/dish on glass coverslips pre-coated with poly-L-Lysine (Sigma). They were maintained in Neurobasal medium + B27 supplement. Over 99% of the cells were neurons as determined by staining with βIIITubulin antibody. Organotypic hippocampal cultures (OHC) were cultured on porous membranes (0.4 μM, Millipore, Billerica, MA, USA) at the interface of 10% CO2 and culture medium (50% minimal essential medium, 25% Hank’s balanced salt solution, and 25% horse serum). After 5 days in culture, they were treated with the mitotic inhibitors cytosine-β-D-arabinofuranoside, uridine and 5-fluoro-2′-deoxyuridine (Sigma; 0.8 μg/mL) to inhibit glial cell growth. Primary microglia were prepared from cortices obtained from postnatal day 1–2 rat pups and plated at a density of 2 cortices/T-75 cm2 flask in a 10% CO2 atmosphere in DMEM with 1 mM sodium pyuruvate and 20% FBS. At 9 days in culture, the flasks were rotated at 180 rpm for 18 h and the resulting suspended cells were plated onto poly-L-Lysine coated flasks (microglia) as previously described (Laing and Aurelian 2008).
Antibodies and reagents
The generation and specificity of the rabbit ICP10 antibody were described. It recognizes an epitope located within amino acid residues 13–26 that are retained by the mutant ICP10 proteins in ΔRR and ΔPK (Smith et al. 1998; Perkins et al. 2002a; Gober et al. 2006; Golembewski et al. 2007) (Fig. S1). The following antibodies were purchased and used according to the manufacturer’s instructions: CD11b (Mac-1m chain-biotin conjugated; Leinco, St. Louis, MO, USA), neutralizing antibodies to FKN (ND50 1 μg/mL), VEGF (ND50 0.6 μg/mL), TNF-α (ND50 0.75 μg/mL), and IL-10 (ND50 2 μg/mL) and normal rabbit IgG (R&D Systems, Minneapolis, MN, USA), p20 fragment of activated caspase 3 (caspase 3p20), Akt, pAkt, (Cell Signaling Technologies, Beverly, MA, USA), extracellular signal-regulated kinase (ERK1/2) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), βIIITubulin, pERK1/2 (Promega, Madison, WI, USA), and NeuN (Millipore). Secondary antibodies include FITC-labeled anti-mouse IgG (BD Biosciences, San Jose, CA, USA), AlexaFluor 546 labeled anti-goat IgG, AlexaFluor 488, AlexaFluor 546, and Alexa-Fluor 350 labeled anti-rabbit IgG, (Molecular Probes, Eugene, OR, USA), Texas Red conjugated streptavidin, FITC conjugated streptavidin, and Texas Red labeled anti-mouse IgG (Vector, Burlingame, CA, USA). NMDA and MPP+ were purchased from Sigma-Aldrich. The mitogen activated protein kinase kinase (MEK) inhibitor U0126 was purchased from Promega and the phosphoinositide 3-kinase (PI3k) inhibitor LY294002 from Cell Signaling Technologies. Recombinant IL-10 and TNF-α were purchased from R&D Systems.
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence were done as previously described (Smith et al. 2000; Gober et al. 2006; Laing et al. 2006; Golembewski et al. 2007; Wales et al. 2007, 2008; Laing and Aurelian 2008). For immunoblotting, cells were lysed with radioimmunoprecipitation (RIPA) buffer; 20 mM Tris-HCl (pH 7.4), 0.15 mM NaCl, 1% Nonidet P-40 (Sigma), 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate] supplemented with protease and phosphatase inhibitor cocktails (Sigma) and proteins were resolved by SDS–polyacrylamide gel electrophoresis and transferred to Immobilon-P (Millipore). Non-specific binding was blocked by incubation [1 h, 25°C] in TNT buffer [0.01 M Tris-HCl (pH 7.4), 0.15 M NaCl, 0.05% Tween 20] containing 5% bovine serum albumin and the blots were exposed (overnight; 4°C) to primary antibodies followed by secondary antibodies conjugated to horseradish peroxidase (Cell Signaling). Quantitation was by densitometric scanning with the Bio-Rad GS-700 imaging densitometer (Bio-Rad, Hercules, CA, USA). For immunofluorescence, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 [in 0.1% sodium citrate buffer (2 min; 25°C)] and blocked with 5% normal goat serum and 5% bovine serum albumin (30 min; 25°C). They were incubated with primary antibody (18 h; 4°C), washed in phosphate-buffered saline (PBS) with 0.1% Tween 20 and exposed to fluorochrome-labeled secondary antibodies (1 h; 37°C). Slides were mounted in Vectashield with 4′,6′-diamidino-2-phenylindole (DAPI) (Vector) and visualized with an Olympus (Center Valley, PA, USA) BX50 fluorescent microscope. Staining cells were counted in five randomly selected fields (at least 250 cells/field) or five distinct areas of the striatum (22 μm2 each).
LacZ expression
ICP10PK positive cells in ΔRR infected OHC were identified by staining with the green fluorescent β-galactosidase substrate, C12-fluorescein di-β-D-galactopyranoside (C12FDG; Molecular Probes), according to manufacturer’s instructions. Staining cells were counted in three randomly selected microscopic fields (at least 200 cells) and the % positive cells was calculated relative to the total number of cells visualized by permeabilizing the culture with 5% Triton X-100 (30 s) followed by DAPI staining (Gober et al. 2006).
Ethidium homodimer staining and TUNEL
Dead cells were measured by staining with Ethidium homodimer (EtHD; Molecular Probes). The positive cells were counted in three randomly selected microscopic fields (at least 250 each) and the % calculated relative to DAPI staining cells, as described (Gober et al. 2006). The In situ Cell Death Detection kit (Roche) was used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, according to the manufacturers’ instructions. Neuroprotection was calculated as previously described (Gober et al. 2006) according to the formula: % protection = [(NMDA-(ΔRR-B)/NMDA] × 100; where NMDA is % EtHD+ or TUNEL+ cells in cultures given NMDA alone, ΔRR is % EtHD+ or TUNEL+ cells in cultures given conditioned media from ΔRR infected hippocampal cultures (NCMΔRR), and B is the % dead cells at the time of virus infection (background).
Ras activation
The Ras activation kit (Upstate Biotechnology, Temecula, CA, USA) was used according to manufacturer’s instructions. Briefly, cells (1 × 107) were resuspended in 1 mL lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal non-ionic detergent, 10 mM MgCl2, and 2% glycerol) with protease and phosphatase inhibitor cocktails (Sigma) and lysed by pipetting five times on ice. Cell debris was removed by centrifugation (5 min, 14 000 g, 4°C) and the supernatants were incubated (45 min at 4°C) with agarose beads (10 μg) bound to a glutathione S-transferase fusion protein that corresponds to the Raf-1 Ras binding domain (RBD). As a positive control, cell extracts were incubated (30 min; 30°C) with 100 μM guanosine 5′-O-[gamma-thio]triphosphate (GTPγS) prior to the Ras binding domain-agarose beads in order to simulate Ras activation. The beads were washed three times with lysis buffer, resuspended in 2× Laemmli buffer with 2 μM dithiothreitol and resolved by SDS–polyacrylamide gel electrophoresis (10% poly-acrylamide gels). After transfer to Immobilon-P membranes, immunoblotting was done with Ras monoclonal antibody provided by the manufacturer (0.05 μg/mL).
Cell-to-cell interaction assay
The assays schematically represented in Fig. S2 were used to examine neuron-neuron and bidirectional neuron-microglia communications. To generate neuronal conditioned media (NCM), hippocampal cultures were infected with ΔRR or ΔPK (moi = 5) or mock infected with PBS, washed extensively and re-incubated in fresh medium for 48 h. The NCM were collected at this time and cleared of cell debris by centrifugation (11 400 g; 30 min) and potentially remaining virus was inactivated by treatment with UV light (Sylvania G15 T8 bulb at a distance of 17 cm), as described (Perkins et al. 2002a; Laing et al. 2006). The NCM were named according to the infection regimen used to generate them, NCMmock, NCMΔRR and NCMΔPK for PBS, ΔRR and ΔPK, respectively. To measure neuron-neuron cross-talk, uninfected (naïve) hippocampal cultures were treated with NMDA (5 μM; 3 h), extensively washed and cultured (24 h) in Neurobasal medium + B27 containing NCM (1 : 1 dilution). They were examined for apoptosis by TUNEL or staining with antibody to activated caspase 3 (caspase 3p20) (Fig. S2a). To assay for bidirectional neuron-microglia cross-talk, primary microglia cultures were grown (48 h) in DMEM-10% FBS containing NCM (1 : 1 dilution) and their conditioned media [microglia conditioned media (MCM)] were cleared of cell debris by centrifugation (11 400 g; 30 min) and assayed for the ability to inhibit NMDA-induced apoptosis in uninfected hippocampal cultures (Fig. S2b).
ELISA
Conditioned media (NCM and MCM) were assayed for VEGF, FKN, TNF-α and IL-10 using ELISA kits, according to manufacturer’s instructions (R&D Systems) For factor neutralization, the conditioned media were incubated (1 h; 37°C) with the respective neutralizing antibodies at a concentration 10-fold higher than the ND50 (6, 10, 7.5, and 20 μg/mL for VEGF, FKN TNF-α and IL-10, respectively) or with normal IgG (10 μg/mL) used as control. Neutralization was confirmed by ELISA.
Animals
Adult (8 weeks old) 129S6 SV/EV male mice were obtained from Taconic (Germantown, NY, USA). They were housed at the University of Maryland, Baltimore on a 12 h light/dark cycle with food and water ad libitum. All procedures were performed in accordance with the Animal Care and Use Committee. 129S6 SV/EV mice were used for intrastriatal injection of NMDA (40.5 nmol/0.5 μL) or NMDA + ΔRR (2500 pfu) as previously described (Golembewski et al. 2007). Animals were killed 48 h later and the brains were removed for analysis.
Statistical analyses
Analysis of variance (ANOVA) was performed with Sigma Stat version 3.1 for Windows (Systat Software, Point Richmond, CA, USA).
Results
ΔRR protects uninfected neurons from excitotoxic injury through ICP10PK expression
ΔRR-infected (ICP10PK+) neurons survive various death signals both in culture and in animal models (Perkins et al. 2002a,b, 2003; Gober et al. 2006; Laing et al. 2006, 2008; Golembewski et al. 2007; Wales et al. 2007, 2008). Because neurons are known to constitutively release toxic or protective soluble factors depending on their health status (He and Sun 2007; Salmina 2009), we wanted to know whether the surviving ICP10PK+ neurons release factors that protect surrounding uninfected neurons from death stimuli (bystander activity). In a first series of experiments, OHC were infected with 105 pfu of ΔRR and treated with NMDA (50 μM) for 3 h. Forty-eight hours later, they were assayed for cell death by EtHD staining and for ICP10PK expression by staining with ICP10 antibody or the fluorescent LacZ substrate C12FDG (Gober et al. 2006). Mock and ΔPK-infected OHC were studied in parallel and served as controls. In the ΔRR-infected OHC, neuroprotection (calculated as described in Materials and Methods) was respectively 77.8 ± 2.6%, 71.7 ± 2.9% and 65.4 ± 3.1% for the CA3, CA1 and DG regions. These levels were significantly higher than the % ICP10PK+ cells [67.2 ± 4.8% (p < 0.01), 60.5 ± 5.2% (p < 0.001) and 25.9 ± 4.6% (p < 0.001) for the CA3, CA1 and DG, respectively], suggesting that ΔRR can protect uninfected neurons from NMDA-induced death (Fig. 1a). Protection is due to ICP10PK expression, because it was not seen for the ΔPK-infected OHC (5.2 ± 1.5%, 7.6 ± 2.7%, and 3.5 ± 2.9% neuroprotection for CA3, CA1, and DG, respectively) although the ICP10PK deleted protein was expressed equally well, as previously described (Gober et al. 2006).
Fig. 1.

ICP10PK-mediated neuroprotection includes a robust bystander component. (a) OHC mock infected with PBS or infected with ΔRR (105 pfu) were treated with NMDA (50 μM; 3 h) and assayed for ICP10PK expression by staining with C12FDG and for cell death by staining with EtHD. Cells were counted in five randomly selected fields (at least 250 cells/field) and the % staining cells was calculated for each region of the hippocampus (CA1, CA3, and DG). Results are expressed as mean % ICP10PK+ cells ± SD and % neuroprotection ± SD calculated as described in Materials and Methods. **p < 0.01, ***p < 0.001 neuroprotection vs. ICP10PK+ cells. (b) Brain sections from animals given intrastriatal injections of NMDA (40.5 nmol/0.5 μL) mixed with PBS or with ΔRR (2500 pfu) as previously described (Golembewski et al. 2007) were stained in double immunofluorescence with AlexaFluor 594-labeled antibody to ICP10PK and FITC-labeled antibody to NeuN, a neuronal marker. The mean number ICP10PK+/NeuN+ cells or NeuN+ cells ± SD in a 22 μM2 field was calculated relative to animals given PBS alone (mock-treated). ***p < 0.001 ICP10PK+/NeuN+ cells vs. total NeuN+ cells.
In a second series of experiments to examine whether ICP10PK can protect surrounding uninfected neurons, brain sections from animals given intrastriatal injections of NMDA together with ΔRR, were stained with antibodies to ICP10 and NeuN (neuronal marker) in double immunofluorescence. Consistent with our previous findings (Golembewski et al. 2007), NMDA caused significant neuronal loss (29.5 ± 8.3 and 73.1 ± 4.8 NeuN+ cells/22 μm2 in treated and untreated animals, respectively) that was counteracted by co-delivery of ΔRR (68.4 ± 6.5 NeuN+ cells/22 μm2). However, only one-half of the NeuN+ cells in the ΔRR-treated animals (37.2 ± 5.9 cells) co-stained with ICP10 antibody (Fig. 1b), supporting the interpretation that ICP10PK+ neurons protect surrounding uninfected neurons.
ICP10PK+ neurons release neuroprotective factors
To examine whether ICP10PK+ neurons release soluble factors that protect uninfected neurons, conditioned media from ΔRR-infected hippocampal cultures (NCMΔRR) were assayed for the ability to inhibit NMDA-induced apoptosis of uninfected cultures, as schematically represented in Fig. S2(a). Neuronal conditioned media from mock- (NCMmock) or ΔPK- (NCMΔPK) infected cultures served as controls. The % NMDA-induced apoptotic (TUNEL+) cells was significantly (p < 0.001) reduced by exposure to NCMΔRR, and neuroprotection was robust (36.4 ± 3.4%). NCMmock and NCMΔPK did not protect (Fig. 2a). Similar results were obtained when apoptosis was measured by caspase 3 activation, as determined by staining with antibody to the activated caspase 3p20 fragment (data not shown). The bystander activity of ICP10PK extends to other toxicity paradigms, as evidenced by the finding that NCM from ΔRR-infected neuronally differentiated PC12 cells protected uninfected PC12 cells from MPP+ (5 mM; 24 h) induced apoptosis (Wales et al. 2008), but protection was not seen for NCMmock or NCMΔPK (Fig. 2b). The data indicate that ICP10PK+ neurons produce soluble factors that protect uninfected neurons from various death stimuli.
Fig. 2.

ICP10PK+ neurons release soluble factors that protect uninfected neurons. (a) Primary hippocampal cultures were infected with ΔRR or ΔPK [multiplicity of infection (MOI) = 5]. Their conditioned media (NCMΔRR and NCMΔPK, respectively) were collected at 48 h post-infection (pi), cleared of remaining virus by UV treatment and mixed (1 : 1 ratio) with Neurobasal medium + B27. Uninfected hippocampal cultures treated with NMDA (5 μM; 3 h) were grown (24 h) in the absence or presence of the NCM containing media and assayed for apoptosis by TUNEL. Data are expressed as mean % TUNEL+ cells/total cell number (determined by DAPI staining) or % neuroprotection, calculated as described in Materials and Methods) ± SD. ***p < 0.001 vs. NMDA-treated cultures. (b) Neuronally differentiated PC12 cells were infected with ΔRR or ΔPK (MOI = 1) and the respective NCM (NCMΔRR and NCMΔPK) were collected 48 h later and mixed (1 : 1 ratio) with NGF (100 ng/mL) supplemented Neurobasal medium + B27. The NCM-containing media were used to culture (24 h) neuronally differentiated PC12 cells that had been treated with MPP+ (5 mM; 24 h) and the cultures were stained by TUNEL. Results are expressed as in (a). **p < 0.01 vs. MPP+ treated cultures.
Neuronal release of soluble protective factors depends on the activation of Ras signaling pathways
Having seen that ICP10PK+ neurons release neuroprotective factors, we wanted to know whether this is associated with activation of the Ras signaling pathways that had been previously implicated in the survival of ICP10PK+ neurons (Smith et al. 2000; Perkins et al. 2002a, 2003). Two series of experiments were done. First, ΔRR infected neuronally differentiated PC12 cells that produce soluble neuroprotective factors (Fig. 2b) were assayed for Ras activation as described in Materials and Methods. Mock- and ΔPK-infected PC12 cells that do not release neuroprotective factors were studied in parallel and served as controls. The levels of activated (GTP-bound) Ras were 4-fold higher in ΔRR- than mock- or ΔPK-infected PC12 cells (Fig. 3a), indicating that production of neuroprotective soluble factors is associated with Ras activation.
Fig. 3.

ICP10PK-induced release of soluble neuroprotective factors depends on the activation of Ras signaling pathways. (a) Neuronally differentiated PC12 cells were mock infected or infected with ΔRR or ΔPK (MOI = 1) and cell extracts were assayed for Ras activation as described in Materials and Methods. The data are presented as fold activation relative to mock-treated cells. The positive control (cont) is incubation with GTPγS provided by the manufacturer. (b) Primary hippocampal cultures were treated with NMDA (5 μM; 3 h), infected with ΔRR (MOI = 5) and cultured in the absence or presence of the MEK inhibitor U0126 (U0, 20 μM) or the PI3-K inhibitor LY294002 (LY, 100 μm). Cell extracts obtained at 48 h post-infection (p.i.) were immunoblotted with antibodies to activated (phosphorylated) AKT (pAKT) or ERK1/2 (pERK1/2), using antibodies to total AKT and total ERK1/2 as controls. Blots were stripped between each probing. (c) Data from (b) were analyzed by densitometric scanning and the results are expressed as the ratio of phosphorylated/total protein. (d) NCM collected from the cultures in (b) were mixed (1 : 1 ratio) with Neurobasal medium + B27 and used to culture uninfected hippocampal neurons that had been treated with NMDA. Twenty-four hours later, the cultures were assayed for cell death by TUNEL. Data are presented as mean % TUNEL+ cells ± SD. ***p < 0.001, *p < 0.05 compared to NCMPBS and ***p < 0.001 compared to NCMΔRR.
In the second series of experiments to examine the role of the Ras signaling pathways in the release of soluble neuroprotective factors, hippocampal cultures were mock-infected or infected with ΔRR in the presence or absence of the MEK inhibitor U0126 (20 μM) or the PI3-K inhibitor LY294002 (100 μM). Cell extracts collected at 48 h p.i. were immunoblotted with antibodies to phosphorylated (activated) ERK (pERK1/2) or Akt (pAkt) using antibodies to total ERK1/2 or Akt as controls, and the corresponding NCM (NCMmock and NCMΔRR) were assayed for their ability to inhibit NMDA-induced apoptosis of uninfected hippocampal cultures. The levels of pERK1/2 and pAkt were significantly higher in extracts from ΔRR than mock-infected cells (2.8 and 2.1-fold, respectively), an increase that was respectively inhibited by U0126 and LY294002 (Fig. 3b and c). Pathway activation is responsible for the release of soluble neuroprotective factors, because: (i) NCM generated by infection with ΔRR in the presence of U0126 or LY294002 (NCMΔRR+U0126 or NCMΔRR+LY294002) did not reduce the % NMDA-induced TUNEL+ cells in uninfected hippocampal cultures and (ii) pERK1/2 and pAkt were not increased in ΔPK-infected cultures and they did not release neuroprotective factors (Fig. 3d).
ΔRR infected neurons release increased levels of VEGF and FKN
Because neurons constitutively release VEGF and FKN and they were implicated in neuroprotection (Jin et al. 2000; Svensson et al. 2002; Mizuno et al. 2003; Limatola et al. 2005), we wanted to know whether NCMΔRR differs from the non-protective NCM in terms of the presence and/or levels of these factors. Consistent with previous reports, we found that NCMmock contained both VEGF and FKN (192.6 ± 11.2 and 404.1 ± 56.4 pg/mL, respectively), but their levels were 2- to 2.5-fold higher in NCMΔRR (396.6 ± 62.9 and 1025.75 ± 44.2 pg/mL for VEGF and FKN, respectively). The levels of VEGF and FKN in NCMΔPK, NCMΔRR+U0126 or NCMΔRR+LY294002 were similar to those seen in NCMmock (Fig. 4a and b), indicating that increased VEGF and FKN release depends on ICP10PK-induced MEK/ERK and/or PI3-K/Akt activation. Increased VEGF and FKN production/release was also seen in ΔRR treated animal models of neurodegeneration (Fig. S3).
Fig. 4.
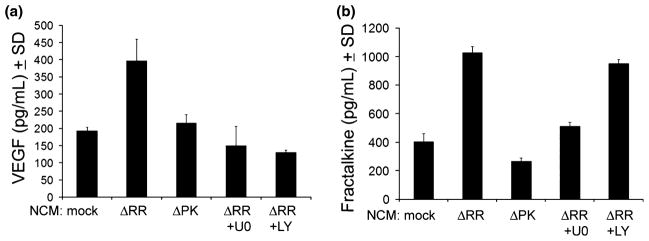
ICP10PK+ hippocampal cultures release VEGF and FKN. Primary hippocampal cultures were mock infected with PBS or infected with ΔPK or ΔRR (MOI = 5), the latter in the absence or presence of the MEK inhibitor U0126 (U0, 20 μM) or the PI3-K inhibitor LY294002 (LY, 100 μm) and the NCM collected at 48 h p.i. were assayed for VEGF (a) and FKN (b) by ELISA.
VEGF functions in neuron-neuron communication
To examine whether VEGF and/or FKN are responsible for the neuroprotective activity of NCMΔRR, the NCM were treated (1 h; 37°C) with neutralizing antibodies to VEGF (6 μg/mL) or FKN (10 μg/mL), and assayed for their ability to inhibit NMDA-induced apoptosis in uninfected hippocampal cultures. ELISA of the antibody-treated NCMΔRR confirmed VEGF and FKN neutralization. VEGF neutralization inhibited the ability of NCMΔRR to decrease the % NMDA-induced TUNEL+ cells (65.7 ± 2.7 and 60.2 ± 7.5 for untreated and VEGF neutralized NCMΔRR, respectively), but neuroprotection was not decreased by FKN neutralization (Fig. 5a). The data indicate that NCMΔRR-mediated neuroprotection includes neuron-neuron communication through increased VEGF release.
Fig. 5.

NCMΔRR functions through VEGF-mediated neuron-neuron cross-talk and microglia modulation. (a) NCM were generated from hippocampal cultures mock-infected with PBS (NCMmock) or infected with ΔRR (NCMΔRR). The NCMΔRR were treated (1 h; 37°C) with neutralizing antibody to VEGF (nVEGF Ab; 6 μg/mL) and/or neutralizing antibody to FKN (nFKN Ab; 10 μg/mL) and VEGF and FKN neutralization was confirmed by ELISA. The NCM were mixed (1 : 1 ratio) with Neurobasal medium + B27 and used to culture uninfected hippocampal neurons that had been treated with NMDA (5 μM; 3 h). Twenty-four hours later, the cultures were assayed for cell death by TUNEL. The data are presented as the mean % TUNEL+ cells ± SD. ***p < 0.001 relative to NCMmock. ***p < 0.001 relative to NCMΔRR. (b) Primary microglia were cultured in medium containing NCMmock, NCMΔRR or NCMΔPK (1 : 1 ratio) and the microglia conditioned media (MCM) were collected 48 h later. The MCM were mixed with Neurobasal medium + B27 (1 : 1 ratio) and used to culture uninfected primary hippocampal cultures that had been treated with NMDA (5 μM; 3 h). Twenty-four hours later, the hippocampal cultures were assayed for cell death by TUNEL. Data are mean % TUNEL+ cells ± SD. ***p < 0.001 relative to NMDA. (c) Primary microglia were cultured (48 h) with NCMΔRR, NCMΔPK or NCM from mock-infected and NMDA-treated (5 μM; 3 h) hippocampal cultures (NCMNMDA). The cells were extensively washed and mixed with uninfected primary hippocampal cultures that had been treated or not with NMDA (5 μM; 3 h). The mixed cultures were fixed 24 h later and stained in triple immunofluoresence with AlexaFluor 350 labeled βIII-tubulin antibody, AlexaFluor 488 labeled caspase 3p20 antibody (activated caspase 3), and Texas Red-labeled CD11b antibody (microglia marker). Turquoise staining represents caspase 3p20+ staining neurons. Data are presented as the mean % caspase 3p20+/βIII-tubulin+ cells ± SD. The % caspase 3p20+/CD11b+ cells was less than 4%.
NCMΔRR stimulates microglia to become neuroprotective
To examine whether microglia contribute to neuroprotection by NCMΔRR, primary microglia cultures were cultured in medium containing NCMΔRR and their conditioned media (MCM) were collected 48 h later and assayed for their ability to inhibit NMDA-induced apoptosis of uninfected hippocampal cultures as schematically represented in Fig. S2(b). NCMmock and NCMΔPK that do not contain high levels of FKN and NCMΔRR in which FKN was neutralized as described above, served as controls. The % NMDA-induced TUNEL+ cells (66.2 ± 2.3) was significantly decreased by MCM from microglia given NCMΔRR (21.0 ± 4.4%; p < 0.001), but not by MCM from microglia given NCMmock, NCMΔPK or NCMΔRR in which FKN was neutralized (Fig. 5a and b). Neuroprotection, calculated as described in Materials and Methods, was 61.3 ± 2.4%. This is an approximately 2-fold higher neuroprotection level than that seen for NCMΔRR (Fig. 2a), indicative of amplification through FKN-mediated bidirectional neuron-microglia cross-talk.
To verify that the neuroprotective potential of the MCM is not an artifact because of residual NCM in the MCM or the use of TUNEL to measure cell death, uninfected hippocampal cultures treated with NMDA were co-cultured (at a 1 : 10 ratio) with NCMΔRR-treated microglia that had been extensively washed before cell mixing and the mixed cultures were stained in double immunofluorescence with antibodies to βIII tubulin and caspase 3p20 (activated caspase 3). Supporting the conclusion that protection of uninfected neurons includes bidirectional neuron-microglia cross-talk, the % cells with co-localized staining (apoptotic neurons) was significantly (p < 0.001) lower in cultures containing NCMΔRR-treated microglia (27.3 ± 11.2%) than those in which the microglia had been treated with NCMmock or NCMΔPK (39.1 ± 7.9% and 41.2 ± 5.2%, respectively) (Fig. 5c).
FKN in NCMΔRR alters the balance of IL-10/TNF-α released by microglia
Having seen that microglia respond to NCMΔRR with the release of neuroprotective factors, we wanted to know whether this is because of FKN-mediated paracrine activity in microglia that express the FKN receptor (CX3R1). We focused on the release of inflammation-regulatory cytokines, because these are associated with neuronal survival or toxicity and they are not present in the NCM (data not shown). Specifically, we studied TNF-α, a pro-inflammatory cytokine that is released by activated microglia (Clausen et al. 2008) and is associated with neurodegeneration (Cacci et al. 2005; Laing et al. 2006; Golembewski et al. 2007; Clausen et al. 2008; Whitney et al. 2009) and IL-10 an anti-inflammatory cytokine that was shown to be neuroprotective (Milligan et al. 2008). We found that microglia cultured with NCMΔRR had higher levels of released IL-10 (153.8 ± 13.8 pg/mL) and lower levels of released TNF-α (27.6 ± 9.6 pg/mL) than those given NCMmock (2.2 ± 1.0 and 82.9 ± 18.9 pg/mL for IL-10 and TNF-α, respectively). Similar results were obtained for microglia given NCM that had been generated by infection with ΔRR in the presence of NMDA (NCMΔRR+NMDA), but the levels of released IL-10 were not increased in microglia given NCMΔPK (7.1 ± 1.4 pg/mL) or NCMNMDA (0.0 ± 0.0 pg/mL) while those of TNF-α were significantly increased (541.4 ± 17.1 and 551.9 ± 22.6 pg/mL for NCMΔPK and NCMNMDA, respectively) (Fig. 6a and b). We conclude that the altered release of IL-10 and TNF-α by microglia given NCMΔRR is due to FKN, because this alteration was not seen for microglia given NCMΔRR+U0126 (7.4 ± 3.1 and 450.8 ± 104.6 pg/mL for IL-10 and TNF-α, respectively) (Fig. 6a and b) or NCMΔRR in which FKN was neutralized (16.4 ± 1.9 and 393.8 ± 16.3 pg/mL for IL-10 and TNF-α, respectively), while it was still seen for microglia given NCMΔRR+LY29402 (139.3 ± 27.6 and 41.9 ± 11.2 pg/mL for IL-10 and TNF-α, respectively) (Fig. 6c and d). Collectively, the data indicate that the increased levels of FKN in NCMΔRR modulate microglia to release increased levels of IL-10 and decreased levels of TNF-α, thereby attenuating the neuroinflammatory activity of the extracellular milieu.
Fig. 6.

Microglia stimulation by NCMΔRR is through FKN-induced modulation of IL-10 and TNF-α release. NCM were generated from primary hippocampal cultures mock infected with PBS in the absence (NCMmock) or presence of NMDA (5 μM; 3 h) (NCMNMDA) and from hippocampal cultures infected with ΔRR in the absence (NCMΔRR) or presence of the MEK inhibitor U0126 (U0, 20 μM) (NCMΔRR+U0) or PI3-K inhibitor LY294002 (LY, 100 μm) (NCMΔRR+LY). NCM were also collected from hippocampal cultures infected with ΔRR and treated with NMDA (5 μM; 3 h) (NCMΔRR+NMDA). All NCM were mixed (1 : 1 ratio) with microglia growth medium and used to culture primary microglia. The microglia conditioned media (MCM) were collected 48 h later, and assayed for IL-10 (a) or TNF-α (b) by ELISA. (c,d) Primary microglia were cultured with NCMmock, NCMΔRR or NCMΔRR that had been treated (1 h; 37°C) with neutralizing antibodies to VEGF (nVEGF; 6 μg/mL) or FKN (nFKN; 10 μg/mL) and their MCM were assayed for IL-10 (c) or TNF-α (d) by ELISA.
FKN-modulated cytokine release defines microglia-mediated neuroprotection
Having seen that microglia given NCMΔRR release increased levels of IL-10, we wanted to know whether this altered cytokine balance contributes to the neuroprotective potential of the MCM. As expected, MCM from NCMΔRR-treated microglia reduced the % NMDA-induced TUNEL+ cells (61.4 ± 4.8 and 28.6 ± 1.4 for untreated and MCM-treated respectively), a reduction that was not seen for MCM from microglia cultured with NCMmock or NCMΔPK (65.6 ± 4.1% and 61.4 ± 4.8% TUNEL+ cells, respectively) (Fig. 7a). The neuroprotective potential of the NCMΔRR-induced MCM was equally reduced by neutralization of FKN or IL-10 (46.4 ± 1.9% and 47.3 ± 2.3% TUNEL+ cells, respectively). It was virtually abrogated by neutralization of both factors (54.9 ± 3.1% TUNEL+ cells) (Fig. 7b), indicating that FKN contributes to the neuroprotective activity of the MCM both through IL-10 induction and independently. We assume that the IL-10 independent contribution involves TNF-α decrease, because the neuroprotective activity of the NCMΔRR-induced MCM was significantly decreased by IL-10 neutralization and virtually abrogated by the addition of rTNF-α, while NCMΔPK-induced MCM acquired neuroprotective activity by TNF-α neutralization and further by the addition of rIL-10 (Fig. 7c). Collectively, the data indicate that ICP10PK uses a multi-targeted strategy to rescue neurons, underscoring its great advantage over single therapies.
Fig. 7.

The IL-10/TNF-α balance in MCM from NCMΔRR-stimulated microglia defines their neuroprotective potential. (a) MCM generated from microglia stimulated with NCMmock, NCMΔRR or NCMΔPK were mixed with Neurobasal medium + B27 and used to culture uninfected primary hippocampal cultures that had been treated or not with NMDA (5 μM; 3 h). Twenty-four hours later the hippocampal cultures were assayed for cell death by TUNEL. Data are mean % TUNEL+ cells ± SD and % neuroprotection calculated as described in Materials and Methods. ***p > 0.001 vs. NCMmock-induced MCM. (b) MCM from microglia stimulated with NCMΔRR as in (a) were treated (1 h; 37°C) with neutralizing antibodies to VEGF (nVEGF; 6 μg/mL), FKN (nFKN; 10 μg/mL), and/or IL-10 (nIL-10; 20 μg/mL) and assayed for their ability to protect uninfected primary hippocampal cultures from NMDA-induced apoptosis (determined by TUNEL 24 h later). Normal IgG (10 μg/mL) was used as control for the neutralizing antibodies. ***p < 0.001 vs. NCMΔRR-induced MCM. (c) Uninfected hippocampal cultures treated with NMDA (5 μM; 3 h) were grown (24 h) with various MCM and assayed by TUNEL. Untreated cultures (Panel 1) and cultures treated with NMDA and grown in the absence of MCM (Panel 2) served as controls. The MCM were from microglia given NCMΔRR (Panel 3) or NCMΔPK (Panel 4). Panel 5 represents MCM from microglia given NCMΔRR in which IL-10 was neutralized as in (b) and rTNF-α (500 pg/mL) was added. Panel 6 represents MCM from microglia given NCMΔPK in which TNF-α was neutralized (1 h; 37°C; 7.5 μg/mL) and rIL-10 (150 pg/mL) was added. Results are expressed as mean %TUNEL cells ± SD. ***p < 0.001 vs. NMDA treated cultures. ***p < 0.001 vs. MCM from microglia given NCMΔRR.
Discussion
Excessive NMDA receptor activation, a process known as excitotoxicity, causes neuronal injury/apoptosis in stroke victims, limiting a patient’s cognition, communication skills, mobility and independence. The development of therapeutic strategies to attenuate the neurological symptoms of acute injury is the subject of major research interest. However, effective treatments remain elusive, due at least in part, to the complex cell-to-cell interactions that regulate neuron cell life/death decisions. Neuronal and glial cell dysfunction can drastically alter the neuronal milieu from one that supports neuronal function to one that initiates or exacerbates neuronal death (He and Sun 2007; Salmina 2009). Microglia are particularly important in this context, acting as pathology sensors that are activated by neuronal inputs and release chemical mediators that help the CNS recover from injury or cause a severe self-propagating inflammatory cycle.
Gene therapy is considered a promising strategy to rescue neurons from death stimuli, thereby attenuating the neurological symptoms of acute injury. However, clinical challenges include identification of the target genes and the modulation of cell-to-cell communications such as to create a self-propagating input-output cycle of chemical mediators that inhibit the progression of acute and chronic neurodegeneration. Development of multiple-target strategies to rescue neurons, including those that surround the treated ones, is particularly desirable. We report for the first time that the HSV-2 gene ICP10PK delivered by the growth compromised vector ΔRR has robust bystander activity that is mediated by increased release of VEGF and FKN from ICP10PK+ neurons through activation of Ras-dependent signaling pathways. The higher levels of released VEGF and FKN contribute to neuroprotection via neuron-neuron and bidirectional neuron-microglia communication, the latter involving FKN-induced release of IL-10 and decreased release of TNF-α (Fig. 8). The following comments seem pertinent with respect to these findings.
Fig. 8.

Schematic representation of ICP10PK-mediated protection of uninfected neurons. ICP10PK infection of primary hippocampal cultures activates Ras and its downstream signaling pathways MEK/ERK and PI3-K/Akt. Activation of both pathways stimulates increased release of VEGF; activation of the MEK/ERK pathway also stimulates increased release of FKN. VEGF protects hippocampal cultures from NMDA-induced apoptosis through direct neuron-neuron cross-talk. FKN has paracrine activity in microglia, inducing them to release increased levels of IL-10 and decreased levels of TNF-α. This altered cytokine balance protects uninfected hippocampal cultures from NMDA-induced apoptosis (bidirectional neuron-microglia cross-talk).
The construction and properties of the ΔRR vector for ICP10PK and the ICP10PK deleted control vector ΔPK were previously described (Smith et al. 1998, 2000; Gober et al. 2006). Both are growth compromised and are not toxic in the CNS, but only ΔRR protects infected neurons from caspase-dependent and independent programmed cell death. Protection is via ICP10PK-mediated activation of Ras signaling pathways and the resulting modulation of apoptosis-regulatory proteins (Perkins et al. 2002a,b, 2003; Gober et al. 2006; Laing et al. 2006, 2008; Golembewski et al. 2007; Wales et al. 2007, 2008). To activate Ras, ICP10PK binds the GDP/GTP exchange factor SOS (son of sevenless) (complexed to the adaptor protein Grb2) and phosphorylates (and, thereby, inhibits) the negative Ras regulator Ras-GAP (Smith et al. 1994, 2000; Nelson et al. 1996). The current studies follow on previous findings that the % hippocampal cells protected from HSV-1 induced apoptosis by ICP10PK transfection includes a robust bystander component (Perkins et al. 2003), and were designed to examine the mechanism of such activity.
We found that both in OHC and in animal models, the % hippocampal neurons that survived NMDA-induced apoptosis after infection with ΔRR (measured by inhibition of both TUNEL and caspase 3 activation) was significantly (p < 0.01 to p < 0.001) higher than that expressing ICP10PK, and similar results were obtained in the MPP+ paradigm of neurodegeneration. Protection of uninfected neurons is mediated by soluble factors released by the ΔRR-infected neurons through ICP10PK-mediated activation of the Ras-dependent signaling pathways MEK/ERK and PI3-K/Akt. Indeed: (i) ΔRR-infected neuronal cultures had high levels of activated Ras (Ras-GTP), ERK1/2 (pERK1/2) and Akt (pAkt) and their NCM (NCMΔRR) contained neuroprotective factors, and (ii) both pathway activation and factors release were inhibited by the MEK inhibitor U0126 and the PI3-K inhibitor LY29402. The Ras signaling pathways were not activated in ΔPK-infected hippocampal cultures that did not release soluble neuroprotective factors.
While uninfected neurons constitutively release VEGF and FKN, the levels of these factors were significantly increased in NCMΔRR and their neutralization interfered with the protection of uninfected neurons. The increased release of VEGF was inhibited by ΔRR infection in the presence of U0126 or LY29402, indicating that it was both MEK/ERK and PI3-K/Akt-dependent. By contrast, the increased release of FKN from ΔRR-infected hippocampal cultures was only inhibited by U0126, indicating that it was only MEK/ERK-dependent. At present, we still do not know whether the higher levels of VEGF and FKN released by ΔRR-infected hippocampal cultures reflect increased transcription and/or facilitated release. Activator protein-1 transcription factors are up-regulated by ICP10PK through activation of the MEK/ERK and PI3-K/Akt pathways (Smith et al. 2000; Perkins et al. 2002a, 2003; Gober et al. 2005; Laing et al. 2008), potentially contributing to VEGF expression (Berra et al. 2000). On the other hand, the specificity protein 1 transcription factor, which regulates expression of the A Disintegrin And Metalloprotease protease that facilitates FKN release from the neuronal membrane (Biber et al. 2008), is downstream of MEK/ERK activation (Kuwahara et al. 2007) potentially contributing to FKN release. Ongoing studies are designed to address this question. Notwithstanding, the finding that VEGF neutralization significantly reduced the ability of NCMΔRR to inhibit NMDA-induced apoptosis of uninfected hippocampal cultures, indicates that the increased levels of VEGF released by ΔRR-infected neurons function through direct neuron-neuron communication. Supporting this interpretation, NMDA-induced toxicity was not reduced by NCMΔRR+U0126 or NCMΔRR+LY29402 that did not contain increased levels of VEGF.
By contrast to VEGF, the increased levels of FKN in NCMΔRR contributed to the protection of uninfected neurons through regulation of bidirectional neuron-microglia cross-talk, providing a 2-fold amplification of the neuroprotective potential seen for VEGF. Indeed: (i) NCMΔRR stimulated primary microglia cultures to release soluble neuroprotective factors, an activity that was lost upon FKN neutralization, and (ii) microglia given NCMΔRR+U0126 that did not contain increased levels of FKN did not release neuroprotective factors but these were released by microglia given NCMΔRR+LY29402 that were positive for increased levels of FKN. Neuroprotection by MCM from microglia given NCMΔRR is not an artifact resulting from residual NCM levels, because neuroprotection was also seen in mixed cultures in which the NCMΔRR-treated microglia were extensively washed in order to remove the NCM before they were mixed with the hippocampal cultures.
Significantly, NCM that do not protect hippocampal cultures from death stimuli, directly or through bidirectional neuron-microglia cross-talk, contain low levels of VEGF and FKN, suggesting that the neuroprotective activity of VEGF and FKN is dose-dependent. Increased levels of VEGF and FKN are likely to have better access to the receptors, thereby providing autocrine and paracrine activities in the local microenvironment. However, the minimal dose that is neuroprotective is unknown. Also, we cannot exclude the possibility that VEGF and FKN produced in response to ICP10PK-mediated activation of the Ras signaling pathways facilitates non-synaptic communication (Adler et al. 2006) for example by causing paracrine neurotransmission (Bunin and Wightman 1999) and/or by activating receptors located outside of the synapse (Harris and Pettit 2008). Presumably, the ability of ICP10PK to facilitate increased release of both of these neuroprotective factors contributes to its robust bystander activity for uninfected surrounding neurons. In this context, it is important to point out that VEGF and FKN also seem to contribute to the ICP10PK-mediated protection of uninfected neurons in animal models, as evidenced by the findings that: (i) the % VEGF+ motor neurons in spinal cords from ΔRR-treated G93A SOD1 rats, and the % FKN+ neurons in brain sections from ΔRR treated kainic acid-exposed animals were higher than those seen in mock-treated animals, and (ii) the % ICP10PK+ neurons were significantly lower than those positive for VEGF or FKN.
How do microglia contribute to the protection of uninfected neurons? Our data indicate that microglia are stimulated to produce neuroprotective factors in response to NCMΔRR through FKN-mediated paracrine effects that include increased release of IL-10 and decreased release of TNF-α. The involvement of the microglia is likely related to the preferential expression of the FKN receptor in these cells (Tarozzo et al. 2003). However, to the extent of our knowledge, this is the first report that FKN can stimulate microglia to produce increased levels of the anti-inflammatory cytokine IL-10. The beneficial effects of IL-10 could be because of direct neuronal effects, for example by increasing survival (Froen et al. 2002; Kremlev and Palmer 2005) or inhibiting apoptosis (Bachis et al. 2001), or/and it could be because of an indirect effect that involves reduction of the oxidative stress mediated by microglia deactivation. However, the neuroprotective activity of the MCM from NCMΔRR-treated microglia was totally lost only when both IL-10 and FKN were neutralized, suggesting that FKN has an additional neuroprotective activity, other than increased IL-10 release. We assume that this independent activity involves decreased release of TNF-α, because: (i) TNF-α levels were lower in MCM from microglia given NCMΔRR, than NCMmock, NCMΔPK, or NCMΔRR+U0126, and (ii) TNF-α levels were significantly increased when the microglia were given NCMΔRR treated with FKN (but not VEGF) neutralizing antibody, or NCMNMDA.
The conclusion that the balance of IL-10/TNF-α released by the microglia plays a determinant role in the protection of uninfected neurons is supported by the finding that the neuroprotective potential of the MCM from NCMΔRR-treated microglia was decreased by IL-10 neutralization and further by addition of rTNF-α, while the opposite was true for MCM from NCMΔPK-treated microglia, the neuroprotective activity of which was increased by TNF-α neutralization and further by the addition of rIL-10. It is consistent with previous findings that overproduction of proinflammatory cytokines by microglia contributes to the pathophysiologic changes seen in neurologic disorders, and microglia pre-treatment with IL-10 is effective in blocking lipopolysaccharide-mediated production of TNF-α (Kim and de Vellis 2005). The mechanism of ICP10PK mediated protection of uninfected neurons is schematically represented in Fig. 8. Preliminary data suggest that the altered IL-10/TNF-α balance functions in neuroprotection through modulation of nuclear factor kappa-light-chain-enhancer of activated B cells p65 nuclear translocation (Hunot et al. 1997), but final conclusions must await the results of ongoing studies to address this question.
Collectively, our data indicate that neurons surviving apoptosis through expression of ΔRR-delivered ICP10PK, release increased levels of VEGF and FKN that protect uninfected neurons from apoptosis both through neuron-neuron and bidirectional neuron-microglia communications. The latter involves increased release of IL-10 and decreased release of TNF-α by the FKN-treated microglia. Although VEGF, FKN and IL-10 were previously shown to have neuroprotective potential, their use as independent therapies is limited by the finding that they can also contribute to neurotoxicity. Indeed, the neuroprotective potential of endogenous VEGF is controversial (Ferrari et al. 2006; Nicoletti et al. 2008; Benton et al. 2009) and exogenously delivered VEGF caused a potent and therapeutically undesirable immune response, was trapped by circulating receptor and did not cross the blood-brain barrier (Storkebaum et al. 2005). Concerns were also raised by the association of microglia-derived FKN with inflammation-related neuropathic pain (Milligan et al. 2008) and cerebral ischemia (Dénes et al. 2008), as well as the apparent association of IL-10 (Rentzos et al. 2009) and FKN-induced microglial activation (Shan et al. 2009) with the development of Parkinson’s disease. Moreover, these factors do not directly address potential problems related to residual activity of pro-inflammatory factors, such as TNF-α, the levels of which are increased by neurotoxic stimuli. Our data show that instead of delivering these molecules independently to the brain extracellular space as where they may have undesirable effects, we can use ΔRR to cause their release in a concerted and appropriately balanced pattern that restores homeostasis. We suggest that ΔRR-delivered ICP10PK is a multiple-targeted strategy to rescue neurons. It has the distinct advantage that in addition to protecting the infected neurons, it modulates them to release neuroprotective soluble factors in a balanced proportion such as to create a self-propagating cycle of neuronal inputs and release of chemical mediators that inhibit the progression of acute and chronic neurodegeneration through protection of uninfected neurons.
Supplementary Material
Acknowledgments
These studies were supported by NINDS, National Institutes of Health (NIH) public health service grant NS45169. We thank Dr. Edna Pereira for providing the organotypic hippocampal cultures.
Abbreviations used
- DAPI
4′6′-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- ERK
extracellular regulated kinase
- EtHD
ethidium homodimer
- FBS
fetal bovine serum
- FKN
fractalkine
- MCM
microglia conditioned media
- MEK
mitogen activated protein kinase kinase
- NCM
neuronal conditioned media
- OHC
organotypic hippocampal cultures
- PBS
phosphate-buffered saline
- PI3-K
phosphoinositide 3-kinase
- PK
protein kinase
- RR
ribonucleotide reducatse
- SDS
sodium dodecyl sulfate
- TNF-α
Tumor necrosis factor
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- VEGF
vascular endothelial growth factor
Footnotes
The authors declare no conflict of interests.
Additional Supporting Information may be found in the online version of this article:
Figure S1. Schematic representation of the ICP10 proteins in the virus mutants ΔRR and ΔPK.
Figure S2. Schematic representation of cell-to-cell communication assay.
Figure S3. VEGF and FKN are expressed by ΔRR-infected neurons in vivo.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Adler M, Geller E, Chen X, Rogers T. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2006;7:E865–E870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Colangelo AM, Vicini S, Doe PP, De Bernardi MA, Brooker G, Mocchetti I. Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J Neurosci. 2001;21:3104–3112. doi: 10.1523/JNEUROSCI.21-09-03104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton R, Maddie M, Gruenthal M, Hagg T, Whittemore S. Neutralizing endogenous VEGF following traumatic spinal cord injury modulates microvascular plasticity but not tissue sparing or functional recovery. Curr Neurovasc Res. 2009;6:124–131. doi: 10.2174/156720209788185678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E, Pagès G, Pouysségur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- Biber K, Vineta J, Boddeke H. Neuron-microglia signaling: chemokines as versatile messengers. J Neuoimmunol. 2008;198:69–74. doi: 10.1016/j.jneuroim.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Bunin M, Wightman R. Paracrine neurotransmission in the CNS: involvement of 5-HT. Trends Neurosci. 1999;22:377–382. doi: 10.1016/s0166-2236(99)01410-1. [DOI] [PubMed] [Google Scholar]
- Cacci E, Claasen J, Kokaia Z. Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res. 2005;80:789–797. doi: 10.1002/jnr.20531. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen B, Lambertsen K, Babcock A, Holm T, Dagnaes-Hansen F, Finsen B. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation. 2008;5:46. doi: 10.1186/1742-2094-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács K. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc Natl Acad Sci USA. 2006;103:17260–17265. doi: 10.1073/pnas.0605556103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstreuter F, Lucius R, Mentlein R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J Neuroimmunol. 2002;132:93–98. doi: 10.1016/s0165-5728(02)00315-6. [DOI] [PubMed] [Google Scholar]
- Froen JF, Munkeby BH, Stray-Pedersen B, Saugstad OD. Interleukin-10 reverses acute detrimental effects of endotoxin-induced inflammation on perinatal cerebral hypoxia-ischemia. Brain Res. 2002;942:87–94. doi: 10.1016/s0006-8993(02)02700-2. [DOI] [PubMed] [Google Scholar]
- Gober MD, Wales SQ, Hunter JC, Sharma BK, Aurelian L. Stress up-regulates neuronal expression of the herpes simplex virus type 2 large subunit of ribonucleotide reductase (R1; ICP10) by activating activator protein 1. J NeuroVirol. 2005;11:329–336. doi: 10.1080/13550280591002423. [DOI] [PubMed] [Google Scholar]
- Gober MD, Laing JM, Thompson S, Aurelian L. The growth compromised HSV-2 mutant DeltaRR prevents kainic acid-induced apoptosis and loss of function in organotypic hippocampal cultures. Brain Res. 2006;1119:26–39. doi: 10.1016/j.brainres.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golembewski E, Wales S, Aurelian L, Yarowsky P. The HSV-2 protein ICP10PK prevents neuronal apoptosis and loss of function in an in vivo model of neurodegeneration associated with glutamate excitotoxicity. Exp Neurol. 2007;203:381–393. doi: 10.1016/j.expneurol.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes E, Papa L, Hao T, Rockwell P. The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Mol Cell Biochem. 2007;205:179–190. doi: 10.1007/s11010-007-9542-2. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Recruiting extrasynaptic NMDA receptors augments synaptic signaling. J Neurophysiol. 2008;99:524–533. doi: 10.1152/jn.01169.2007. [DOI] [PubMed] [Google Scholar]
- He F, Sun Y. Glial cells more than support cells? Int J Biochem Cell Biol. 2007;39:661–665. doi: 10.1016/j.biocel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci USA. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Mao X, Greenberg D. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Kremlev SG, Palmer C. Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J Neuroimmunol. 2005;162:71–80. doi: 10.1016/j.jneuroim.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kuwahara I, Lillehoj EP, Koga T, Isohama Y, Miyata T, Kim KC. The signaling pathway involved in neutrophil elastase stimulated MUC1 transcription. Am J Respir Cell Mol Biol. 2007;37:691–698. doi: 10.1165/rcmb.2007-0072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing JM, Aurelian L. DeltaRR vaccination protects from KA-induced seizures and neuronal loss through ICP10PK-mediated modulation of the neuronal-microglial axis. Genet Vaccines Ther. 2008;6:1. doi: 10.1186/1479-0556-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing JM, Gober MD, Golembewski EK, Thompson S, Gyure K, Yarowsky P, Aurelian L. Intranasal administration of the growth-compromised HSV-2 vector DeltaRR prevents kainate-induced seizures and neuronal loss in rats and mice. Mol Ther. 2006;13:870–881. doi: 10.1016/j.ymthe.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing JM, Golembewski EK, Wales SQ, Liu J, Jafri M, Yarowsky P, Aurelian L. Growth-compromised HSV2 vector [delta]RR protects from N-methyl-D-Aspartate- induced neuronal degeneration through redundant activation of the MEK/ERK and PI3K/Akt survival pathways, either one of which overrides apoptotic cascades. J Neuro Res. 2008;86:378–391. doi: 10.1002/jnr.21486. [DOI] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Watkins LR. Glia in pathological pain: a role for fractalkine. J Neuroimmunol. 2008;198:113–120. doi: 10.1016/j.jneuroim.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L, Ajmone-Cat MA, De Berardinis MA, De Simone R. Microglial activation in chronic neurodegenerative diseases: roles of apoptotic neurons and chronic stimulation. Brain Res Rev. 2005;48:251–256. doi: 10.1016/j.brainresrev.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Functional roles of microglia in the central nervous system. Hum Cell. 1998;11:141–155. [PubMed] [Google Scholar]
- Nelson J, Zhu J, Smith C, Kulka M, Aurelian L. ATP and SH3 binding sites in the protein kinase of the large subunit of herpes simplex virus type 2of ribonucleotide reductase (ICP10) J Biol Chem. 1996;271:17021–17027. doi: 10.1074/jbc.271.29.17021. [DOI] [PubMed] [Google Scholar]
- Nicoletti J, Shah S, McCloskey D, Goodman J, Elkady A, Atassi H, Hylton D, Rudge J, Scharfman H, Croll S. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience. 2008;151:232–241. doi: 10.1061/j.neuroscience.2007.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D, Pereira EFR, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol. 2002a;76:1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D, Yu Y, Bambrick L, Yarowsky P, Aurelian L. Expression of herpes simplex virus type 2 protein ICP10 PK rescues neurons from apoptosis due to serum deprivation or genetic defects. Exp Neurol. 2002b;174:118–122. doi: 10.1006/exnr.2001.7849. [DOI] [PubMed] [Google Scholar]
- Perkins D, Pereira EFR, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein bag-1. J Virol. 2003;77:1292–1305. doi: 10.1128/JVI.77.2.1292-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzos M, Nikolaou C, Andreadou E, Paraskevas GP, Rombos A, Zoga M, Tsoutsou A, Boufidou F, Kapaki E, Vassilopoulos D. Circulating interleukin-10 and interleukin-12 in Parkinson’s disease. Acta Neurol Scand. 2009;119:332–337. doi: 10.1111/j.1600-0404.2008.01103.x. [DOI] [PubMed] [Google Scholar]
- Salmina A. Neuron-glia interactions as therapeutic targets in neurodegeneration. J Alzheimers Dis. 2009;16:485–502. doi: 10.3233/JAD-2009-0988. [DOI] [PubMed] [Google Scholar]
- Schiera G, Proia P, Alberti C, Mineo M, Savettieri G, Di Liegro I. Neurons produce FGF2 and VEGF and secrete them at least in part by shedding extracellular vesicles. J Cell Mol Med. 2007;11:1384–1394. doi: 10.1111/j.1582-4934.2007.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S, Hong-Min T, Yi F, et al. NEW evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson’s disease. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.03.004. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Sköld M, Kanje M. Vascular endothelial growth factor in central nervous system injuries – a vascular growth factor getting nervous? Curr Neurovasc Res. 2008;5:246–259. doi: 10.2174/156720208786413451. [DOI] [PubMed] [Google Scholar]
- Smith CC, Luo JH, Hunter JC, Ordonez JV, Aurelian L. The transmembrane domain of the large subunit of HSV-2 ribonucleotide reductase (ICP10) is required for protein kinase activity and transformation-related signaling pathways that result in ras activation. Virology. 1994;200:598–612. doi: 10.1006/viro.1994.1223. [DOI] [PubMed] [Google Scholar]
- Smith CC, Peng T, Kulka M, Aurelian L. The PK domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is required for immediate-early gene expression and virus growth. J Virol. 1998;72:9131–9141. doi: 10.1128/jvi.72.11.9131-9141.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Nelson J, Aurelian L, Gober M, Goswami B. Ras-GAP binding and phosphorylation by herpes simplex virus type 2 RR1 PK (ICP10) and activation of the Ras/MEK/MAPK mitogenic pathway are required for timely onset of virus growth. J Virol. 2000;74:10417–10429. doi: 10.1128/jvi.74.22.10417-10429.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Svensson B, Peters M, Konig HG, Poppe M, Levkau B, Rothermundt M, Arolt V, Kogel D, Prehn JHM. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J Cereb Blood Flow Metab. 2002;22:1170–1175. doi: 10.1097/01.wcb.0000037988.07114.98. [DOI] [PubMed] [Google Scholar]
- Tarozzo G, Bortolazzi S, Crochemore S, Chen S, Lira A, Abrams J, Beltramo M. Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res. 2003;73:81–88. doi: 10.1002/jnr.10645. [DOI] [PubMed] [Google Scholar]
- Wales S, Li B, Laing J, Aurelian L. The herpes simplex virus type 2 gene ICP10PK protects from apoptosis caused by nerve growth deprevation through inhibtion of caspase-3 activation and XIAP-upregulation. J Neurochem. 2007;103:365–379. doi: 10.1111/j.1471-4159.2007.04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wales S, Laing J, Chen L, Aurelian L. ICP10PK inhibits calpain-dependent release of apoptosis-inducing factor and programmed cell death in response to the toxin MPP+ Gene Ther. 2008;15:1397–1409. doi: 10.1038/gt.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney N, Eidem T, Peng H, Huang Y, Zheng J. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.