

INTRODUCTION
The Biopharmaceutics Classification System (BCS), based on aqueous solubility and intestinal permeability, has been widely used to predict drug absorption during pharmaceutical development (1). Recently, the system has also been employed in regulatory practice to determine if waiver of in vivo bioavailability or bioequivalence studies (i.e., biowaivers) can be granted for an immediate-release solid oral dosage form (2). In this regard, the US Food and Drug Administration (FDA) currently allows biowaivers for BCS class I (highly soluble and highly permeable) drugs formulated in immediate release, rapidly dissolving drug products (2). Since the publication of the FDA Guidance on BCS in 2000, however, there has been continued interest in the possible extension of BCS-based biowaivers, particularly for class III drugs that exhibit high solubility and low permeability (3–5).
One of the major challenges to the allowance of biowaivers for BCS class III drugs is related to the potential effects of excipients on drug absorption (6). Historically, excipients were considered inert substances that could be used mainly in the manufacture of drug products as diluents, fillers, binders, lubricants, coatings, solvents, and dyes (7). However, with the advances in pharmaceutical science, some “active” excipients have been found to be capable of influencing drug absorption or bioavailability through a variety of mechanisms, such as modification in solubility/dissolution, change in intestinal permeability (including transporters), and modulation of gastrointestinal (GI) motility (8–20). To further investigate the effects of “active” excipients on the absorption of BCS class III drugs, this paper examines those that may modulate GI motility and affect the transit time of drugs in the gut.
In a recent FDA contract study, sorbitol was shown to decrease ranitidine absorption, which was attributed to an increased GI fluid volume from the osmotic load of sorbitol, resulting in enhanced GI motility and reduced ranitidine transit time in the small intestine, a primary absorption site for the drug (19). Similarly, mannitol (an isomer of sorbitol) was reported to decrease the bioavailability of cimetidine in solution or as chewable tablets (10). Both ranitidine and cimetidine are BCS class III drugs that exhibit site-dependent absorption characteristics (19). Polyethylene glycol (PEG) 400, a commonly used excipient for enhancing drug solubility, was also found to accelerate small intestine transit and adversely influence the absorption of ranitidine (15,16,20). It is noted that all of these excipients (sorbitol, mannitol, and PEG 400) are osmotically active at the amounts relevant to those used in pharmaceutical formulations (21).
From a regulatory perspective, to extend BCS-based biowaivers or allow biowaivers for minor changes in manufacturing and/or formulation, it is essential to know the threshold level of an “active” excipient incorporated in the formulation that would influence the bioavailability of the drug and/or change the outcome of bioequivalence. In the case of osmotically “active” excipients, sorbitol has been shown to decrease the bioavailability of ranitidine in a linear dose–response manner (19). However, the influence of PEG 400 on ranitidine absorption was reported to be concentration dependent (16). The aims of this article were to (a) review the dose–response relationship of various osmotically active excipients on drug absorption, (b) determine the threshold levels of these excipients in affecting bioavailability or bioequivalence, and (c) discuss the regulatory implications of using these excipients in the formulation with a poorly permeable drug.
COLLIGATIVE PROPERTY OF OSMOTICALLY ACTIVE EXCIPIENTS
The effect of an osmotically active excipient on drug absorption has been hypothesized to come from the osmotic pressure exerted by the excipient that accelerates small intestine transit, thereby decreasing drug absorption time and reducing its bioavailability (9,16,19). If this theory is true, the direct (osmotic) effect of such an excipient on small intestine transit time should be a function of the number of molecules in the solution, i.e., a colligative property, regardless of the type of solute (i.e., excipient) present in the medium. In turn, the extent of reduction in drug absorption, which is secondary to the acceleration of intestinal transit, should be proportional to the osmotic potential of the excipient solution, again depending on the number of the molecules dissolved in vivo.
To verify the colligative property of osmotically active excipients, we conducted a search in the scientific literature for in vivo investigations that studied the osmotic effect of such excipients on GI motility. Two studies were found; one was done with mannitol and the other with PEG 400 (9,16). In both studies, the osmotic effect of the “active” excipient was demonstrated by the reduction in small intestinal transit time, which was measured using gamma scintigraphy. To express the osmotic potential of these excipients, we computed the ratio of each excipient’s mass (weight) to its molecular weight, assuming a constant fluid volume at the absorption site. The assumption was made based on the considerations that the GI tract is an open dynamic system, and it is difficult to estimate the fluid volume at the absorption site although a fixed amount of water was generally given with the drug in the study. Using the GI transit time data obtained from the two literature studies, we plotted the resultant osmotic potential versus small intestinal transit time (percent control). As shown in Fig. 1, a linear relationship exists between the small intestinal transit time and osmotic potential of both mannitol and PEG 400. All the data points fell on the same straight line regardless of the type of excipient present, demonstrating the colligative property of these solutions (Fig. 1). In contrast, such linear relationship was not found when the small intestinal transit time was plotted against excipient weight (Fig. 2)
Fig. 1.
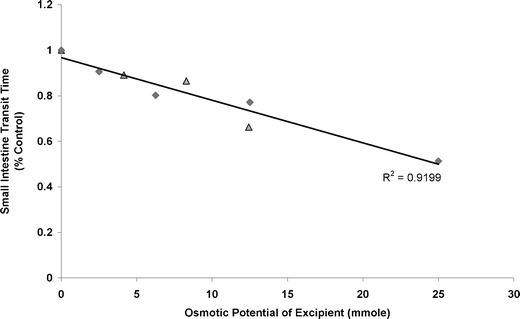
Relationship of small intestine transit time and osmotic potential of excipients, mannitol (triangle) and PEG 400 (diamond)
Fig. 2.
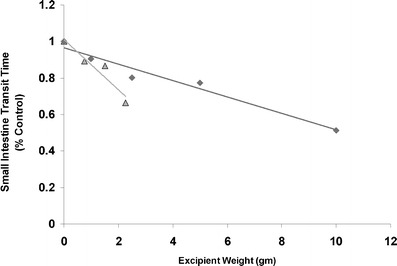
Relationship of small intestine transit time and weight of excipients, mannitol (triangle) and PEG 400 (diamond)
DOSE–RESPONSE RELATIONSHIP OF OSMOTICALLY ACTIVE EXCIPIENTS
Our literature search has revealed three types of excipients that are osmotically active and may affect drug absorption in the GI tract when incorporated in drug products. These excipients mainly consist of sugar alcohols, polyethylene glycols (PEG), and sodium acid pyrophosphate (SAPP), as described below.
Sugar Alcohols
Most sugar alcohols, including mannitol, sorbitol, maltitol, xylitol, and lactitol, are poorly absorbed and thus are osmotically active in the lumen (22–26). Since sugar alcohols are commonly used as diluents or sweetening agents in oral dosage forms, it is important to understand the potential impact of these excipients on the absorption of co-administered drugs, especially for those that possess poor intestinal permeability.
The dose–response relationship of sugar alcohols on drug absorption has previously been studied for sorbitol and mannitol (10,19,22). Table I summarizes the results of these studies, in which the bioavailability of a test drug or compound was assessed either by the total area under the plasma concentration–time curve (AUC) or cumulative urinary excretion (percent dose). As shown, the bioavailability of ranitidine was reduced by 7.2%, 25%, and 45.5% when the amount of sorbitol increased from 1.25 g to 2.5 and 5 g. In contrast, no such adverse effects were found with the addition of sorbitol to metoprolol solution or theophylline tablets, indicating the fact that the osmotic effect of sorbitol has little impact on the bioavailability or bioequivalence of BCS class I drugs. Similar osmotic effects were observed with mannitol (Table I). As depicted, administration of 2.264 g mannitol decreased the bioavailability of cimetidine by ∼30%, irrespective of the dosage form or drug product. Likewise, mannitol reduced the intestinal uptake of d-xylose and l-rhamnose in a dose-dependent manner (Table I).
Table I.
Summary data for bioavailability/bioequivalence studies with sorbitol and mannitol
| Excipient/drug product | Excipient | Drug | ||||||
|---|---|---|---|---|---|---|---|---|
| Weight (g) | Concentration (%w/v) | Osmotic potential (mmol) | Dose (mg) | AUC (ng h/ml) | Urine (% dose) | Bioavailability (% control) | BCS class | |
| Sorbitol (25) | ||||||||
| Ranitidine | 0 (control) | 0 | 0 | 150 | 2685 | NA | 100 | III |
| Solution | 1.25 | 8.33 | 6.86 | 150 | 2493 | NA | 92.8 | |
| 2.5 | 16.67 | 13.72 | 150 | 2014 | NA | 75.0 | ||
| 5 | 33.33 | 27.45 | 150 | 1464 | NA | 54.5 | ||
| Metoprolol | 0 | 0 | 0 | 50 | 316.4 | NA | 100 | I |
| Solution | 5 | 33.33 | 27.45 | 50 | 292.7 | NA | 92.5 | |
| Theophylline | 0 | 0 | 0 | 240 | 80.9 | NA | 100 | I |
| Tablet | 10 | 66.67 | 54.89 | 240 | 86.2 | NA | 106.6 | |
| Mannitol (10,28) | ||||||||
| Cimetidine | 0 | 0 | 0 | 200 | 4679 | NA | 100 | III |
| Solution | 2.264 | 1.132 | 12.43 | 200 | 3337 | NA | 71.3 | |
| Cimetidine | 0 | 0 | 0 | 200 | 4490 | NA | 100 | III |
| Chewable Tablet | 2.264 | 1.132 | 12.43 | 200 | 3119 | NA | 69.5 | |
| d-Xylose | 0 | NA | 0 | 500 | NA | 37.6 | 100 | |
| Solution | 5 | NA | 27.45 | 500 | NA | 31.6 | 84.0 | |
| 10 | NA | 54.89 | 500 | NA | 29.2 | 77.7 | ||
| 20 | NA | 109.78 | 500 | NA | 21.1 | 56.1 | ||
| l-Rhamnose | 0 | NA | 0 | 1000 | NA | 14.7 | 100 | |
| Solution | 5 | NA | 27.45 | 1000 | NA | 9.6 | 65.3 | |
| 10 | NA | 54.89 | 1000 | NA | 7.1 | 48.3 | ||
| 20 | NA | 109.78 | 1000 | NA | 5.7 | 38.8 | ||
To determine the threshold level of sugar alcohols in affecting drug absorption, we obtained the dose–response relationship by plotting the osmotic potential of these excipients versus bioavailability of co-administered drugs (normalized with the controls used in the individual studies). As shown in Fig. 3, an apparent linear dose–response relationship was observed for sorbitol solutions with ranitidine. The data points for mannitol/cimetidine also fell on the same line as those for sorbitol/ranitidine, presumably due to the fact that mannitol is an isomer of sorbitol, and both ranitidine and cimetidine share similar absorption kinetics (19,27). It appears that our assumption of a constant fluid volume for the excipient at the absorption site in vivo is valid since the linear dose–response relationship holds regardless of whether the drug product was in solution or tablets. Accordingly, the extent of influence in drug absorption by an “active” excipient can be readily obtained based on its dose–response relationship (such as shown in Fig. 3) so long as the acceptable criterion of bioavailability or bioequivalence has been established.
Fig. 3.
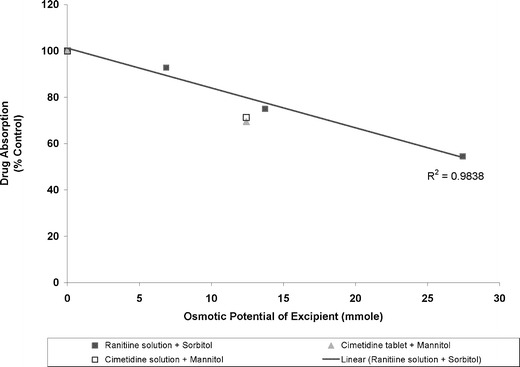
Relationship between osmotic potential of sorbitol/mannitol and absorption of ranitidine/cimetidine compared to the control
It should be noted that the quantitative dose–response relationship for sorbitol/mannitol on ranitidine/cimetidine absorption may not be extrapolated to other compounds that exhibit different absorption characteristics. This is illustrated in Fig. 4 where the osmotic potential of mannitol was plotted against the intestinal absorption of two monosaccharides, d-xylose and l-rhamnose. As shown, ingestion of mannitol reduced l-rhamnose absorption to a greater extent than d-xylose. While the absorption of d-xylose decreased linearly with the increase in osmotic load of mannitol, a curvilinear relationship was found for l-rhamnose. It has been reported that d-xylose is largely absorbed in the jejunum by carrier-mediated transport whereas l-rhamnose is absorbed throughout the small intestine by nonmediated permeation (22). Hence, apart from the osmotic effect, the influence of an osmotically active excipient on intestinal absorption of a compound may also rely on the absorption characteristics (e.g., absorption mechanism and absorption site) of the compound under study. The curvilinear relationship observed for mannitol and l-rhamnose might be explained by a saturation of osmotic effect on the absorption of l-rhamnose when an excess of mannitol (>10 g) was included.
Fig. 4.
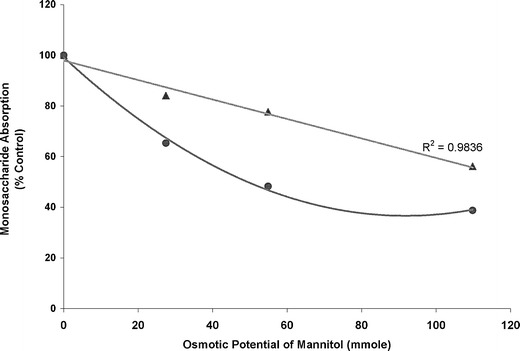
Relationship between osmotic potential of mannitol and absorption of two monosaccharides compared to the control: d-xylose (triangle) and l-rhamnose (circle)
Polyethylene Glycols
The dose–response relationship of PEGs on drug absorption and bioavailability is more complicated as compared to sugar alcohols. PEGs are known to exist in a variety of molecular weight grades, ranging from 200 to 35,000 (7,28). The low molecular weight PEGs (<600) are liquids in nature and commonly employed as a vehicle to enhance the solubility and bioavailability of poorly water-soluble drugs (29). However, recent studies have revealed that PEG 400 at the doses relevant to pharmaceutical formulations stimulates GI motility and accelerates small intestine transit, leading to lower drug bioavailability (15,16,20). Like sorbitol and mannitol, PEGs cannot be absorbed in the intestine and thus retains fluid by osmotic action. In this context, it was suspected that PEGs might interfere with the absorption of co-administered drugs through several mechanisms, including dilution, modification of solubility, direct action on intestinal mucosa, and acceleration of intestinal transit time (30). Additionally, PEGs have been demonstrated to interact with certain transporters, particularly efflux transporters such as P-glycoprotein (P-gp) (28).
Table II provides the summary data of three bioavailability studies previously conducted on ranitidine with PEG 400 (15,16,20). In these studies, various amounts of PEG 400 were added to ranitidine solutions (in orange juice or water) and administered to healthy volunteers. Absolute bioavailability was determined in study I via both intravenous and oral administration, whereas in study II and III cumulative urinary excretion was used for bioavailability assessment. The results of these studies are shown in Table II. For study I, 10 g of PEG 400 decreased the absolute bioavailability of ranitidine capsules by 31% in male subjects. Similarly, for study II (also in male subjects), 2.5 and 5 g of PEG 400 yielded an approximately 38% reduction in the bioavailability of ranitidine solution. In contrast, for study III, at a dose of 0.5–1.5 g, PEG was found to enhance ranitidine bioavailability in male, but not female subjects (Table II). For male subjects, ranitidine absorption was increased with the dose of PEG 400 over the range of 0.5–1.5 g, peaking at 0.75 g followed by a gradual decline to the baseline at 1.5 g. However, for female subjects, reduced ranitidine bioavailability was found in the presence of PEG 400 between 0.5 and 1.5 g.
Table II.
Summary Data for Bioavailability Studies on Ranitidine (150 mg) with PEG 400
| Drug product study/subjects | PEG 400 | Ranitidine | |||||
|---|---|---|---|---|---|---|---|
| Weight (g) | Concentration (% w/v) | Osmotic potential (mmol) | Absolute bioavailability (%) | Urinary Excretion in 24 hrs (mg) | Bioavailability (% control) | BCS class | |
| Ranitidine | III | ||||||
| Capsules | |||||||
| Study I (17) | 0 (Control) | 0 | 0 | 51 | NA | 100 | |
| Men | 10 | 6.67 | 25 | 35 | NA | 69 | |
| Ranitidine | III | ||||||
| Solutions | 0 | 0 | 0 | NA | 34 ± 16 | 100 | |
| Study II (21) | 1.0 | 0.67 | 2.5 | NA | 48 ± 11 | 141 | |
| Men | 2.5 | 1.67 | 6.25 | NA | 21 ± 5 | 62 | |
| 5.0 | 3.33 | 12.5 | NA | 21 ± 5 | 62 | ||
| Study III (26) | 0 | 0 | 0 | NA | 35 ± 8 | 100 | |
| Men | 0.5 | 0.33 | 1.25 | NA | 47 ± 14 | 134 | |
| 0.75 | 0.5 | 1.875 | NA | 57 ± 12 | 163 | ||
| 1.0 | 0.67 | 2.5 | NA | 52 ± 13 | 149 | ||
| 1.25 | 0.83 | 3.125 | NA | 50 ± 12 | 143 | ||
| 1.5 | 1.0 | 3.75 | NA | 37 ± 12 | 106 | ||
| Study III | 0 | 0 | 0 | NA | 38 ± 12 | 100 | |
| Women | 0.5 | 0.33 | 1.25 | NA | 29 ± 11 | 76 | |
| 0.75 | 0.5 | 1.875 | NA | 35 ± 7 | 92 | ||
| 1.0 | 0.67 | 2.5 | NA | 33 ± 12 | 87 | ||
| 1.25 | 0.83 | 3.125 | NA | 33 ± 8 | 87 | ||
| 1.5 | 1.0 | 3.75 | NA | 33 ± 5 | 87 | ||
The observed gender difference in ranitidine bioavailability has been attributed, in part, to the potential interplay between the osmotic effect of PEG 400 on small intestine transit and its modulation of intestinal permeability via inhibition of P-gp efflux transport and/or alteration on membrane fluidity (16,20). Specifically, it was postulated that the increased ranitidine bioavailability in male subjects with PEG 400 at low doses (e.g., 0.5–1.5 g) was due to the ability of this excipient to modulate intestinal permeability, an absorption-enhancing mechanism that was, however, overshadowed at higher doses (≥2.5 g) by the marked acceleration of transit time in the small intestine (16,20). On the other hand, female subjects might have higher proportion of gut efflux transporters including breast cancer resistance protein (BCRP) that could contribute to the differences in bioavailability observed in this study (20). The effects of PEG 400 on BCRP have yet to be studied. Because of all the complicating factors, it is difficult to obtain the net dose–response relationship of PEG 400 specific to its osmotic action on drug absorption.
PEG 4000 and PEG 400 may share similar properties of osmotic action and affect drug absorption in the GI tract. This can be supported by the literature data on chlorthalidone, amoxicillin, and digoxin (30–32). As shown in Table III, addition of chlorthalidone powder to 10% PEG 4000–water solution (treatment C) decreased the AUC and urinary excretion of chlorthalidone compared to other treatments without PEG 4000. While PEG 4000 at a low dose of 2.95 g reduced the amoxicillin AUC by 8.4%, a high dose of 20 g resulted in a 30% decrease in the AUC of digoxin (Table III). Both chlorthalidone and amoxicillin have low intestinal permeability; the former is a BCS class IV drug and the latter a class III drug (33). However, digoxin may be a BCS class II drug (33,34). As shown in Fig. 5, there is a linear relationship between the osmotic potential of PEG 4000 and reduction in the bioavailability of drugs tested over the dose range of 2.95–10 g for PEG 4000. The intestinal transport of amoxicillin was mediated by both passive diffusion and active transport through a dipeptide carrier (30). Digoxin is known to be a sensitive P-gp substrate in vitro (35); however, its role as an in vivo probe substrate of P-gp has been questioned recently (36).
Table III.
Summary Data for Bioavailability/Bioequivalence Studies with PEG 4,000
| Excipient | Drug | Bioavailability | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug product | Weight (g) | Concentration (% w/v) | Osmotic potential (mmol) | Dose (mg) | AUC (mg h/l) | Urine (mg) | BCS class | AUC (% control) | Urine (% control) |
| Chlorthalidone (37) | IV | ||||||||
| Tx. A/Tablet I | 0 (Control) | 0 | 0 | 50 | 293 ± 67 | 18.3 ± 2.5 | – | – | |
| Tx. B/Tablet IIa | 0 | 0 | 0 | 50 | 336 ± 53 | 22.1 ± 5.6 | – | – | |
| Tx. C/PEG 4,000- | 10 | 10 | 2.5 | 50 | 278 ± 71 | 16.6 ± 3.4 | 94.9 (C/A) | 90.7 (C/A) | |
| water solution | 82.7 (C/B) | 75.1 (C/B) | |||||||
| 91.1 (C/D) | 92.7 (C/D) | ||||||||
| Tx. D/Solution–suspension | 0 | 0 | 0 | 50 | 305 ± 74 | 17.9 ± 3.2 | – | – | |
| Amoxicillin (36) | III | ||||||||
| Tablet | 0 | 0 | 0 | 1000 | 47.8 ± 8.2 | NA | 100 | NA | |
| 2.95 | 1.97 | 0.74 | 43.8 ± 6.8 | NA | 91.6 | NA | |||
| Digoxin (38) | |||||||||
| Tablet | 0 | 0 | 0 | 0.5 | 28.3 ± 6.3 | NA | II | 100 | NA |
| Powder | 20 | – | 5 | 0.5 | 19.9 ± 4.8 | NA | 70.3 | NA | |
Tx treatment, tablets I and II represent two different brands
Fig. 5.
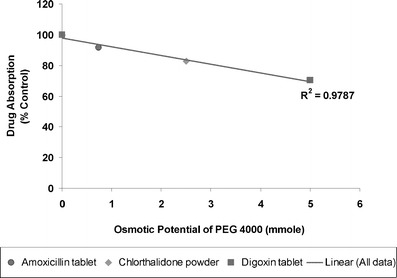
Relationship between osmotic potential of PEG 4,000 and bioavailability of various drugs compared to the control
Unlike PEG 400, there is a lack of information regarding the potential interactions between PEG 4000 and P-gp transporter, although reports are available with respect to other PEG grades or derivatives (37–39). For example, the efflux transport of rhodamine 123 (a P-gp substrate) was found to be significantly decreased when incubated with PEG 400, 2,000, and 20,000 in Caco-2 cell lines (37). The inhibitory effects of these PEGs for the intestinal P-gp function were concentration-dependent over the range of 0.1–20% (v/v or w/v). Other PEG derivatives such as PEG-32 lauric glycerides, PEG-35 castor oil, PEG-20 stearate, PEG-50 stearate, PEG-12 stearate, and PEG-660 12-hydroxystearate were also found to inhibit P-gp function in a logarithmic fashion that is dependent on excipient concentration (38). In another study using Caco-2 cell monolayers, PEG 300 and 400 showed an interaction with P-gp, whereas PEG 200 did not (39). It is unknown if these study results could be extrapolated to PEG 4000. However, as shown in Fig. 5, a linear dose–response relationship holds between the osmotic potential of PEG 4000 and extent of reduction in drug bioavailability, which might indicate minimal impact of potential interactions between PEG 4000 and P-gp in these studies.
Sodium Acid Pyrophosphate
SAPP is commonly used as a food additive and mostly found in bakery products (40). SAPP can also be employed as an excipient for effervescent formulations to provide rapid disintegration of pharmaceutical products. In a relative bioavailability study on ranitidine, it was found that co-administration of 1,132 mg (5.1 mmol) SAPP significantly reduced ranitidine absorption (54% based on AUC), which was further shown to be in parallel to a 56% decrease in small intestine transit time using scintigraphic imaging (41).
Regulatory Implications
Evidently, the presence of ‘active’ excipients in pharmaceutical formulations will have implications for regulatory decisions on biowaivers. In the USA, a biowaiver can be granted for an oral solution containing an active ingredient in the same concentration and dosage as an approved product (42). Biowaivers may also be allowed for minor changes to formulation or manufacturing (43). With the introduction of BCS, biowaivers may be given for a BCS class I drug formulated in an immediate-release, rapidly dissolving drug product (2). A major premise underlying all regulations on biowaivers is that no inactive ingredient (such as excipient) included in the product may significantly affect absorption of the active drug ingredient (42). The issue of “active” excipients is particularly relevant for BCS class III drugs because these compounds often exhibit site-dependent absorption properties and transit time through specific regions of the upper intestine may be critical for drug absorption (6). The presence of an “active” excipient in one formulation (but not in another) or in varying amounts between formulations may result in different bioavailability or bioinequivalence. Hence, it is essential to identify the presence of an “active” excipient in a drug product and determine if there is a potential interaction between the excipient and drug in the formulation. This is of paramount importance in the cases where formulation changes occur during drug development for both innovator and generic firms.
Proper characterization of the dose–response relationship for “active” excipients on drug absorption/bioavailability will allow for the optimal use of these excipients during drug development and product manufacturing. However, it may be difficult to elucidate the dose–response relationship for some “active” excipients, such as PEGs, that exhibit multiple mechanisms for their effects. In addition to the action of osmotic pressure on GI transit time, PEGs can alter integral/transporter proteins in the intestinal membrane that may trigger signaling processes, resulting in the changes in intestinal permeability and motility. The dose–response relationship can be further complicated for excipients that do not form true molecular solutions but instead form aggregates or micelles, as with the case of surfactants. More research is necessary to understand the mechanistic pathways by which an “active” ingredient changes the intestinal permeability.
Similarly, it may not be clear as to how some sugar alcohols and SAPP exert their osmotic action and change the intestinal motility although some have suspected that these “active” excipients might cause a dragging effect across the membrane due to changes in water flux (44). Further research may also be needed to determine how the neuro-immuno-hormonal control of GI tract could be altered by “active” excipients. Apart from lactose intolerance, there are good examples such as Celiac and Crohn’s disease where immune and/or inflammatory responses may affect drug absorption due to changes in the morphology of microvilli along the intestinal tract. Hyperosmotic conditions could have a significant impact on microvilli as well. In addition, poorly absorbed agents may stimulate colonic activity via cholecystokinin, a peptide hormone present in the GI tract that could influence intestinal motility (45).
Conclusions
While most excipients available on the market are devoid of pharmacological action, there is possibility that some “active” excipients may be present in the formulation that alters drug bioavailability and/or bioequivalence. As demonstrated in this study, many sugar alcohols, PEG 400/4,000, and SAPP exemplify such excipients that can influence the absorption of poorly permeable drugs via their osmotic loads in the small intestine.
The current study confirms the colligative property of osmotically active excipients in that the number of excipient molecules (not concentration) in solution will determine their osmotic pressure in vivo, which results in the acceleration of small intestinal transit and in turn, reduction of drug absorption, and bioavailability. In addition to the osmotic action, this study further demonstrates that the effect of osmotically active excipients on drug absorption is dependent on where and how the drug is absorbed in the GI tract. There are evidences that these excipients have more impact on the absorption and bioavailability of poorly permeable compounds as opposed to the compounds with high permeability. A possible explanation for this finding is that poorly permeable drugs tend to stay longer in the gut lumen, and thus, their absorption is prone to be affected by the osmotic action of these “active” excipients. Indeed, in the present study, most drugs that yielded lower bioavailability upon addition of these excipients are either BCS class III (cimetidine, ranitidine, and amoxicillin) or class IV (chlorthalidone) drugs. In contrast, osmotically active excipients exerted little influence on the absorption of BCS class I drugs such as metoprolol and theophylline.
From a regulatory viewpoint, the importance of recognizing the presence of an “active” excipient in a formulation cannot be overemphasized during drug development for both generic and innovator companies. As illustrated in this paper, a better understanding of the dose–response relationship for an “active” excipient can certainly facilitate its optimal use in formulations and further provide opportunities of biowaivers while ensuring product quality and performance. Further research is needed to elucidate the mechanistic pathways for the effects of an “active” excipient on drug absorption and bioavailability/bioequivalence.
References
- 1.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. August 2000. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070246.pdf. Accessed 9 Aug 2012.
- 3.Blume HH, Schug BS. The biopharmaceutics classification system (BCS): class III drugs—better candidates for BA/BE waiver? Eur J Pharm Sci. 1999;9:117–121. doi: 10.1016/S0928-0987(99)00076-7. [DOI] [PubMed] [Google Scholar]
- 4.Polli JE, Yu LX, Cook JA, Amidon GL, et al. Summary workshop report: bioequivalence, biopharmaceutics classification system and beyond. AAPS J. 2008;10:373–379. doi: 10.1208/s12248-008-9040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stavchansky S. Scientific perspectives on extending the provision for waivers of in vivo bioavailability and bioequivalence studies for drug products containing high solubility-low permeability drugs (BCS Class 3) AAPS J. 2008;10:300–305. doi: 10.1208/s12248-008-9030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, Shah VP, Lesko LP, Chen M-L, Lee VHL, Hussain AS. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19:921–925. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 7.Rowe RC, Sheskey PJ, Cook WG, Fenton ME. Handbook of pharmaceutical excipients. 7. London: Pharmaceutical Press; 2012. [Google Scholar]
- 8.Adkin DA, Davis SS, Sparrow RA, Huckle PD, Philips AJ, Wilding IR. The effects of pharmaceutical excipients on small intestinal transit. Br J Clin Pharmacol. 1995;39:381–387. doi: 10.1111/j.1365-2125.1995.tb04466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adkin DA, Davis SS, Sparrow RA, Huckle PD, Phillips AJ, Wilding IR. The effect of different concentrations of mannitol in solution on small intestinal transit: implications for drug absorption. Pharm Res. 1995;12:393–396. doi: 10.1023/A:1016256619309. [DOI] [PubMed] [Google Scholar]
- 10.Adkin DA, Davis SS, Sparrow RA, Huckle PD, Wilding IR. The effect of mannitol on the oral bioavailability of cimetidine. J Pharm Sci. 1995;84:1405–1409. doi: 10.1002/jps.2600841205. [DOI] [PubMed] [Google Scholar]
- 11.Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–1572. doi: 10.1023/A:1016268311867. [DOI] [PubMed] [Google Scholar]
- 12.Nerurkar MM, Burton PS, Borchardt RT. The use of surfactants to enhance the permeability of peptides through Caco-2 cells by inhibition of an apically polarized efflux system. Pharm Res. 1996;13:528–534. doi: 10.1023/A:1016033702220. [DOI] [PubMed] [Google Scholar]
- 13.Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A, Winnike R, Coffin M. Vitamin E-TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res. 1999;16:1812–1817. doi: 10.1023/A:1018939006780. [DOI] [PubMed] [Google Scholar]
- 14.Rege BD, Yu LX, Hussain AS, Polli JE. Effect of common excipients on Caco-2 transport of low permeability drugs. J Pharm Sci. 2001;90:1776–1786. doi: 10.1002/jps.1127. [DOI] [PubMed] [Google Scholar]
- 15.Basit AW, Podczeck F, Newton JM, Waddington WA, Ell PJ, Lacey LF. Influence of polyethylene glycol 400 on the gastrointestinal absorption of ranitidine. Pharm Res. 2002;19:1368–1374. doi: 10.1023/A:1020315228237. [DOI] [PubMed] [Google Scholar]
- 16.Schulze JD, Waddington WA, Ell PJ, Parsons GE, Coffin MD, Basit AW. Concentration-dependent effects of polyethylene glycol 400 on gastrointestinal transit and drug absorption. Pharm Res. 2003;20:1984–1988. doi: 10.1023/B:PHAM.0000008046.64409.bd. [DOI] [PubMed] [Google Scholar]
- 17.Villalobos AP, Gunturi SR, Heavner GA. Interaction of polysorbate 80 with erythropoietin: a case study in protein–surfactant interactions. Pharm Res. 2005;22:1186–1194. doi: 10.1007/s11095-005-5356-7. [DOI] [PubMed] [Google Scholar]
- 18.Hermeling S, Jiskoot W, Crommelin DJA, Schellekens H. Reaction to the paper: interaction of polysorbate 80 with erythropoietin: a case study in protein-surfactant interactions. Pharm Res. 2006;23:641–644. doi: 10.1007/s11095-006-9573-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen ML, Straughn AB, Sadrieh N, Meyer M, Faustino PJ, Ciavarella AB, Meibohm B, Yates CR, Hussain AS. A modern view of excipient effects on bioequivalence: case study of sorbitol. Pharm Res. 2007;24:73–80. doi: 10.1007/s11095-006-9120-4. [DOI] [PubMed] [Google Scholar]
- 20.Ashiru DA, Patel R, Basit AW. Polyethylene glycol 400 enhances the bioavailability of a BCS Class III drug (ranitidine) in male subjects but not females. Pharm Res. 2008;25:2327–2333. doi: 10.1007/s11095-008-9635-y. [DOI] [PubMed] [Google Scholar]
- 21.U.S. Food and Drug Administration, Center for Drug Evaluation and Research, Division of Labeling and Program Support, Office of Generic Drugs. Inactive ingredient search for approved drug products. http://www.accessdata.fda.gov/scripts/cder/iig/index.cfm. Accessed 13 Aug 2012
- 22.Menzies IS, Jenkins AP, Heduan E, Catt SD, Segal MB, Creamer B. The effect of poorly absorbed solute on intestinal absorption. Scand J Gastroenterol. 1990;25:1257–1264. doi: 10.3109/00365529008998562. [DOI] [PubMed] [Google Scholar]
- 23.Riley SA, Kim M, Sutcliffe F, Kapas M, Rowland M, Turnberg LA. Effects of a non-absorbable osmotic load on drug absorption in healthy volunteers. Br J Clin Pharmacol. 1992;34:40–46. doi: 10.1111/j.1365-2125.1992.tb04105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain NK, Rosenberg DB, Ulahannan MJ, Glasser MJ, Pitchumoni CS. Sorbitol intolerance in adults. Am J Gastroenterol. 1985;80:678–681. [PubMed] [Google Scholar]
- 25.Niwa H, Hikichi N, Sakurai E, Ueda M, Fukush G. Effects of maltitol or mannitol on gastrointestinal absorption of drugs. Yakugaku Zasshi J Pharm Soc. 1980;100:1118–1126. doi: 10.1248/yakushi1947.100.11_1118. [DOI] [PubMed] [Google Scholar]
- 26.Oku T, Nakamura S. Threshold for transitory diarrhea induced by ingestion of xylitol and lactitol in young male and female adults. J Nutr Sci Vitaminol. 2007;53:13–20. doi: 10.3177/jnsv.53.13. [DOI] [PubMed] [Google Scholar]
- 27.Grammatte T, Desoky EE, Klotz U. Site-dependent small intestinal absorption of ranitidine. Eur J Clin Pharmacol. 1994;46:253–259. doi: 10.1007/BF00192558. [DOI] [PubMed] [Google Scholar]
- 28.Goole J, Lindley DJ, Roth W, Carl SM, Amighi K, Kauffmann J-M, Knipp GT. The effects of excipients on transporter-mediated absorption. Int J Pharm. 2010;393:17–31. doi: 10.1016/j.ijpharm.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 29.Yalkowsky SH. Techniques of solubilization of drugs. New York: Marcel Dekker; 1981. [Google Scholar]
- 30.Padoin C, Tod M, Brion N, Louchahi K, Gros VL, Petitjean O. Pharmacokinetics of amoxicillin co-administered with a saline-polyethylene glycol solution in healthy volunteers. Biopharm Drug Dispos. 1995;16:169–176. doi: 10.1002/bdd.2510160302. [DOI] [PubMed] [Google Scholar]
- 31.Williams RL, Blume CD, Lin ET, Holford NHG, Benet LZ. Relative bioavailability of chlorthalidone in humans: adverse influence of polyethylene glycol. J Pharm Sci. 1982;71:533–535. doi: 10.1002/jps.2600710514. [DOI] [PubMed] [Google Scholar]
- 32.Ragueneau I, Poirier J-M, Radembino N, Sao AB, Funck-Brentano C, Jaillon P. Pharmacokinetic and pharmacodynamic drug interaction between digoxin and macrogol 4000, a laxative polymer, in healthy volunteers. Br J Clin Pharmacol. 1999;48:453–456. doi: 10.1046/j.1365-2125.1999.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transporter/absorption/elimination interplay and development of a Biopharmaceutics Drug Disposition Classification System. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 34.Emami J. In vitro-in vivo correlation: from theory to application. J Pharm Pharm Sci. 2006;9:169–189. [PubMed] [Google Scholar]
- 35.Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi JG, Zhang Y, Yeleswaram S. The relevance of assessment of intestinal P-gp inhibition using digoxin as an in vivo probe substrate. Nat Rev Drug Discov. 2011;10:75. doi: 10.1038/nrd3028-c1. [DOI] [PubMed] [Google Scholar]
- 37.Shen Q, Lin Y, Handa T, Doi M, Sugie M, Wakayama K, Okada N, Fujita T, Yamamoto A. Modulation of intestinal P-glycoprotein function by polyethylene glycols and their derivatives by in vitro transport and in situ absorption studies. Int J Pharm. 2006;313:49–56. doi: 10.1016/j.ijpharm.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 38.Wang S-W, Monagle J, McNulty C, Putnam D, Chen H. Determination of P-glycoprotein inhibition by excipients and their combinations using an integrated high-throughput process. J Pharm Sci. 2004;93:2755–2767. doi: 10.1002/jps.20183. [DOI] [PubMed] [Google Scholar]
- 39.Ashiru-Oredope DAI, Patel N, Forbes B, Patel R, Basit AW. The effect of polyoxyethylene polymers on the transport of ranitidine in Caco-2 cell monolayers. Int J Pharm. 2011;409:164–168. doi: 10.1016/j.ijpharm.2011.02.059. [DOI] [PubMed] [Google Scholar]
- 40.Evaluation of the health aspects of phosphates as food ingredients. Report PB-262 651, Life Sciences Research Office, Fed Am Soc Exp Biol. 1975.
- 41.Koch KM, Parr AF, Tomlinson JJ, Sandefer EP, Digenis GA, Donn KH, Powell JR. Effect of sodium acid pyrophosphate on ranitidine bioavailability and gastrointestinal transit time. Pharm Res. 1993;10:1027–1030. doi: 10.1023/A:1018918907670. [DOI] [PubMed] [Google Scholar]
- 42.Title 21 Code of Federal Regulations (CFR) Part 320.22, Office of Federal Register, National Archives and Records Administration, US Government Printing Office, Washington, 2011.
- 43.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products—general considerations. March 2003. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070124.pdf. Accessed 16 Aug 2012.
- 44.Hammel HT, Schlegel WM. Osmosis and solute-solvent drag: fluid transport and fluid exchange in animals and plants. Cell Biochem Biophys. 2005;42:277–345. doi: 10.1385/CBB:42:3:277. [DOI] [PubMed] [Google Scholar]
- 45.Guyton AC, Hall JE. Textbook of medical pysiology. 9. Philadelphia: Saunders; 1996. [Google Scholar]