

Abstract
This review provides an introduction to stable isotope dilution mass spectrometry (MS) and its emerging applications in the analysis of membrane transporter proteins. Various approaches and application examples, for the generation and use of quantitation reference standards—either stable isotope-labeled peptides or proteins—are discussed as they apply to the MS quantitation of membrane proteins. Technological considerations for the sample preparation of membrane transporter proteins are also presented.
KEY WORDS: membrane protein, membrane proteome, membrane transporter, quantitative proteomics, stable isotope dilution, stable isotope labeling, mass spectrometry
INTRODUCTION
Transmembrane proteins, defined as having one or more membrane spanning domains, comprise about 25% of the eukaryotic proteome, and are involved in many important biological structures, functions, and pathways (1). They are particularly important in their roles for cell homeostasis. Thus, membrane proteins are targets for the development of new diagnostic and therapeutic approaches. Overall, they form about 70% of all known drug targets (2). Membrane proteins have been scrutinized by many different analytical methods to further understand their structure and behavior. X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy are two powerful methods that have been unable to fully explain the behavior of these species, in particular for transmembrane proteins. Analytical challenges arise in the form of poor crystallization, relatively low abundance, and the large mass of these proteins. Immunoaffinity-based analysis has also been used for membrane proteins; however, it relies on the development of target-specific antibodies. The development of these immuno-grade antibodies can be difficult and expensive for membrane proteins. With the advancement of instrumentation and new methodologies, mass spectrometry (MS) is increasingly expanding its utility in the quantitative analysis of complex protein or proteome samples (3). Like X-ray and NMR analysis, MS-based quantitation of transmembrane proteins needs to deal with the same analytical challenges coming from intrinsic characteristics of these proteins. The requirement of elaborate sample preparation and the reproducibility of MS analysis are restraints for quantitative MS. Stable isotope dilution (SID), which uses a stable isotope-labeled internal standard (IS), is a common means of MS-based quantitation and results in high accuracy and precision. SID when coupled to a multiple reaction monitoring (MRM) workflow in tandem MS, is the pillar of targeted quantitation of proteins (4,5). With enhanced sensitivity and selectivity, MRM-MS also becomes an emerging technology for clinical applications (6–8).
This review provides an observation on the workflow and challenges in published literature for SID-MS of membrane protein quantitation. The review includes SID approaches developed at both protein and peptide levels, experimental designs, and sample preparation techniques. Two major MS strategies, nontargeted and targeted quantitation, will be discussed as the major measurement platforms.
MEMBRANE TRANSPORTERS IN DISEASE AND THERAPEUTICS
Cells maintain stable internal conditions (homeostasis) via controlling the passage of substances through cell membranes (9). Membrane transporters are integral membrane proteins that help maintain homeostasis by controlling the movement of ions, molecules, and even macromolecules across biological membranes. Therefore, membrane transporters are important in transferring essential ions and nutrients, to drug absorption, and for the exportation of cellular waste products (10–14). The transporter classification system classifies the great variety of membrane transporters according to their function and phylogeny (15).
Membrane Transporters in Disease
A multitude of diseases are caused by an absent or malfunctioning membrane transporter. A noncomprehensive list of disease-related transporters studied using MS is presented in Table I (21).
Table I.
Diseases Associated with Deficient Function of Membrane Transporters
| Disease | Gene | Affected area | Substrate transported | References |
|---|---|---|---|---|
| Acrodermatitis enteropathica | SLC39A4 | Skin | Zinc | (16) |
| Autosomal recessive congenital ichthyoses | ABCA12 | Skin | Lipids | (17) |
| Cystic fibrosis | CFTR | Mostly epithelial cells | Chloride ion, other anions | (18) |
| Cystinuria | SLC3A1 | Kidneys | Cystine, dibasic amino acids | (19) |
| De vivo | SLC2A1 | Blood–brain barrier | Glucose | (20) |
| Hartnup | SLC6A19 | Kidneys and intestine | Neutral amino acids | (20) |
| Tangier | ABCA1 | Systemic | Precursor to high density lipids | (20) |
Membrane Transporters in Drug and Biomarker Discovery/Screening
Quantitative analysis of the membrane subproteome can help elucidate disease mechanisms and drug targets. As the membrane proteome is increasingly explored and understood, the possibility increases to find new potential drug targets (22). A recent article studies the targeting of ABBC2 in the apical membrane (23). The gene ABBC2 encodes for a multidrug resistant transporter (MRP) called MRP2. MRPs make less effective treatment of disease; drugs are cleared or denied entry by these transporters (24). Breast cancer resistance protein is the MRP, which is responsible for biliary clearance of many drugs. Its expression level was measured by liquid chromatography (LC)-MS/MS in livers with variant alleles (18). In the biomarker area, one of the great desires for a potential biomarker is ease of measurement. This not only refers to the analytical skills needed to do a quality measurement but also easy access. Thus, biomarkers are usually studied in bodily fluids: urine, blood, cerebral spinal fluid, etc. This being said, membrane proteins (for noncirculating cells) as biomarkers would require looking at the cell membrane via biopsy; however, if the membrane protein (or part of it) were shed and released into the blood, then it could possibly be measured in that matrix (25). The direct measurement of a cell's membrane protein is still being evaluated for developing potential biomarkers. Two recent studies showcase this point: gastric cancer biomarkers (26) and lymph node metastasis and lung adenocarcinoma (27).
MS ANALYSIS OF MEMBRANE TRANSPORTERS
Sample Preparation and Separation
A number of challenges exist for MS-based quantitation of membrane transporters. The plasma membrane needs to be disrupted, typically using detergents to retrieve the proteins. Relative low abundance of the proteins further makes the quantitation difficult. Various methods for enriching membrane proteins are investigated (28).
Enrichment of plasma membrane proteins can be achieved by exploiting the amine group of lysine residues on extracellular loops of the proteins. Amine groups form covalent bonds with a separation enabling reagent, typically an active ester–biotin molecule. Followed by cell lysis, the biotin-tagged proteins can be pulled down by avidin onto solid supports. Examples for such enrichment include studies of ovarian cancer (29) and cancer metastasis (30). A metal affinity column can be used to enrich poly-histidine fusion proteins in a similar way (31); engineered cells need to be used, however. Cationic particles have also been utilized to enrich membrane proteins, and the proteins are collected in sheets of membranes coated with the particles (32,33). This is a global and fast method for membrane protein enrichment, and it can also allow for automation based on properties of the particles.
Due to the hydrophobic nature of membrane proteins, sodium dodecyl sulfate–polyacrylamide gel electrophoresis is a good choice for protein separation. Detergents can be used to solubilize proteins and then readily removed via subsequent gel washing. The separation method can be further integrated with in-gel protein digestion by specific proteases (e.g., trypsin) and LC separation of the resulting peptides. This workflow is often referred to as GeLC. When gel casting and running conditions are controlled, large membrane proteins can be well separated from smaller proteins and be enriched in a narrow, high molecular weight region of the gel. In addition, the sample complexity can be further reduced by centrifugation fractionation prior to electrophoresis: zonal centrifugation followed by ultracentrifugation (34), sucrose gradient (35), and zonal centrifugation with a commercial kit (36). These three examples use precipitation after centrifugation to retrieve the protiens. Extraction with different organic solvents can also be used (37).
An online LC system coupled to a mass spectrometer is routinely used for biological applications. However, it requires prior sample preparation when dealing with membrane transporter proteins in complex biological samples. Most LC techniques use complementary separation steps to overcome challenges in membrane protein MS analysis. When MS-based protein quantitation involves labeling techniques, buffers are additional source of potential interferences in the sample preparation steps (26,38,39). Digestion of membrane proteins is also an important experimental step that should be optimized (40).
Prefractionation of peptides following proteolytic digestion decreases sample complexity at the peptide level, resulting in improvement of detection and quantitation of low abundance membrane proteins. Multidimensional LC separations currently are common practice in analysis of proteome digests. Applying tandem separations using two stationary phases (columns) with orthogonal characteristics helps decrease the sample complexity to a great extent (41). The most common approaches involve online or offline strong cation exchange (SCX) coupled with reverse-phase chromatography as in the multidimensional protein identification technology (42). The first dimension of chromatography is typically based on a salt gradient or pH fractionation using SCX or isoelectric point-based separation using isoelectric focusing (43). Chromatography is preferably performed at nanoliters per minute flow rates, in order to obtain good quantitation limits for low abundance proteins like membrane transporters (26,44–46). Fractionation of peptide mixtures of proteome digests also decreases suppression of ion generation of particular peptides in the ionization source of a mass spectrometer.
Mass Spectrometry
Targeted mass spectrometric quantitation is applicable to quantify previously identified protein targets in a mixture. In the analysis, the number of analyte targets is typically small. Thus, targeted MS quantitation is fast, sensitive, and specific (47). In contrast, nontargeted MS quantitation measures analytes in a global, unbiased manner; many peptides in a proteome digest are identified and quantified together. Most of the nontargeted MS quantitation as in shotgun proteomics is based on data dependent analysis (DDA). In this tandem MS method, one survey scan is performed to screen the precursor ions, and when the precursor ions pass the defined criteria by the user, a subsequent MS/MS experiment is iniatiated to record fragments of the precursor ions. Abundant peptides yield large numbers of precursor ions and are readily detected based on DDA criteria (48,49). One advantage of nontargeted MS approaches is that there is no need for extensive method design and optimization of instrument parameters before doing the experiment.
MRM or selected reaction monitoring MS is a gold standard for MS quantitation of target peptides and proteins in complex biological samples. Due to the high level of selectivity and sensitivity, it is the method of choice in targeted quantitative proteomics (50). Therefore, most of the SID-MS-based absolute and relative quantitation is implemented in MRM-MS. Two tandem steps of m/z selection of precursor and fragment ions, forming one particular gas-phase transition, drastically reduce the chance of the signal interference from off-target species; MRM-MS is focused on only specific target peptides. By limiting the number of target analytes, one can decrease the time of a full analysis cycle in a mass spectrometer. Additionally, it also increases the ions dwell time in the mass analyzer; therefore, data with higher quality is achievable. The best instrumental platform for MRM-MS analysis is a triple quadrupole tandem mass spectrometer (51–56). Figure 1 illustrates the mechanism of an MRM analysis in a triple quadrupole mass spectrometer. The mass selection (filtering) capability of a quadrupole mass analyzer is the key of the MRM analysis mode. Some of the commonly used mass spectrometers for MRM-MS and similar analysis include: TSQ triple quadrupole (57,58) and ion trap (56) from Thermo-Finnegan, triple quadrupole from Agilent (59) and QTrap from ABSciex (44,60).
Fig. 1.
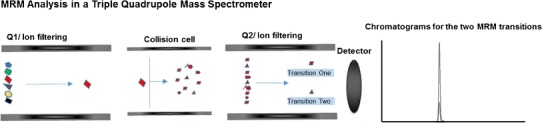
MRM analysis using a triple quadrupole mass spectrometer for two preselected transitions
MS can be used in both high throughput and multiplexed quantitation (42,47). The significance of these two analytical merits is best exemplified in the context of the biomarker discovery pipeline (61,62). In the early stages of biomarker discovery, one is dealing with thousands of proteins, but with only a few numbers of biological samples to identify biomarker candidates. To identify potential candidates, there is a need for quantitative methods with multiplexing power, i.e., analyzing many different analytes in a single experiment. This is typically done by nontargeted MS for quantitative profiling of small sets of samples. The MRM-MS strategy can then be applied to validate the candidates via multiplexed, targeted quantitative analysis, in which hundreds of transitions can be set for quantitative measurements of the biomarker candidates, with high sensitivity and confidence (62,63). Validation of the selected peptide candidates can also be done by MRM-MS, but with a varied focus on limited number of analyte targets in many samples (64).
MS QUANTITATION VIA STABLE ISOTOPE DILUTION
The use of an internal standard (IS) is vital to quantitative MS analysis. Protein quantitation in general is based on measurements of MS signals generated from surrogate peptides; these serve as signatures for precursor proteins. Because MS analyzes molecules based on their mass/charge ratios, stable isotope analogues are well suited as internal standards for MS quantitation. Multiple steps in sample preparation and intrinsic difficulties in membrane protein manipulation make the use of internal reference standards essential for accurate quantitation. High preanalysis property similarities between analyte molecules and their stable isotope analogues are required. In short, both types of molecules need to behave in the same manner before MS analysis, including coelution in the chromatographic system and the same ionization efficiency in a mass spectrometer. Although proteins are quantified by MS analysis of surrogate peptides, isotopic internal standards can be added in either forms of peptides or proteins. Compared to the addition of peptide standards, the use of protein standards allows for earlier incorporation into sample preparation workflows, thus the increased accuracy in quantitation. The IS approach is a better choice to avoid differential analyte loss than the use of a calibration curve (55,65). Table II summarizes the different SID technologies and their respective membrane protein applications in literature.
Table II.
Applications of SID Approaches in Transmembrane Protein Quantitation
| Technology | Selected target membrane protein/transporter | Sample | Reference |
|---|---|---|---|
| AQUA | Rhodopsin | Rod outer segments (ROS) and HEK 293 cells | (55) |
| ABCA7, ABCB4, ABCC1, ABCC3 and ABCC4 | Human platelets and megakaryocytic progenitor cells | (44) | |
| OATP2B1 (SLCO2B1) | Human platelets | (60) | |
| Multidrug resistance-associated protein 2 (MRP2/ABCC2) |
Bile canalicular membrane in human liver tissues | (6) | |
| Human breast cancer resistance protein (BCRP/ABCG2) | Bile canalicular membrane in human liver tissues | (8) | |
| MDR1a, MRP4, BCRP, 4F2HC, ASCT2, GLUT1, MCT1, LAT1, OAT3, OATP2, OATPF and TAUT | Mouse brain capillary endothelial cells | (58) | |
| BSEP, MRP2, MRP3, MRP6, BCRP, ABCG5, ABCG8, 4F2HC, NAT, GAT2, GLUT1, MCT1, NTCP, OATP1 and OATP2 | Mouse liver | (58) | |
| MRP2, MRP4, BCRP, 4F2HC, ASCT2, MCT1, OAT1, OAT3, OATP1, TAUT and MATE1 | Mouse kidney | (58) | |
| (MDR1), (BCRP), (MRP4), (GLUT1), GLUT3/14, (MCT1), MCT8 | Monkey’s BBB | (66) | |
| ABCA2, ABCA3, ABCA6,ABCB1/MDR1, ABCC1/MRP1, ABCC4/MRP4, ABCG2/BCRP, SLC2A1/GLUT1, SLC3A2/4F2hc, SLC16A1/MCT1, SLC29A1/ENT1 and SLC38A1/ATA1, | Human cerebral Microvascular endothelial cell hCMEC/D3 |
(67) | |
| BCRP, MDR1, ABCA2, ABCA8, EAAT1, GLUT, GLUT3/14, 4F2hc, BGT1, CAT1 and MCT1 | Human brain micro vessel (BBB) | (45) | |
| 18O | CFTR | HT29 and BHK cells | (68) |
| Golgi membrane protein 1, moesin, Calmyrin, Annexin IV and Epidermal growth factor receptor pathway substrate 8 (EPS8) | Human cholangiocarcinomas | (69) | |
| CD13, carbonic anhydrase IX, potassium-transporting ATPase, and SDF-1 | B16F10 melanoma cells | (70) | |
| C-type cytochromes OmcA and MtrC | Shewanella oneidensis MR-1 | (71) | |
| ICAT™ | CD45, squalene synthetase, SQS, FTFD | HL-60 | (56) |
| DRMMs and non-DRMMs | Rat basophilic leukemia cells (RBL-2H3) | (72) | |
| iTRAQ™ | SLC3A2 and SLC7A5 | MKN7 cancerous cell vs HFE145 healthy cell | (26) |
| ITMAP Voltage dependent anion channel 1 Voltage dependent anion channel 2 |
Pancreatic ZG membranes | (73) | |
| AdeABC and RND : AdeA (A1S_1751,1752) and AdeB (A1S_1750 BFMS: BFMS (A1S_0749) OMPs: A1S_0884, 3297, and 3317 |
A. baumannii DU202 | (74) | |
| Membrane proteins from plasma membrane, ER, golgi, vacuole, and mitochondria | Arabidopsis thaliana | (46) | |
| Connnexin 46, connnexin 50 and aquaporin 0 (AQP0) BASP-1, paralemmim 1 and vimentin |
Lens cells | (75) | |
| TMT™ | COX2, CADM3 and BASI | WT and PrP-KO cerebellar granule neurons (CNG) | (57) |
| SILAC | Uncharacterized protein 2760762, voltage-dependent anion selective channel protein 1 | High metastatic PC3M-LN4 and low metastatic PC3M | (76) |
| CD133/Prominin-1, Glypican-4, Neuroligin-4, ErbB2, Glycoprotein M6B, and PTPRZ | Human embryonic stem cells (hESH) | (77) | |
| Facilitated glucose transporter- member 1, 4 F2 cell-surface antigen heavy chain and large neutral amino acids transporter small subunit 1 | Human breast cancer mitoxantrone resistant MCF-7 (MXR MCF-7) and MCF-7 | (78) |
Signature Peptide Selection Criteria
A signature peptide is representative and unique to the target protein. There are a number of criteria that should be considered in selecting the best possible signature peptide. These criteria slightly vary depending on the applied quantitation methodology. For example, the peptides containing cysteine residues usually are avoided in the derivatization-free approaches, whereas they are required in cysteine derivatization-based approaches. Table III shows a list of the criteria (58,79)
Table III.
Common Criteria for Signature Peptides
| Case | Criteria | Effect |
|---|---|---|
| Generic | Optimum length: 6–16 amino acids | Peptide uniqueness and ionization efficiency |
| Generic | No methionine, cysteine or tryptophan residues | Avoiding uncontrolled chemical modifications during sample preparation |
| Generic | No post-translational modifications | Avoiding mass variation and changes in stoichiometry |
| Polymorph proteins | No single nucleotide polymorphism | Avoiding sequence variation |
| Membrane transporters | No transmembrane region | Digestion efficiency |
| Tryptic digestion | No successive Arg or Lys | Digestion efficiency |
Use of Stable Isotope Labeled Peptide Standards
A peptide internal standard is a heavy isotope counterpart of a signature peptide. Stable isotope-labeled peptides are commonly used as quantitation references for signature peptides whose concentrations are stoichiometrically equivalent to those of target proteins. The labeled peptides can be made via either derivatization-free approaches or derivatization-based methods. Derivatization-free approaches are relatively simple and easy to use. While derivatization-based methods are cost effective and allow for analysis of increased numbers of samples in a single analysis, however, this comes at the expense of increased sample complexity due to chemical derivatization of the sample. Labeled peptides are introduced to samples after protein digestion (Fig. 2). In comparison, stable isotope-labeled protein standards are introduced after protein/proteome samples being collected.
Fig. 2.
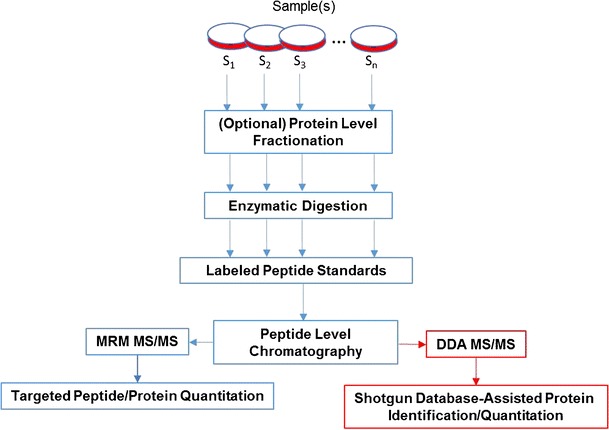
General workflow of targeted and nontargeted (red lines) proteome quantitation using peptide standards
Derivatization-Free Methods
AQUA Peptides
Peptides containing stable isotope-labeled amino acids, the so-called absolute quantitation (AQUA) peptides, are chemically synthesized and used for the absolute quantitation of proteins and their modifications (51). Heavy amino acids used for synthesis mostly carry 13C or 15N in order to obtain the minimal retention time deviation from that of the native peptide in reverse phase chromatography. 2H are less expensive and can also be incorporated in less hydrophobic residues of the synthetic peptides (55,80). Several reports use the AQUA strategy to analyze membrane proteins and membrane transporters. Absolute quantitation of rhodopsin, a prototype of the G protein-coupled receptor superfamily of transporters, was reported (58). Rhodopsin was quantified with two methods; spiked-in labeled peptide standards and generating an external standard calibration curve. The results were comparable with each other. In addition, AQUA was applied for quantifying p-glycoprotein, as a drug transporter, in two systems: Caco-2 cells and human embryonic kidney-multidrug resistance 1 (HEK-MDR1) vesicles (59). The quantitative results showed a relationship between the level of the p-glycoprotein expression measured by LC-MRM-MS and its functionality determined by 3H-labeled N-methyl quinidine (NMQ) uptake by the cell. The expression of the ATP-binding cassette (ABC) transporters was comprehensively profiled using the AQUA approach, in the human platelet membrane proteome (44). The atorvastatin drug uptake level was investigated by measuring the level of anion transporting polypeptide family (OATP2B1) expression in human platelets and megakaryocytes differentiated from CD34+ stem cells, using VLLQTLR as the signature peptide to OATP2B1 (60).
In a clinical study, the expression difference of MRP2/ABCC2 transporter was measured in 51 hepatic tissue samples from a liver bank. A synthetic, isotope-labeled peptide (LTIIPQDPILFSGSL [13C615N1] R) was used to quantify the corresponding surrogate signature peptide for MRP2. It was found that the MRP2 expression level is neither age (7–61 years old) nor sex dependent (6). In a similar study, concentration of BCRP/ABCG2 was measured for samples with a range of age, sex, and genotype, using the AQUA strategy (8). The selected signature peptide was SSLLDVLAAR. This peptide differs from the signature peptide, ENLQFSAALR, used in an earlier study of the same protein (81). It is claimed that the selection of the new peptide resulted in quantitation with improved sensitivity. The hepatic expression of BCRP also was found neither sex nor age (7–70 years old) dependent.
Absolute quantitation of 36 membrane transporters in mouse kidney, liver, and brain tissues was conducted and 216 MRM transitions were monitored in a single MS analysis (58). The same research group later studied drug disposition on a model system of cynomolgus monkeys. They profiled the membrane protein expression and its change in the monkey and compared the results with a previous blood–brain barrier (BBB) microvessel study in mice (66). Later on, they investigated the membrane transporters in human cerebral microvascular endothelial cells (hCMEC/D3) as a human model for BBB (67). They quantified 12 membrane transporters in the membrane fractions and the whole cell lysate separately and compared those to human umbilical vein endothelial cells. Comprehensive quantitation of the human BBB transporters and receptors in human brain microvessels from seven male humans (16–77 years old) was reported in a different study (45), and the results were extensively compared to that of the mouse study (58).
AQUA strategy is fast and relatively cheap and simple. Additionally, it is applicable for quantitation of proteins independent of their origins. However, since it is added after protein preparation and digestion, the prior differential sample loss and the varying protein digestion efficiency compromise the quantitation accuracy and precision. When many peptides are targeted for quantitation, the cost of labled peptides as the references increases.
18O-Labeled Peptides
Another approach for derivatization-free production of labeled peptides uses the method of enzymatic 18O labeling of peptides (82). 18O is specifically incorporated into the carboxylate group at the C-termini of the peptides via protease catalysis. This is a global strategy in which virtually all peptides in a sample (e.g., the membrane proteome digest) can be labeled and used as quantitation references. The simplicity and global applicability of 18O-labeled peptides make the technique attractive for biomarker discovery and clinical applications. Caution should be taken, however, to avoid the back-exchange reaction, via e.g., complete removal of enzymes used for the 18O labeling. Protein membrane enrichment together with an 18O-labeled quantitation reference was used to screen differential membrane protein expression in normal human biliary tract tissue and xenografts from human cholangiocarcinomas, in a quest for potential cancer biomarkers (69). A number of novel biomarker candidates were proposed, such as golgi membrane protein 1, moesin, and calcium and integrin binding protein (Calmyrin). Furthermore, this study confirmed the previously reported overexpressed membrane proteins. 18O-labeled peptides were also utilized to study the effect of hypoxia on membrane protein expression (70). Relative quantitation of the membrane proteins in hypoxic and normoxic B16F10 melanoma cells showed that 44% of the membrane protein subset is significantly upregulated in the hypoxic condition; among them were aminopeptidase N (CD13) and stromal cell derived factor I (SDF-1). Another study reported the use of 18O-labeled peptides for the differential membrane protein expression in a wild type Shewanella oneidensis MR-1 and a type II secretion protein (gspD) deleted (ΔgspD) mutant (71). It is believed that type II secretion system (T2SS) in this organism is impaired due to ΔgspD mutation. A group of membrane proteins that were translocated during the secretion mechanism had altered abundance and therefore were identified as potential substrates for T2SS (71).
When signature peptides are selected for a target protein, previously nonlabeled synthetic peptides can also be labeled with 18O via acid-catalyzed exchange (83,84). However, it should be noted that compared to the enzyme-catalyzed exchange, the acid labeling method should be only used for signature peptides with small numbers of carboxylate groups and that do not have significant side reactions under acidic conditions. An acid-labeled peptide has been utilized as the quantitation standard to measure cystic fibrosis transmembrane conductance regulator in human intestinal (HT29) and baby hamster kidney (BHK) cell lines (68). The signature peptide was NSILTETLHR, and the IS was prepared as NSILTETLHR-18O4 via acid catalysis (68).
Derivatization-Based Methods
Peptides Labeled with ICAT™
Isotope Coded Affinity Tag (ICAT) reagents carry three major functional groups, each designed for a specific purpose: a reactive group which targets specific peptides for effective derivatization, an isotope coded linker which acts as a mass tag, and an affinity tag for downstream enrichment of tagged peptides (52), which makes the ICAT strategy especially useful for low abundant proteins quantitation. Light and heavy ICAT reagents can be used to separately derivatize two protein digests (e.g., control/treated or healthy/diseased samples). The heavy linker differs from the light by the designed mass difference. A thiol reactive group is designed to react with cysteine residues in peptides. The affinity tag is biotin, which allows for enrichment of labeled peptides using avidin pull-down strategies. In a study, ICAT was utilized to characterize and quantify 491 proteins in microsomal fractions of native and in vitro differentiated human promyelocytic leukemia (HL-60) cell lines (56). The two native and 12-phorbol 13-myristate acetate (PMA) treated microsomal fractions were labeled with D0 and D8 ICAT reagents, respectively. It was revealed that certain membrane associated proteins and membrane associated signal transduction proteins show significantly lower expression levels in the PMA-treated cell line. ICAT reagents not only allow for SID-based MS quantitation but also sample complexity reduction, which further improves quantitation precision and accuracy. In another study, the combination of ICAT and 18O labeling was utilized to compare the efficacy of two different detergents, Triton X-100 and Brij-96, for extracting detergent-resistant membrane microdomain (DRMM) proteins (72). The study in rat basophilic leukemia cells (RBL-2H3) showed that Triton X-100 more effectively extracted both DRMM and non-DRMM proteins.
Peptides Labeled with iTRAQ™/TMT™ Reagents
Other common derivatization methods for labeling peptides with stable isotopes include Isobaric Tagging for Relative and Absolute Quantitation (iTRAQ ™) and Tandem Mass Tagging (TMT ™). Both of these methods allow for the analysis of multiple samples in a single experiment. iTRAQ reagents are a multiplexed (four- and eight-plex) set of isobaric reagents that are reactive to the N-termini of peptides. All the iTRAQ reagents in a set are isobaric, but upon fragmentation in a mass spectrometer, the derivatized peptides produce small mass ions, differing in mass/charge ratios (i.e. 114, 115, 116, and 117). The intensities of these ions report the relative quantities of the same peptide in different samples (53). The TMT strategy operates under the same principle (54).
The iTRAQ approach was utilized to quantify SLC3A2 as a gastric cancer biomarker in HFE145 and MKN7 cell lines (healthy and cancerous), respectively. Tryptic peptides from membrane protein fractions were labeled with the iTRAQ reagents. The ratio of the differentially labeled peptides (the ratio of a protein in different samples by implication) was measured by quantifying the peak areas under m/z 114 (HFE145) and 116 (MKN7). Expression levels of solute carrier family of transporters, such as SLC7A5 and SLC3A2, were compared for the two cell lines (26). Furthermore, a four-plexed iTRAQ strategy was implemented to study a multidrug resistant Acinetobacter baumannii (74). A specific clinical isolate, A. baumanii DU202 that is resistant to selected antibiotics was chosen to study the cell wall and membrane proteome in two different culture conditions (with and without antibiotics). A total of 484 proteins were identified and 62.4% of them were plasma membrane proteins in the outer, inner, or periplasmic areas of the membrane. Quantitative analysis was also reported. Combined use of four- and eight-plex iTRAQ reagents was employed to study the aging process and its effect on the cell membrane bound macromolecules on lens (75). A number of integral membrane proteins were quantified in a targeted manner. Major lens proteins, such as crystalline and cytoskeletal proteins, were found to become tightly bound to the membrane of older fiber cells. Another three-plexed iTRAQ approach was taken to study pancreatic zymogen granule membranes to distinguish the intrinsic membrane proteins from the peripheral proteins in the rat pancreata zymogen granule membranes (73). TMT™ reagents have also been employed to study the membrane protein expression of wild type and PrP-knockout (PrP-KO) cerebellar granule neurons (57). Three membrane proteins, COX2, CADM3, and BASI, were found to be significantly downregulated in PrP-KO neurons. Three samples, derivatized using three different TMT™ reagents, were quantified in parallel by MRM-MS.
Use of Stable Isotope-Labeled Protein or Proteome Standards
Addition of stable isotope-labeled internal standards at any earlier stage of sample preparation workflow leads to a more reliable and accurate quantitation. Addition of the stable isotope standard allows for normalization of differential sample loss in different preparation steps (Fig. 3). Therefore, studies that take this analytical advantage are emerging. Generation of stable isotope-labeled proteins has practical limitations in design and development. They are often more costly than making labeled peptide standards. A few methods, including stable isotope labeling by amino acids in cell culture (SILAC) and protein standard absolute quantitation that use whole protein internal standards, are established for MS quantitation of proteins. SILAC is commonly used and labels all proteins in a proteome by adding heavy amino acids in the cell culture media (85). Proteome-wide labeling of the SILAC method makes it suitable for global relative quantitation studies. However, this approach only fits to quantitation of proteome, originated from cultured cells and not tissues or biopsies. The SILAC approach was applied to investigate the proteome expression of the metastatic phase of prostate cancer. PC3M-LN4 cells cultured in SILAC media containing 13C6 lysine and PC3M cells in normal media; these cell lines represent a pair of high and low metastatic potential models. Among a total of 444 identified proteins, membrane proteins such as voltage-dependent anion-selective channel protein 1 and an uncharacterized protein with GenBank accession number of 22760762 were pinpointed as potential biomarkers (76). The SILAC technology was also used for a comparative study of the undifferentiated and differentiated human embryonic stem cells (hESH) in their expression levels of membrane proteins (77). In another study, membrane protein changes in breast cancer cells upon acquiring drug resistance were quantified with the SILAC assistance (78). Secreted proteins with stable isotope labels, including the shedded membrane proteins, were prepared via the SILAC approach and used as internal standards for relative and absolute quantitation (86). Stable isotope-labeled, full-length proteins are recently being synthesized in vitro as reference standards for MS quantitation (87). However, their use for quantitative measurements can be limited for membrane transporter proteins, partly due to the poor solubility and typically large size of the proteins.
Fig. 3.
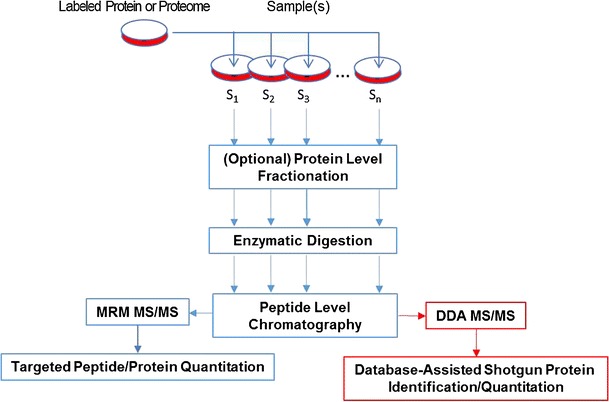
General workflow of targeted and nontargeted (in red) proteome quantitation using protein/proteome standards
The SILAC strategy was adapted to quantify proteins in tissue samples. In the so-called super SILAC approach, multiple SILAC cell lines of HMEC, MCF7, HCC2218, HCC1599, and HCC1937 were pooled to prepare a single protein standard to study breast cancer tissue (88). A related approach quantified tissue and biopsy samples using stable isotope labeling of mammals (SILAM) (89). Quantitation of dystrophin in human muscle biopsy was successfully performed using SILAM tissues of mice which were fed with custom diet containing 13C6-lysine (90). These methods are suitable for quantifying membrane transporters in tissues and biopsies.
Conclusion
MS is arguably the most promising technology for absolute quantitation of proteins. Applications of SID-based quantitative MS to transmembrane protein research are emerging. Successful MS analysis of transmembrane proteins requires effective sample preparations: protein enrichment and fractionation, proteolytic digestion, and peptide separations. The fundamental principle of SID-MS technology is the minimization of differential analyte loss among samples and the use of an internal standard for quantitation normalization. Challenges in preparing transmembrane protein samples, mainly due to their high hydrophobicity and molecular weight, necessitate quantifying the proteins through their surrogate signature peptides. It is convenient and cost effective to use stable isotope-labeled peptides as quantitation references for MS analysis. Use of reference proteome standards with globally labeled proteins allows for the early addition of a quantitation references into samples, thus improving quantitation accuracy.
Acknowledgments
Financial supports from the Cystic Fibrosis Foundation and the National Cancer Institute/National Institutes of Health are gratefully acknowledged.
Contributor Information
Vahid Farrokhi, Email: vahid.farrokhi@uconn.edu.
Adam J. McShane, Email: adam.mcshane@uconn.edu
Reza Nemati, Email: reza.nemati@uconn.edu.
Xudong Yao, Email: x.yao@uconn.edu.
References
- 1.Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 2010;190:491–500. doi: 10.1083/jcb.201004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boja ES, Rodriguez H. Mass spectrometry-based targeted quantitative proteomics: achieving sensitive and reproducible detection of proteins. Proteomics. 2012;12:1093–1110. doi: 10.1002/pmic.201100387. [DOI] [PubMed] [Google Scholar]
- 5.Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;22:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deo AK, Prasad B, Balogh L, Lai Y, Unadkat JD. Interindividual variability in hepatic expression of the multidrug resistance-associated protein 2 (MRP2/ABCC2): quantification by liquid chromatography/tandem mass spectrometry. Drug Metab Dispos. 2012;40:852–855. doi: 10.1124/dmd.111.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fortin T, Salvador A, Charrier JP, Lenz C, Bettsworth F, Lacoux X, et al. Multiple reaction monitoring cubed for protein quantification at the low nanogram/milliliter level in nondepleted human serum. Anal Chem. 2009;81:9343–9352. doi: 10.1021/ac901447h. [DOI] [PubMed] [Google Scholar]
- 8.Prasad B, Lai Y, Lin Y, Unadkat JD. Interindividual variability in the hepatic expression of the human breast cancer resistance protein (BCRP/ABCG2): effect of age, sex, and genotype. J Pharm Sci. 2013;102:787–793. doi: 10.1002/jps.23436. [DOI] [PubMed] [Google Scholar]
- 9.Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Anderle P, Bussey KJ, Barbacioru C, Shankavaram U, Dai Z, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004;64:4294–4301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- 11.Saier MH, Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34:D181–D186. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busch W, Saier MH., Jr The IUBMB-endorsed transporter classification system. Mol Biotechnol. 2004;27:253–262. doi: 10.1385/MB:27:3:253. [DOI] [PubMed] [Google Scholar]
- 13.Russel FGM. Transporters: importance in drug absorption, distribution, and removal. Enzyme- Transp-Based Drug–Drug Interact. 2010:27–49.
- 14.Lee HJ, Sagawa K, Shi W, Murer H, Morris ME. Hormonal regulation of sodium/sulfate co-transport in renal epithelial cells. Proc Soc Exp Biol Med. 2000;225:49–57. doi: 10.1046/j.1525-1373.2000.22506.x. [DOI] [PubMed] [Google Scholar]
- 15.Saier MH., Jr A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev. 2000;64:354–411. doi: 10.1128/MMBR.64.2.354-411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitt S, Kury S, Giraud M, Dreno B, Kharfi M, Bezieau S. An update on mutations of the SLC39A4 gene in acrodermatitis enteropathica. Hum Mutat. 2009;30:926–933. doi: 10.1002/humu.20988. [DOI] [PubMed] [Google Scholar]
- 17.Akiyama M. ABCA12 mutations and autosomal recessive congenital ichthyosis: a review of genotype/phenotype correlations and of pathogenetic concepts. Hum Mutat. 2010;31:1090–1096. doi: 10.1002/humu.21326. [DOI] [PubMed] [Google Scholar]
- 18.Clancy JP, Jain M. Personalized medicine in cystic fibrosis: dawning of a new era. Am J Respir Crit Care Med. 2012;186:593–597. doi: 10.1164/rccm.201204-0785PP. [DOI] [PubMed] [Google Scholar]
- 19.Chillaron J, Font-Llitjos M, Fort J, Zorzano A, Goldfarb DS, Nunes V, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6:424–434. doi: 10.1038/nrneph.2010.69. [DOI] [PubMed] [Google Scholar]
- 20.Broer S, Wagner CA. Membrane transporter diseases. New York: Kluwer/Plenum; 2003. [Google Scholar]
- 21.Savas JN, Stein BD, Wu CC, Yates JR., 3rd Mass spectrometry accelerates membrane protein analysis. Trends Biochem Sci. 2011;36:388–396. doi: 10.1016/j.tibs.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams R, Worth CL, Guenther S, Dunkel M, Lehmann R, Preissner R. Binding sites in membrane proteins–diversity, druggability and prospects. Eur J Cell Biol. 2012;91:326–339. doi: 10.1016/j.ejcb.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Emi Y, Yasuda Y, Sakaguchi M. A cis-acting five-amino-acid motif controls targeting of ABCC2 to the apical plasma membrane domain. J Cell Sci. 2012;125:3133–3143. doi: 10.1242/jcs.099549. [DOI] [PubMed] [Google Scholar]
- 24.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 25.Tien WS, Chen YT, Wu KP. SecretePipe: a screening pipeline for secreted proteins with competence to identify potential membrane-bound shed markers. J Proteome Res. 2013;12:1235–1244. doi: 10.1021/pr3009012. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Toy W, Choong LY, Hou P, Ashktorab H, Smoot DT, et al. Discovery of SLC3A2 cell membrane protein as a potential gastric cancer biomarker: implications in molecular imaging. J Proteome Res. 2012;11:5736–5747. doi: 10.1021/pr300555y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P, Zeng G, Hu R, Li C, Yi H, Li M, et al. Identification of Flotillin-1 as a novel biomarker for lymph node metastasis and prognosis of lung adenocarcinoma by quantitative plasma membrane proteome analysis. J Proteome. 2012;77:202–214. doi: 10.1016/j.jprot.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 28.Vuckovic D, Dagley LF, Purcell AW, Emili A. Membrane proteomics by high performance liquid chromatography-tandem mass spectrometry: analytical approaches and challenges. Proteomics. 2013;13:404–423. doi: 10.1002/pmic.201200340. [DOI] [PubMed] [Google Scholar]
- 29.Faca VM, Ventura AP, Fitzgibbon MP, Pereira-Faca SR, Pitteri SJ, Green AE, et al. Proteomic analysis of ovarian cancer cells reveals dynamic processes of protein secretion and shedding of extra-cellular domains. PLoS One. 2008;3:e2425. doi: 10.1371/journal.pone.0002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karhemo PR, Ravela S, Laakso M, Ritamo I, Tatti O, Makinen S, et al. An optimized isolation of biotinylated cell surface proteins reveals novel players in cancer metastasis. J Proteome. 2012;77:87–100. doi: 10.1016/j.jprot.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramjeesingh M, Li C, Garami E, Huan LJ, Hewryk M, Wang Y, et al. A novel procedure for the efficient purification of the cystic fibrosis transmembrane conductance regulator (CFTR) Biochem J. 1997;327(Pt 1):17–21. doi: 10.1042/bj3270017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choksawangkarn W, Kim S, Cannon JR, Edwards NJ, Lee SB, Fenselau C. Enrichment of plasma membrane proteins using nanoparticle pellicles: comparison between silica and higher density nanoparticles. J Proteome Res. 2013;12:1134–1141. doi: 10.1021/pr301107x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahbar AM, Fenselau C. Integration of Jacobson's pellicle method into proteomic strategies for plasma membrane proteins. J Proteome Res. 2004;3:1267–1277. doi: 10.1021/pr040004t. [DOI] [PubMed] [Google Scholar]
- 34.Li B, Chang J, Chu Y, Kang H, Yang J, Jiang J, et al. Membrane proteomic analysis comparing squamous cell lung cancer tissue and tumour-adjacent normal tissue. Cancer Lett. 2012;319:118–124. doi: 10.1016/j.canlet.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 35.Nair S, Xavier T, Kumar MK, Saha S, Menon KN. Exploitation of detergent thermodynamics in the direct solubilization of myelin membrane proteins for two-dimensional gel electrophoresis for proteomic analysis. Electrophoresis. 2011;32:3621–3629. doi: 10.1002/elps.201100248. [DOI] [PubMed] [Google Scholar]
- 36.Perdomo AB, Ciccosanti F, Iacono OL, Angeletti C, Corazzari M, Daniele N, et al. Liver protein profiling in chronic hepatitis C: identification of potential predictive markers for interferon therapy outcome. J Proteome Res. 2012;11:717–727. doi: 10.1021/pr2006445. [DOI] [PubMed] [Google Scholar]
- 37.Mitra SK, Walters BT, Clouse SD, Goshe MB. An efficient organic solvent based extraction method for the proteomic analysis of Arabidopsis plasma membranes. J Proteome Res. 2009;8:2752–2767. doi: 10.1021/pr801044y. [DOI] [PubMed] [Google Scholar]
- 38.Yun S, Choi C, Kwon S, Park GW, Cho K, Kwon K, et al. Quantitative proteomic analysis of cell wall and plasma membrane fractions from multidrug-resistant Acinetobacter baumannii. J Proteome Res. 2011;10:459–469. doi: 10.1021/pr101012s. [DOI] [PubMed] [Google Scholar]
- 39.Hogl S, van Bebber F, Dislich B, Kuhn P, Haass C, Schmid B, et al. Label-free quantitative analysis of the membrane proteome of Bace1 protease knock-out zebrafish brains. Proteomics. 2013;13:1519–1527. doi: 10.1002/pmic.201200582. [DOI] [PubMed] [Google Scholar]
- 40.Choksawangkarn W, Edwards N, Wang Y, Gutierrez P, Fenselau C. Comparative study of workflows optimized for in-gel, in-solution, and on-filter proteolysis in the analysis of plasma membrane proteins. J Proteome Res. 2012;11:3030–3034. doi: 10.1021/pr300188b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischer F, Wolters D, Roegner M, Poetsch A. Toward the complete membrane proteome: high coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics. 2006;5:444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 43.Manadas B, English JA, Wynne KJ, Cotter DR, Dunn MJ. Comparative analysis of OFFGel, strong cation exchange with pH gradient, and RP at high pH for first-dimensional separation of peptides from a membrane-enriched protein fraction. Proteomics. 2009;9:5194–5198. doi: 10.1002/pmic.200900349. [DOI] [PubMed] [Google Scholar]
- 44.Niessen J, Jedlitschky G, Grube M, Kawakami H, Kamiie J, Ohtsuki S, et al. Expression of ABC-type transport proteins in human platelets. Pharmacogenet Genomics. 2010;20:396–400. doi: 10.1097/FPC.0b013e32833997b0. [DOI] [PubMed] [Google Scholar]
- 45.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011;117:333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 46.Sadowski PG, Dunkley TP, Shadforth IP, Dupree P, Bessant C, Griffin JL, et al. Quantitative proteomic approach to study subcellular localization of membrane proteins. Nat Protoc. 2006;1:1778–1789. doi: 10.1038/nprot.2006.254. [DOI] [PubMed] [Google Scholar]
- 47.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 49.Martinez-Aguilar J, Chik J, Nicholson J, Semaan C, McKay MJ, Molloy MP. Quantitative mass spectrometry for colorectal cancer proteomics. Proteomics Clin Appl. 2013;7:42–54. doi: 10.1002/prca.201200080. [DOI] [PubMed] [Google Scholar]
- 50.Yocum AK, Chinnaiyan AM. Current affairs in quantitative targeted proteomics: multiple reaction monitoring-mass spectrometry. Brief Funct Genomic Proteomics. 2009;8:145–157. doi: 10.1093/bfgp/eln056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci U S A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 53.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 55.Barnidge DR, Dratz EA, Martin T, Bonilla LE, Moran LB, Lindall A. Absolute quantification of the G protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal Chem. 2003;75:445–451. doi: 10.1021/ac026154+. [DOI] [PubMed] [Google Scholar]
- 56.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stella R, Cifani P, Peggion C, Hansson K, Lazzari C, Bendz M, et al. Relative quantification of membrane proteins in wild-type and prion protein (PrP)-knockout cerebellar granule neurons. J Proteome Res. 2012;11:523–536. doi: 10.1021/pr200759m. [DOI] [PubMed] [Google Scholar]
- 58.Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K, et al. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res. 2008;25:1469–1483. doi: 10.1007/s11095-008-9532-4. [DOI] [PubMed] [Google Scholar]
- 59.Miliotis T, Ali L, Palm JE, Lundqvist AJ, Ahnoff M, Andersson TB, et al. Development of a highly sensitive method using liquid chromatography-multiple reaction monitoring to quantify membrane P-glycoprotein in biological matrices and relationship to transport function. Drug Metab Dispos. 2011;39:2440–2449. doi: 10.1124/dmd.111.040774. [DOI] [PubMed] [Google Scholar]
- 60.Niessen J, Jedlitschky G, Grube M, Bien S, Schwertz H, Ohtsuki S, et al. Human platelets express organic anion-transporting peptide 2B1, an uptake transporter for atorvastatin. Drug Metab Dispos. 2009;37:1129–1137. doi: 10.1124/dmd.108.024570. [DOI] [PubMed] [Google Scholar]
- 61.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics. 2010;9:184–196. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 63.Schiess R, Wollscheid B, Aebersold R. Targeted proteomic strategy for clinical biomarker discovery. Mol Oncol. 2009;3:33–44. doi: 10.1016/j.molonc.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yao X, Bajrami B, Shi Y. Ultrathroughput multiple reaction monitoring mass spectrometry. Anal Chem. 2010;82:794–797. doi: 10.1021/ac9026274. [DOI] [PubMed] [Google Scholar]
- 65.Nanavati D, Gucek M, Milne JL, Subramaniam S, Markey SP. Stoichiometry and absolute quantification of proteins with mass spectrometry using fluorescent and isotope-labeled concatenated peptide standards. Mol Cell Proteomics. 2008;7:442–447. doi: 10.1074/mcp.M700345-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Ito K, Uchida Y, Ohtsuki S, Aizawa S, Kawakami H, Katsukura Y, et al. Quantitative membrane protein expression at the blood–brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci. 2011;100:3939–3950. doi: 10.1002/jps.22487. [DOI] [PubMed] [Google Scholar]
- 67.Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood–brain barrier model. Mol Pharm. 2013;10:289–296. doi: 10.1021/mp3004308. [DOI] [PubMed] [Google Scholar]
- 68.Jiang H, Ramos AA, Yao X. Targeted quantitation of overexpressed and endogenous cystic fibrosis transmembrane conductance regulator using multiple reaction monitoring tandem mass spectrometry and oxygen stable isotope dilution. Anal Chem. 2010;82:336–342. doi: 10.1021/ac902028f. [DOI] [PubMed] [Google Scholar]
- 69.Kristiansen TZ, Harsha HC, Gronborg M, Maitra A, Pandey A. Differential membrane proteomics using 18O-labeling to identify biomarkers for cholangiocarcinoma. J Proteome Res. 2008;7:4670–4677. doi: 10.1021/pr800215n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stockwin LH, Blonder J, Bumke MA, Lucas DA, Chan KC, Conrads TP, et al. Proteomic analysis of plasma membrane from hypoxia-adapted malignant melanoma. J Proteome Res. 2006;5:2996–3007. doi: 10.1021/pr0601739. [DOI] [PubMed] [Google Scholar]
- 71.Zhang H, Brown RN, Qian WJ, Monroe ME, Purvine SO, Moore RJ, et al. Quantitative analysis of cell surface membrane proteins using membrane-impermeable chemical probe coupled with 18O labeling. J Proteome Res. 2010;9:2160–2169. doi: 10.1021/pr9009113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blonder J, Yu LR, Radeva G, Chan KC, Lucas DA, Waybright TJ, et al. Combined chemical and enzymatic stable isotope labeling for quantitative profiling of detergent-insoluble membrane proteins isolated using Triton X-100 and Brij-96. J Proteome Res. 2006;5:349–360. doi: 10.1021/pr050355n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen X, Walker AK, Strahler JR, Simon ES, Tomanicek-Volk SL, Nelson BB, et al. Organellar proteomics: analysis of pancreatic zymogen granule membranes. Mol Cell Proteomics. 2006;5:306–312. doi: 10.1074/mcp.M500172-MCP200. [DOI] [PubMed] [Google Scholar]
- 74.Yun SH, Choi CW, Kwon SO, Park GW, Cho K, Kwon KH, et al. Quantitative proteomic analysis of cell wall and plasma membrane fractions from multidrug-resistant Acinetobacter baumannii. J Proteome Res. 2011;10:459–469. doi: 10.1021/pr101012s. [DOI] [PubMed] [Google Scholar]
- 75.Truscott RJ, Comte-Walters S, Ablonczy Z, Schwacke JH, Berry Y, Korlimbinis A, et al. Tight binding of proteins to membranes from older human cells. Age (Dordr) 2011;33:543–554. doi: 10.1007/s11357-010-9198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Everley PA, Krijgsveld J, Zetter BR, Gygi SP. Quantitative cancer proteomics: stable isotope labeling with amino acids in cell culture (SILAC) as a tool for prostate cancer research. Mol Cell Proteomics. 2004;3:729–735. doi: 10.1074/mcp.M400021-MCP200. [DOI] [PubMed] [Google Scholar]
- 77.Prokhorova TA, et al. Stable isotope labeling by amino acids in cell culture (SILAC) and quantitative comparison of the membrane proteomes of self-renewing and differentiating human embryonic stem cells. Mol Cell Proteomics. 2009;8.5:959–970. doi: 10.1074/mcp.M800287-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rahbar AM, Fenselau C. Unbiased examination of changes in plasma membrane proteins in drug resistant cancer cells. J Proteome Res. 2005;4:2148–2153. doi: 10.1021/pr0502370. [DOI] [PubMed] [Google Scholar]
- 79.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 80.Bajrami B, Farrokhi V, Zhang M, Shehu A, Yao X. Back to deuterium: Utility of 2H-labeled peptides for targeted quantitative proteomics. Int J Mass Spectrom. 2012;312:17–23. doi: 10.1016/j.ijms.2011.05.006. [DOI] [Google Scholar]
- 81.Li N, Palandra J, Nemirovskiy OV, Lai Y. LC-MS/MS mediated absolute quantification and comparison of bile salt export pump and breast cancer resistance protein in livers and hepatocytes across species. Anal Chem. 2009;81:2251–2259. doi: 10.1021/ac8024009. [DOI] [PubMed] [Google Scholar]
- 82.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 83.Niles R, Witkowska HE, Allen S, Hall SC, Fisher SJ, Hardt M. Acid-catalyzed oxygen-18 labeling of peptides. Anal Chem. 2009;81:2804–2809. doi: 10.1021/ac802484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang S, Bobst CE, Kaltashov IA. Pitfalls in protein quantitation using acid-catalyzed O18 labeling: hydrolysis-driven deamidation. Anal Chem. 2011;83:7227–7232. doi: 10.1021/ac201657u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Investig Ophthalmol Vis Sci. 2010;51:3379–3386. doi: 10.1167/iovs.09-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6:2139–2149. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- 88.Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat Methods. 2010;7:383–385. doi: 10.1038/nmeth.1446. [DOI] [PubMed] [Google Scholar]
- 89.McClatchy DB, Yates JR., 3rd Stable Isotope Labeling of Mammals (SILAM) CSH Protocol. 2008 doi: 10.1101/pdb.prot4940. [DOI] [PubMed] [Google Scholar]
- 90.Brown KJ, Marathi R, Fiorillo AA, Ciccimaro EF, Sharma S, Rowlands DS, et al. Accurate quantitation of dystrophin protein in human skeletal muscle using mass spectrometry. J Bioanalysis Biomed. 2012 doi: 10.4172/1948-593X.S7-001. [DOI] [PMC free article] [PubMed] [Google Scholar]