

Abstract
Glyburide (glibenclamide, GLB) is a widely prescribed antidiabetic with potential beneficial effects in central nervous system injury and diseases. In vitro studies show that GLB is a substrate of organic anion transporting polypeptide (OATP) and ATP-binding cassette (ABC) transporter families, which may influence GLB distribution and pharmacokinetics in vivo. In the present study, we used [11C]GLB positron emission tomography (PET) imaging to non-invasively observe the distribution of GLB at a non-saturating tracer dose in baboons. The role of OATP and P-glycoprotein (P-gp) in [11C]GLB whole-body distribution, plasma kinetics, and metabolism was assessed using the OATP inhibitor rifampicin and the dual OATP/P-gp inhibitor cyclosporine. Finally, we used in situ brain perfusion in mice to pinpoint the effect of ABC transporters on GLB transport at the blood–brain barrier (BBB). PET revealed the critical role of OATP on liver [11C]GLB uptake and its subsequent impact on [11C]GLB metabolism and plasma clearance. OATP-mediated uptake also occurred in the myocardium and kidney parenchyma but not the brain. The inhibition of P-gp in addition to OATP did not further influence [11C]GLB tissue and plasma kinetics. At the BBB, the inhibition of both P-gp and breast cancer resistance protein (BCRP) was necessary to demonstrate the role of ABC transporters in limiting GLB brain uptake. This study demonstrates that GLB distribution, metabolism, and elimination are greatly dependent on OATP activity, the first step in GLB hepatic clearance. Conversely, P-gp, BCRP, and probably multidrug resistance protein 4 work in synergy to limit GLB brain uptake.
KEY WORDS: ABC transporters, blood–brain barrier, glyburide, organic anion transporting polypeptide, positron emission tomography
INTRODUCTION
Glyburide (glibenclamide, 5-chloro-N-[2-[4-(cyclohexylcarbamoylsulfamoyl)phenyl]ethyl]-2-methoxybenzamide, abbreviated GLB) is a hypoglycemic agent commonly used in the treatment of noninsulin-dependent diabetes. It inhibits sulfonylurea receptor 1 (SUR1, ABCC8), the regulatory subunit of the pancreatic potassium efflux channel (KATP) of the ATP-binding cassette (ABC) family. Recent reports have revealed that GLB may also have beneficial effects in acute ischemic, traumatic, and inflammatory central nervous system (CNS) injuries and diseases via its affinity for transient receptor potential melastatin 4 in the brain (1,2).
In vitro studies have shown that GLB is a substrate for several transporters belonging to the ABC and solute carrier (SLC) superfamilies, which may control its distribution and pharmacokinetics. Among the ABC transporters, GLB has been identified as a substrate of the breast cancer resistance protein (BCRP, ABCG2) (3–7) and some multidrug resistance proteins (MRP1 and MRP3; ABCC1,3) (3,7). However, GLB transport by P-glycoprotein (P-gp, ABCB1, MDR1) remains controversial (3,7–9). More recently, GLB has also been shown to be a substrate of the organic anion-transporting polypeptide (OATP, SLCO) family. GLB is preferentially transported in vitro by OATP2B1 (10), OATP1A2, and, to a lesser extent, OATP1B1 (11).
The differential subcellular expression and function and cooperative properties of these transporters have added a new layer of complexity to the prediction of their impact on drug pharmacokinetics (12). The uptake of many compounds from the circulating blood to hepatocytes is governed by OATP1B1, OATP1B3, and OATP2B1 at the sinusoidal membrane, which is devoid of P-gp and BCRP. ABC transporters such as P-gp, BCRP, and MRP2 are located in the canalicular membrane and mediate the export of xenobiotics into the bile (12). In a clinical study, the administration of the broad-spectrum OATP inhibitor rifampicin (RFA) resulted in decreased plasma clearance and metabolism of orally administered GLB (13,14). Therefore, we can hypothesize that OATP transporters, metabolic enzymes, and ABC transporters work in a synchronized manner to control GLB distribution in the liver and enable its subsequent metabolism and elimination. Conversely, P-gp, BCRP, and MRP4 (but not MRP1–3) work together at the luminal surface of brain endothelial cells forming the blood–brain barrier (BBB) to restrict the penetration of drugs into the CNS (15,16). Nevertheless, the role of ABC and OATP transporters in the uptake of GLB by the brain and peripheral organs remains to be assessed in vivo.
In the present study, we aimed to evaluate the influence of selected transporter inhibitors on the brain and whole-body distribution of GLB in nonhuman primate. We used [11C]GLB positron emission tomography (PET) imaging to non-invasively observe the distribution of GLB at a non-saturating tracer dose in baboons. Next, we tested the influence of RFA and cyclosporine (CsA) to compare the in vivo consequences of OATP and combined OATP/P-gp inhibition on [11C]GLB distribution. Finally, we used in situ brain perfusion in mice to obtain an in-depth understanding of the influence of P-gp, BCRP, MRP, and OATP transporters separately and in combination on the permeability of the BBB to GLB.
MATERIALS AND METHODS
Animals
All animal use procedures were in strict accordance with the recommendations of the European Community (86/609/CEE) and the French National Committees (law 87/848) for the care and use of laboratory animals. PET studies were carried out on two adult Papio anubis baboons (14 and 16 kg in weight). Adult male Fvb mice (30–40 g, 7–11 weeks old) were obtained from Janvier (Genest, France). A dual P-gp/BCRP-deficient mouse strain Mdr1a; Mdr1b; Bcrp(−/− ; −/− ; −/−) (derived from Fvb mice) was bred in-house from progenitors obtained from the laboratory of Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, The Netherlands).
Chemicals
PSC833 (valspodar) was a gift from Novartis (Switzerland), GF120918 (elacridar) was from GSK (Collegeville, PA, USA), and MK-571 from Merck Frosst (Kirkland, Canada). Fumitremorgin C (FTC) and GLB were purchased from Sigma-Aldrich (France). CsA, rifampicin (RFA), and pantoprazole were administered using the commercial drugs Sandimmun® (Novartis, France), Rifadine® (Sanofi-Aventis, France), and Inipomp® (Nicomed, France), respectively.
[3H]GLB (2 TBq/mmol) was purchased from PerkinElmer (France). [11C]GLB was prepared as previously reported (17). Desmethyl glyburide, the precursor for carbone-11 labeling, was obtained in one chemical step by treating GLB with a 1 M solution of BBr3 (4 eq.) in dichloromethane at low temperatures (−90 to −20°C). Carbon-11 labeling of GLB was performed using a TRACERLab FX-C Pro synthesizer (GEMS, France) and consisted of (1) the trapping at −10°C of [11C]MeOTf in acetone (0.3 mL) containing the precursor (0.5–0.8 mg) and aqueous 3 N NaOH (5 μL), (2) heating at 110°C for 2 min, (3) dilution in 1.0 mL of the HPLC mobile phase and purification using semi-preparative reversed-phase HPLC (Waters Symmetry® C-18: eluant, CH3CN/H2O/TFA—45/55/0.1 (v/v/v); flow rate, 5 mL/min; detection, at 254 nm), and (4) SepPak®Plus C-18-based formulation for i.v. injection. Starting from a 74-GBq cyclotron-produced [11C]carbon dioxide batch, 2.1 to 4.0 GBq of [11C]GLB, >99% radiochemically pure and ready-to-inject, were obtained in 40 min. Specific radioactivity ranged from 75 to 150 GBq/μmol. The log P and log D7.4 values measured for [11C]GLB were 2.09 and 1.82, respectively.
PET Imaging in Baboons
Two hours before PET acquisition, animals received ketamine (10 mg/kg; i.m.). After being intubated, baboons were artificially ventilated, maintained anesthetized with 66% N2O/1% isoflurane and infused i.v. with 250 mL/h 0.9% NaCl. Experiments were performed on a HR+ Tomograph (Siemens Healthcare, Knoxville TS, USA) for brain PET studies, and a Biograph-6® PET-CT scanner (Siemens Healthcare) for whole-body exams. Animals were injected i.v. with 222 to 444 MBq [11C]GLB. The time period of the investigation was limited to 60 min due to the fast radioactive decay of carbon-11 (radioactive half-life = 20.4 min). The total amount of GLB injected ranged from 7 to 45 nmol (i.e., 3.5 to 22 μg). We examined whether [11C]GLB distribution was modified by selected OATP- and/or ABC transporter inhibitors. RFA (an OATP inhibitor; 8.6 mg/kg, i.v.) was infused over 30 min immediately before [11C]GLB injection. CsA (a dual P-gp/OATP inhibitor; 15 mg/kg/h, i.v.) and pantoprazole (a dual P-gp/BCRP inhibitor; 30 mg/kg/h, i.v.) were infused 30 min before beginning and during PET scanning. Each condition was tested at least twice in two different baboons to take interindividual variability into consideration (Table I). Heart rate, end-tidal pCO2, and rectal temperature were continuously monitored. Time–activity curves were generated by calculating the mean radioactivity in selected volumes of interest (VOIs) drawn from PET or CT images. Standard uptake values (SUV) were generated by correcting the radioactivity in the VOIs for carbon-11 decay, attenuation, injected dose, and animal weight. The area under the curve (AUC; SUV.min) was calculated from 0 to 60 min using a linear trapezoidal method.
Table I.
Area Under the Time–Activity Curve (AUC; SUV.min) from 0 to 60 min in Plasma, and AUCtissue/AUCplasma Ratio in Selected Organs Determined Using PET Imaging in Baboons Following [11C]Glyburide Injection
| Control | Rifampicin (8.6 mg/kg) | Cyclosporine (15 mg/kg/h) | Pantoprazole (30 mg/kg/h) | ||
|---|---|---|---|---|---|
| Plasma | AUCplasma | 140 ± 19 (n = 3 + 2) | 688 (516; 860) | 643 ± 191 (n = 2 + 2) | 233 (223; 243) |
| Liver | AUCliver | 1846 ± 60 (n = 2 + 1) | 685 (592; 779) | 554 (584; 523) | |
| AUCliver/AUCplasma | 14.0 ± 1.4 (n = 2 + 1) | 1.0 (1.1; 0.9) | 0.8 (0.9; 0.6) | ||
| Renal cortex | AUCrenal cortex | 363 ± 160 (n = 2 + 1) | 633 ± 34 (608; 657) | 495 (577; 413) | |
| AUCrenal cortex/AUCplasma | 2.9 ± 1.0 (n = 2 + 1) | 1.0 (1.18; 0.76) | 0.7 (0.92; 0.47) | ||
| Brain | AUCbrain | 4.5 ± 1.0 (n = 3 + 2) | 11.5 (12.9; 10.1) | 17.2 ± 3.7 (n = 2 + 2) | 8.1 (7.7; 8.6) |
| AUCbrain/AUCplasma | 0.032 ± 0.005 (n = 3 + 2) | 0.018 (0.025; 0.012) | 0.029 ± 0.013 (n = 2 + 2) | 0.035 (0.035; 0.035) | |
| Myocardium | AUCmyocardium | 44.5 ± 5.8 (n = 2 + 1) | 136 (123; 148) | 122 (105; 138) | |
| AUCmyocardium/AUCplasma | 0.34 ± 0.03 (n = 2 + 1) | 0.20 (0.23; 0.17) | 0.16 (0.16; 0.16) | ||
| Pancreas | AUCpancreas | 46.2 ± 25.0 (n = 2 + 1) | 191 (209; 171) | 326 (302; 350) | |
| AUCpancreas/AUCplasma | 0.36 ± 0.21 (n = 2 + 1) | 0.30 (0.41; 0.20) | 0.44 (0.48; 0.40) | ||
| Lungs | AUClungs | 30.0 ± 6.0 (n = 2 + 1) | 87 (81; 93) | 114 (115; 112) | |
| AUClungs/AUCplasma | 0.18 ± 0.05 (n = 2 + 1) | 0.12 (0.13; 0.12) | 0.13 (0.16; 0.11) |
When n ≥ 3, data are means ± SD and the number of experiments in each baboon is described (n = n (baboon 1) + n (baboon 2)). Individual values are specified when n = 1 + 1. Values in bold characters are significantly different from controls
AUC area under the time–activity curve
[11C]GLB Plasma Analysis
During PET acquisition, carbon-11 radioactivity was estimated in arterial plasma obtained from blood samples drawn from the femoral artery. Plasma samples obtained at 5, 15, 30, and 60 min were deproteinized with acetonitrile and injected into an UV/radioactive-HPLC system. [11C]GLB was separated from its radiometabolites using a C18 semipreparative (10 × 250 mm, 10 μm) Atlantis® column (Waters, France). The mobile phase consisted of 10 mM ammonium acetate in purified water (A) and acetonitrile (B). The gradient elution of B from 20% at 0 min to 90% at 12 min was applied to the column. Radioactivity in urine samples was also measured and analyzed using this radio-HPLC system. The [11C]GLB fraction bound to plasma protein was measured before and during CsA, RFA, or pantoprazole infusion using a validated ultrafiltration method (MPS Micropartition Microcon®-YM-10 membrane from Millipore, France). When possible, urine samples were drawn using a urethral catheter at the end of the scan, counted for radioactivity, and analyzed using HPLC.
Cell Culture and Accumulation Assay
The stably transfected Madin–Darby Canine Kidney cell lines MDCKII-hMDR1 and MDCKII-hBCRP were a gift from Dr. Alfred H. Schinkel (National Cancer Institute, The Netherlands). Culture and western blot analyses were performed as previously described (18). Cells were seeded onto 24-well plates (3.105 cells/well) and allowed to grow for 3 days. On the day of the experiment, the culture medium was removed and cell monolayers washed and incubated with a buffer containing ∼3.7 kBq/mL [3H]GLB with or without transport inhibitors. Full inhibition of P-gp and BCRP was obtained using PSC833 5 μM (18,19) and FTC 10 μM (5,18), respectively. GF120918 5 μM was used to assess the effects of the inhibition of both P-gp and BCRP (20). After 1 h of incubation, the monolayers were washed twice and cells lysed using 500 μL of Triton X-100 0.1% overnight. Samples from each vial were mixed with Ultima Gold® scintillation cocktail (Perkin Elmer) and counted to estimate tritium radioactivity.
In situ Brain Perfusion in Mice
Mice were anesthetized with ketamine (140 mg/kg, i.p.) + xylazine (8 mg/kg, i.p.). In situ brain perfusion was performed as previously described (21). Briefly, the right common carotid artery was exposed and the external carotid artery was ligated at the level of the bifurcation of the common and internal carotid arteries. The right common carotid artery was catheterized and the catheter connected to a syringe containing the perfusion fluid (protein-free Krebs carbonate-buffered physiological saline). The thorax was opened, the heart was cut, and perfusion started immediately at a flow rate of 2.5 mL/min. Each mouse was perfused for 90 s. The perfusion fluid contained [3H]GLB (11 kBq/mL; 5.5 nM) and the vascular integrity marker 14C-sucrose (3.7 kBq/mL). The intrinsic transport rate (Kin; in microliter per gram per second, n = 4–6) for [3H]GLB was calculated as previously reported (21). The brain vascular volume was calculated using the distribution of 14C-sucrose, which does not measurably cross the BBB in a short time. This distribution volume was used to check BBB integrity and enabled us to correct [3H]GLB brain activity based on the vascular content. The apparent distribution volume of [3H]GLB (Vbrain; in microliter per gram) and its transport rate or brain clearance rate were calculated from tritium radioactivity in the right hemisphere (Kin = Vbrain/90 s ; in microliter per gram per second). Selected transporter inhibitors were added to the perfusion fluid to assess the role of transporters on GLB transport at the luminal BBB. P-gp and BCRP were fully and selectively inhibited using PSC833 5 μM and FTC 10 μM, respectively. The broad-spectrum inhibitors MK-571 50 μM (22,23) and RFA 60 μM (13) were used to inhibit MRP and OATP, respectively. GF120918 was used at 5 and 10 μM to test the effect of the combined inhibition of both P-gp and BCRP at the BBB. The intrinsic transport rate (Kin; in microliter per gram per second, n = 4–6) for [3H]GLB was also measured in P-gp/BCRP-deficient mice.
Statistical Analysis
Comparisons between PET values were carried out using a linear mixed-effect model on log-transformed data. In this model, “treatment” (CsA, RFA, or pantoprazole) was the fixed and “baboon” the random factor. If the estimation of the variance of the random factor was negative, it was removed from the model. A one-tailed test was performed to compare treatment with control conditions. The p values obtained by this analysis were then adjusted using the Holm–Bonferroni procedure to take into account multiple comparisons, using WinNonlin 5.3 software (Pharsight, USA). One-way analysis of variance followed by post hoc tests was performed for multiple comparisons for in vitro and in situ experiments. Data are presented as means ± standard deviations.
RESULTS
PET Study in Baboons
Pretreatment with RFA or CsA i.v. increased ∼5-fold the area under the time–activity curve (AUC) for radioactivity in plasma between 0 and 60 min (Table I). Under control conditions, [11C]GLB represented 70% of plasma radioactivity 60 min post-injection, the remaining 30% being polar radiometabolites. No radiometabolite could be detected after CsA or RFA pretreatment under our analytical conditions. The dose of pantoprazole used (30 mg/kg/h) was well tolerated. This treatment reduced [11C]GLB metabolism (Fig. 1) and increased AUCplasma ∼1.7-fold (Table I). [11C]GLB plasma protein binding was 99.9% and did not change after treatment with CsA, RFA, or pantoprazole.
Fig. 1.
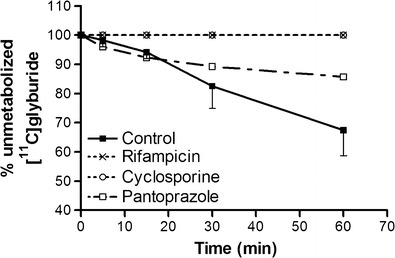
Influence of cyclosporine, rifampicin, and pantoprazole on [11C]glyburide metabolism. Data are presented as mean percentages ± SD of unmetabolized [11C]glyburide in plasma vs. time after i.v. injection of [11C]glyburide in baboons under control conditions and after rifampicin (8.6 mg/kg), cyclosporine (15 mg/kg/h), and pantoprazole (30 mg/kg/h) treatment
Under control conditions in baboons, [11C]GLB distribution was characterized by high accumulation in the liver parenchyma (SUVmax ∼38; Figs. 2 and 3). The AUCliver/AUCplasma ratio was drastically reduced by RFA (∼14-fold) and CsA (∼18-fold) with no significant difference between these treatments (Table I). The radioactive content of the gallbladder and bile duct showed high variability between experiments and no evidence of a difference between treatments and control conditions.
Fig. 2.
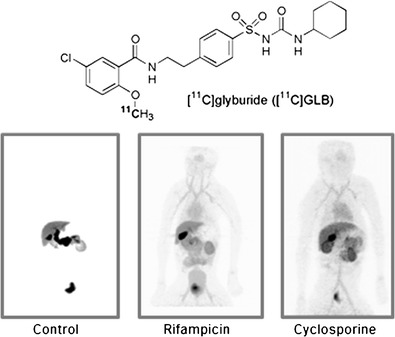
Whole-body PET images obtained 60 min following i.v. [11C]glyburide injection in a baboon under control conditions and after rifampicin (8.6 mg/kg) and cyclosporine (15 mg/kg/h) treatment
Fig. 3.
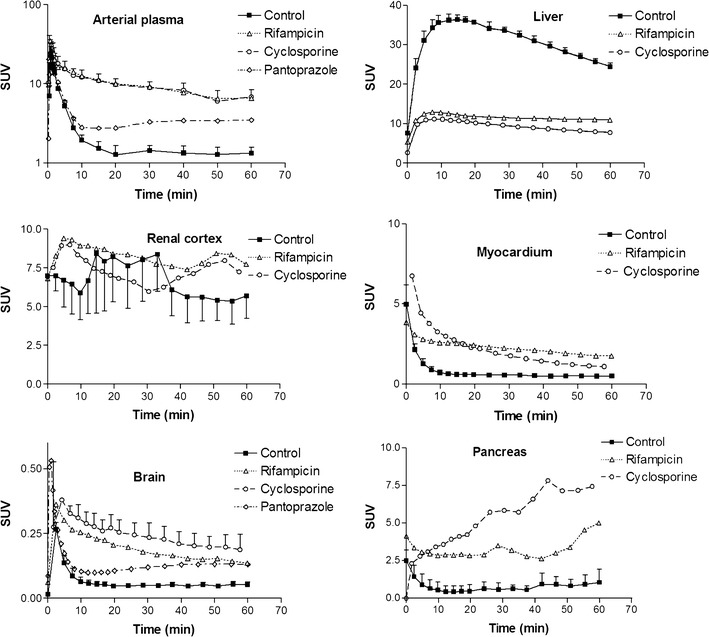
Mean time–activity curves in arterial plasma, liver, renal cortex, heart tissue, brain, and pancreas obtained after [11C]glyburide injection in a baboon under control conditions and after cyclosporine, rifampicin, and pantoprazole treatment
We observed the substantial accumulation of [11C]GLB in the renal cortex of control animals (SUVmax ∼10; Figs. 2 and 3). RFA and CsA did not affect overall renal exposure to [11C]GLB (AUCrenal cortex) (Fig. 3, Table I). However, the AUCrenal cortex/AUCplasma ratio of [11C]GLB was similarly decreased by both treatments (Table I). The amount of radioactivity in the urine varied substantially between and within groups, ranging from 0.1 to 2% of the injected dose. Radio-HPLC analysis of urine samples showed the presence of GLB metabolites; no [11C]GLB was detected under any test conditions.
The AUCmyocardium/AUCplasma ratio of [11C]GLB was decreased by both CsA and RFA. This phenomenon was not observed for the lungs or the pancreas (Table I). The brain uptake of [11C]GLB was low (SUVmax = 0.6, Fig. 3) and the AUCbrain/AUCplasma ratio was not affected by either RFA, CsA, or pantoprazole treatment (Table I).
Cell Accumulation Assay
The lack of a difference between the effects of CsA and RFA treatment on [11C]GLB brain uptake in baboons prompted us to assess the contribution of P-gp compared to BCRP to GLB transport in vitro. We used MDCKII cells transfected with the human isoforms of P-gp (MDR1) and BCRP, considering the homology between humans and monkeys. In MDCKII-MDR1 cells, [3H]GLB uptake was significantly increased by the P-gp inhibitor PSC833 and the dual P-gp/BCRP inhibitor GF120918. No effect of FTC, a specific BCRP inhibitor, was detected. Experiments with BCRP-transfected cells showed that GLB was also transported by human BCRP, since both FTC and GF120918 increased GLB uptake. The effect of PSC833 on MDCKII-BCRP cells was considered to be independent of BCRP and probably due to the presence of endogenous (canine) P-gp expression, which has been previously described in this cell line (5,18) (Fig. 4).
Fig. 4.
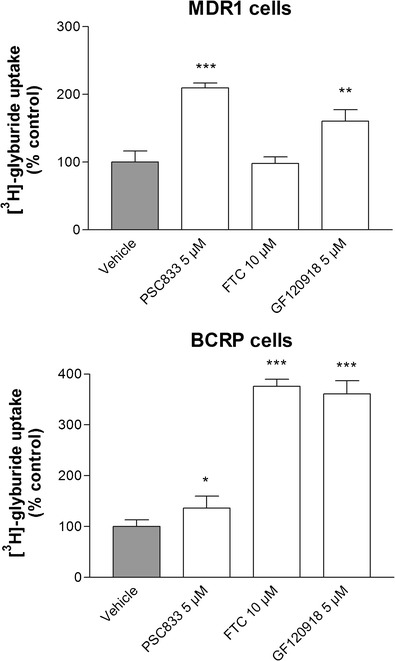
[3H]glyburide uptake in MDCKII-MDR1 and -BCRP cells in the presence of inhibitors of P-gp (PSC833), BCRP (FTC), and both P-gp and BCRP (GF120918). Data are expressed as the percentage of [3H]GLB accumulated in cell monolayers in the presence of the inhibitor tested, compared to control conditions (vehicle) (means ± SD). Significant difference from controls: *p < 0.05; **p < 0.01; ***p < 0.001
In situ Brain Perfusion in Mice
In situ experiments were performed in mice to assess the contribution of ABC and OATP transporters to GLB transport at the luminal membrane of the BBB. The intrinsic brain transport rate of [3H]GLB, Kin, in wild-type mice was 0.50 ± 0.11 μL/g/s. Comparison with the brain clearance rate of diazepam (Kin = 42.3 μL/g/s), a fluid flow marker, measured under the same conditions, indicates that the brain took up ∼1.2% of the perfused [3H]GLB (21). There was no significant increase in brain permeability to [3H]GLB following the full and selective inhibition of either P-gp, BCRP, or MRP. Conversely, even partially reduced P-gp and BCRP transport activity led to a significant increase in [3H]GLB brain uptake, with a maximal ∼3-fold effect in P-gp/BCRP-deficient mice or using GF120918 10 μM. The effect of MRP inhibition by MK-571 was evidenced in P-gp/BCRP-deficient mice but not in wild-type mice. In wild-type mice, co-perfusion with RFA, a chemical inhibitor of OATP, did not significantly influence [3H]GLB brain uptake (Fig. 5).
Fig. 5.
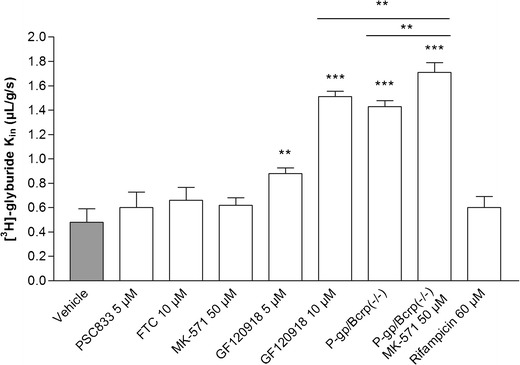
[3H]glyburide transport rate (K in; in microliter per gram per second) measured at the mouse blood–brain barrier using in situ brain perfusion. Data are presented as means ± SD (n = 4–6). Significant difference from controls: **p < 0.01; ***p < 0.001
DISCUSSION
Our PET data demonstrate that OATP transporters are a critical factor controlling the uptake of [11C]GLB by the liver. Indeed, GLB is a substrate for OATP2B1 and OATP1B1, which are expressed at the sinusoidal membrane of hepatocytes. It has been shown that CYP3A4 and, to a lesser extent, CYP2C9 are the main liver enzymes involved in GLB metabolism, resulting in the production of 4-trans-hydroxyglyburide and 3-cis-hydroxyglyburide in vivo (24). CsA and pantoprazole are capable of inhibiting liver CYP3A4 activity (25,26) whereas RFA is not (27). [11C]GLB metabolites could not be detected in the plasma after RFA infusion, in accordance with reduced liver uptake of [11C]GLB. This suggests that the availability of GLB for metabolic enzymes is greatly dependent on OATP activity, which represents the first and most important molecular step in GLB hepatic clearance.
The elimination of radioactivity by the bile duct via the gallbladder was obvious in our PET images (Fig. 2). However, due to great variability between experiments, our PET data regarding the gallbladder cannot provide accurate information as to the differential impact of P-gp inhibition on the biliary clearance of GLB. The time period investigated in the PET experiments, as compared to the half-life of GLB reported in humans (∼4.7 h), is probably too short to accurately explore the GLB elimination phase (14). Nevertheless, the lack of a difference between RFA and CsA treatments on liver and plasma [11C]GLB AUCs suggests that the additional impact of inhibiting P-gp is minor, at least when OATP is already inhibited at the liver. This is in agreement with the sequential and synchronized activity of sinusoidal OATP, cytosolic enzymes, and canalicular P-gp in hepatocytes (12). Furthermore, a synergistic kinetic compensatory mechanism involving P-gp, BCRP, and MRP has been described for some triple P-gp/BCRP/MRP substrates, which could potentially offset the inhibition of P-gp alone (28). Indeed, the combined deletion or inhibition of both P-gp and BCRP is thus necessary to demonstrate their activity (28,29). It has also been reported that Bcrp deficiency alone does not affect systemic GLB clearance in mice (5). Unfortunately, we found no validated and readily available dual P-gp/BCRP inhibitor that could be safely injected in baboons and that did not interact with OATP transporters, to test the hypothesis of a cooperative ABC efflux system with [11C]GLB (30). Instead, we used high-dose pantoprazole (30 mg/kg/h) to inhibit P-gp and BCRP, based on a previous report in rodents (31). This treatment was well tolerated and increased AUCplasma (Table I). However, the impact of pantoprazole on [11C]GLB metabolism does not allow us to conclusively determine the role of ABC transporters in [11C]GLB body distribution and their role at the canalicular membrane of hepatocytes. This requires further investigation with a validated broad-spectrum ABC inhibitor.
The tissue/plasma ratio of [11C]GLB in the renal cortex was reduced by OATP inhibition, suggesting an OATP-mediated uptake of [11C]GLB by the renal parenchyma. However, OATP inhibition did not modify overall kidney exposure (AUCrenal cortex; Table I). In fact, the inhibition of OATP-mediated uptake at the kidney level might have counterbalanced the increase in [11C]GLB AUCplasma. It has been shown that ∼50% of GLB is eliminated in the urine, mainly as metabolites (14,32). Similarly, no unmetabolized [11C]GLB could be detected in the urine of baboons. This was not affected by RFA treatment, suggesting that OATP transporters play a minor role in the tubular excretion or reabsorption of GLB and its metabolites, as previously reported in human (14). This suggests that the initial step in renal secretion, i.e., the entry of the substrate from the blood into renal tubule cells, is not dependent on OATP transporter activity (12). Moreover, PET data from the urinary bladder and the analysis of urine samples failed to show any effect of CsA. Taken together, these data suggest that OATP and P-gp are not critical factors in the renal elimination of GLB.
The distribution of [11C]GLB in the pancreas was low, such that this organ could not be directly seen in PET images (Fig. 2). OATP transporters did not seem to be involved in [11C]GLB uptake by the pancreas, despite the significant expression of these transporters in pancreatic tissue (33). However, the increased pancreatic exposure (AUCpancreas) to [11C]GLB following inhibitor treatment illustrates the consequences of drug–drug interactions between GLB and OATP inhibitors. This has been described in patients in whom the hypoglycemic effects for GLB were enhanced when RFA was co-administered (14). Our results suggest that CsA might have similar effects.
The role of OATP2B1 in the vascular endothelium of human cardiac tissue has been illustrated ex vivo using atorvastatin (34). In the present study, both RFA and CsA similarly decreased [11C]GLB accumulation in the myocardium, suggesting that OATP2B1 influences the cardiac uptake of [11C]GLB, since OATP1B1 and 1A2 are not expressed in this tissue (33,34). To our knowledge, this is the first study to demonstrate the activity of OATP transporters in the heart tissue in vivo. Thus, the influence of the OATP2B1-mediated cardiac uptake of GLB on the efficacy of treatments used for cardioprotective ischemic preconditioning deserves further attention (35).
In contrast to cardiac tissue, GLB brain uptake was not influenced by OATP inhibition in baboons. Recent proteomic analyses of human and monkey brain microvessels have not revealed any OATP transporters involved in GLB transport at the BBB (15,36). Our in situ brain perfusion experiments in mice have confirmed that GLB brain uptake is not influenced by OATP function.
The distribution of GLB in baboon and mouse brains is fairly poor, despite its favorable lipophilicity (LogD7.4 = 1.8). However, several other detrimental physiochemical factors such as the number of heteroatoms, hydrogen bonds, and the high polar surface area could explain the low membrane and BBB permeability of GLB (37). GLB binding to plasma proteins is high (∼99.9%) and contributes to restricting GLB distribution (38). However, the brain perfusion fluid we used in mice did not contain proteins and the intrinsic permeability of the BBB for GLB is low. Our results suggest that GLB transport across the BBB is restricted by cooperation and synergy between several ABC transporters, as single ABC inhibition had no significant effect in mice or baboons. In mice, the combined inhibition of P-gp and BCRP using GF120918 significantly increased GLB transport across the BBB in a dose-dependent manner. Unfortunately, we cannot be sure that P-gp and BCRP were fully inhibited or even modulated at the dose of pantoprazole we used in baboons.
The transport of GLB was enhanced by the MRP inhibitor MK-571 in P-gp/BCRP-deficient mice. This suggests that a MK-571-sensitive efflux transporter, possibly MRP4 (16), could be also involved in GLB transport at the luminal BBB. Finally, in the absence of functional P-gp, BCRP, and MRP4 on the luminal side of the mice BBB, the brain uptake of GLB was increased 3.4-fold, reaching a maximal brain extraction of ∼4%.
The critical role of OATP1A, OATP1B, and OATP2B1 in drug pharmacokinetics has aroused interest in developing specific and noninvasive tools to evaluate the impact of these transporters and the risk of drug–drug interactions (39,40). GLB labeling with carbon-11 permits the original structure of the compound and its transporter substrate properties to be preserved. Our study illustrates how GLB plasma pharmacokinetics and distribution are largely governed by OATP transporters. RFA and CsA have a more pronounced inhibitory effect on OATP1A2, OATP1B1, and OATP1B3 than on OATP2B1 (13,41–44). In the present study, the organs in which [11C]GLB uptake was modified by RFA were more concordant with OATP2B1 distribution than with the pharmacological targets of GLB (45), such as SUR1, which is expressed in the brain, pancreas, and spinal cord (46). However, before [11C]GLB can be used as a probe to study OATP2B1 function in vivo, further investigations are necessary to assess its specificity for other OATP isoforms.
CONCLUSION
In conclusion, the present study demonstrates how OATP transporters control GLB distribution, mainly in the liver, with a subsequent impact on its metabolism, pharmacokinetics, and the exposure of several organs including the pancreas and the brain. The inhibition of P-gp in addition to OATP had no further impact on GLB distribution. Conversely, P-gp, BCRP, and possibly MRP4 work in concert to restrict GLB transport across the BBB. Thus, our results clarify the pharmacological consequences of the interaction between GLB and inhibitors of the OATP and ABC transporters in vivo.
ACKNOWLEDGMENTS
We thank Maria Smirnova, Vincent Brulon, and Amandine Grelier, who contributed to the research. Statistical analyses were performed by Dr. Marcel Debray. The English text was edited by Dr. S. Rasika. Salvatore Cisternino received a grant from the Commissariat à l'énergie atomique et aux énergies alternatives and the Assistance Publique des Hôpitaux de Paris.
Conflict of Interest
The authors have no conflict of interest to declare.
References
- 1.Schattling B, Steinbach K, Thies E, Kruse M, Menigoz A, Ufer F, et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2012;18:1805–1811. doi: 10.1038/nm.3015. [DOI] [PubMed] [Google Scholar]
- 2.Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: a focused review. J Cereb Blood Flow Metab. 2012;32:1699–717. doi: 10.1038/jcbfm.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gedeon C, Behravan J, Koren G, Piquette-Miller M. Transport of glyburide by placental ABC transporters: implications in fetal drug exposure. Placenta. 2006;27:1096–1102. doi: 10.1016/j.placenta.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Cygalova LH, Hofman J, Ceckova M, Staud F. Transplacental pharmacokinetics of glyburide, rhodamine 123, and BODIPY FL prazosin: effect of drug efflux transporters and lipid solubility. J Pharmacol Exp Ther. 2009;331:1118–1125. doi: 10.1124/jpet.109.160564. [DOI] [PubMed] [Google Scholar]
- 5.Zhou L, Naraharisetti SB, Wang H, Unadkat JD, Hebert MF, Mao Q. The breast cancer resistance protein (Bcrp1/Abcg2) limits fetal distribution of glyburide in the pregnant mouse: an Obstetric-Fetal Pharmacology Research Unit Network and University of Washington Specialized Center of Research Study. Mol Pharmacol. 2008;73:949–959. doi: 10.1124/mol.107.041616. [DOI] [PubMed] [Google Scholar]
- 6.Pollex E, Lubetsky A, Koren G. The role of placental breast cancer resistance protein in the efflux of glyburide across the human placenta. Placenta. 2008;29:743–747. doi: 10.1016/j.placenta.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Hemauer SJ, Patrikeeva SL, Nanovskaya TN, Hankins GDV, Ahmed MS. Role of human placental apical membrane transporters in the efflux of glyburide, rosiglitazone, and metformin. Am J Obstet Gynecol. 2010;202:383.e1–383.e7. doi: 10.1016/j.ajog.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gedeon C, Anger G, Piquette-Miller M, Koren G. Breast cancer resistance protein: mediating the trans-placental transfer of glyburide across the human placenta. Placenta. 2008;29:39–43. doi: 10.1016/j.placenta.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Golstein PE, Boom A, van Geffel J, Jacobs P, Masereel B, Beauwens R. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 1999;437:652–660. doi: 10.1007/s004240050829. [DOI] [PubMed] [Google Scholar]
- 10.Satoh H, Yamashita F, Tsujimoto M, Murakami H, Koyabu N, Ohtani H, et al. Citrus juices inhibit the function of human organic anion-transporting polypeptide OATP-B. Drug Metab Dispos. 2005;33:518–523. doi: 10.1124/dmd.104.002337. [DOI] [PubMed] [Google Scholar]
- 11.Koenen A, Köck K, Keiser M, Siegmund W, Kroemer HK, Grube M. Steroid hormones specifically modify the activity of organic anion transporting polypeptides. Eur J Pharm Sci. 2012;47:774–780. doi: 10.1016/j.ejps.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 12.König J, Müller F, Fromm MF. Transporters and drug–drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 13.Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164–172. doi: 10.1053/jhep.2002.34133. [DOI] [PubMed] [Google Scholar]
- 14.Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin's inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85:78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shawahna R, Uchida Y, Declèves X, Ohtsuki S, Yousif S, Dauchy S, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8:1332–1341. doi: 10.1021/mp200129p. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal S, Uchida Y, Mittapalli RK, Sane R, Terasaki T, Elmquist WF. Quantitative proteomics of transporter expression in brain capillary endothelial cells isolated from P-glycoprotein (P-gp), breast cancer resistance protein (Bcrp), and P-gp/Bcrp knockout mice. Drug Metab. Dispos. 2012;40:1164–1169. doi: 10.1124/dmd.112.044719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhnast B, Damont A, Tournier N, Saba W, Valette H, Bottlaender M, et al. Radiosynthesis of [C-11]glyburide for in vivo imaging of BCRP function with PET. J Label Compounds Radiopharm. 2011;54(Suppl 1):S262. [Google Scholar]
- 18.Tournier N, Chevillard L, Megarbane B, Pirnay S, Scherrmann J-M, Declèves X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) Int J Neuropsychopharmacol. 2010;13:905–915. doi: 10.1017/S1461145709990848. [DOI] [PubMed] [Google Scholar]
- 19.Cisternino S, Mercier C, Bourasset F, Roux F, Scherrmann J-M. Expression, up-regulation, and transport activity of the multidrug-resistance protein Abcg2 at the mouse blood–brain barrier. Cancer Res. 2004;64:3296–3301. doi: 10.1158/0008-5472.CAN-03-2033. [DOI] [PubMed] [Google Scholar]
- 20.Sane R, Agarwal S, Mittapalli RK, Elmquist WF. Saturable active efflux by P-glycoprotein and breast cancer resistance protein at the blood–brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther. 2013;345:111–124. doi: 10.1124/jpet.112.199786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cattelotte J, André P, Ouellet M, Bourasset F, Scherrmann J-M, Cisternino S. In situ mouse carotid perfusion model: glucose and cholesterol transport in the eye and brain. J Cereb Blood Flow Metab. 2008;28:1449–1459. doi: 10.1038/jcbfm.2008.34. [DOI] [PubMed] [Google Scholar]
- 22.Mease K, Sane R, Podila L, Taub ME. Differential selectivity of efflux transporter inhibitors in Caco-2 and MDCK-MDR1 monolayers: a strategy to assess the interaction of a new chemical entity with P-gp, BCRP, and MRP2. J Pharm Sci. 2012;101:1888–1897. doi: 10.1002/jps.23069. [DOI] [PubMed] [Google Scholar]
- 23.Xie M, Rich TC, Scheitrum C, Conti M, Richter W. Inactivation of multidrug resistance proteins disrupts both cellular extrusion and intracellular degradation of cAMP. Mol Pharmacol. 2011;80:281–293. doi: 10.1124/mol.111.071134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou L, Naraharisetti SB, Liu L, Wang H, Lin YS, Isoherranen N, et al. Contributions of human cytochrome P450 enzymes to glyburide metabolism. Biopharm Drug Dispos. 2010;31:228–242. doi: 10.1002/bdd.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amundsen R, Åsberg A, Ohm IK, Christensen H. Cyclosporine A- and tacrolimus-mediated inhibition of CYP3A4 and CYP3A5 in vitro. Drug Metab Dispos. 2012;40:655–661. doi: 10.1124/dmd.111.043018. [DOI] [PubMed] [Google Scholar]
- 26.Li X-Q, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32:821–827. doi: 10.1124/dmd.32.8.821. [DOI] [PubMed] [Google Scholar]
- 27.Lau YY, Okochi H, Huang Y, Benet LZ. Pharmacokinetics of atorvastatin and its hydroxy metabolites in rats and the effects of concomitant rifampicin single doses: relevance of first-pass effect from hepatic uptake transporters, and intestinal and hepatic metabolism. Drug Metab Dispos. 2006;34:1175–1181. doi: 10.1124/dmd.105.009076. [DOI] [PubMed] [Google Scholar]
- 28.De Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 29.Lin F, Marchetti S, Pluim D, Iusuf D, Mazzanti R, Schellens JHM, et al. Abcc4 together with Abcb1 and Abcg2 form a robust co-operative drug efflux system that restricts the brain entry of camptothecin analogs. Clin Cancer Res. 2013;19:2084–2095. doi: 10.1158/1078-0432.CCR-12-3105. [DOI] [PubMed] [Google Scholar]
- 30.Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug–drug interactions. J Med Chem. 2012;55:4740–4763. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breedveld P, Pluim D, Cipriani G, Wielinga P, van Tellingen O, Schinkel AH, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 32.Pearson JG, Antal EJ, Raehl CL, Gorsch HK, Craig WA, Albert KS, et al. Pharmacokinetic disposition of 14C-glyburide in patients with varying renal function. Clin Pharmacol Ther. 1986;39:318–324. doi: 10.1038/clpt.1986.46. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2008;23:22–44. doi: 10.2133/dmpk.23.22. [DOI] [PubMed] [Google Scholar]
- 34.Grube M, Köck K, Oswald S, Draber K, Meissner K, Eckel L, et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin Pharmacol Ther. 2006;80:607–620. doi: 10.1016/j.clpt.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Juurlink DN, Gomes T, Shah BR, Mamdani MM. Adverse cardiovascular events during treatment with glyburide (glibenclamide) or gliclazide in a high-risk population. Diabet Med. 2012;29:1524–1528. doi: 10.1111/j.1464-5491.2012.03772.x. [DOI] [PubMed] [Google Scholar]
- 36.Hoshi Y, Uchida Y, Tachikawa M, Inoue T, Ohtsuki S, Terasaki T. Quantitative atlas of blood–brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci. 2013 doi: 10.1002/jps.23575. [DOI] [PubMed] [Google Scholar]
- 37.Clark DE. In silico prediction of blood–brain barrier permeation. Drug Discov Today. 2003;8:927–933. doi: 10.1016/S1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 38.Nanovskaya TN, Patrikeeva S, Hemauer S, Fokina V, Mattison D, Hankins GD, et al. Effect of albumin on transplacental transfer and distribution of rosiglitazone and glyburide. J Matern Fetal Neonatal Med. 2008;21:197–207. doi: 10.1080/14767050801929901. [DOI] [PubMed] [Google Scholar]
- 39.Takashima T, Kitamura S, Wada Y, Tanaka M, Shigihara Y, Ishii H, et al. PET imaging-based evaluation of hepatobiliary transport in humans with (15R)-11C-TIC-Me. J Nucl Med. 2012;53:741–748. doi: 10.2967/jnumed.111.098681. [DOI] [PubMed] [Google Scholar]
- 40.Leonhardt M, Keiser M, Oswald S, Kühn J, Jia J, Grube M, et al. Hepatic uptake of the magnetic resonance imaging contrast agent Gd-EOB-DTPA: role of human organic anion transporters. Drug Metab Dispos. 2010;38:1024–1028. doi: 10.1124/dmd.110.032862. [DOI] [PubMed] [Google Scholar]
- 41.Bruderer S, Aänismaa P, Homery M-C, Häusler S, Landskroner K, Sidharta PN, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 2012;14:68–78. doi: 10.1208/s12248-011-9316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Bruyn T, Fattah S, Stieger B, Augustijns P, Annaert P. Sodium fluorescein is a probe substrate for hepatic drug transport mediated by OATP1B1 and OATP1B3. J Pharm Sci. 2011;100:5018–5030. doi: 10.1002/jps.22694. [DOI] [PubMed] [Google Scholar]
- 43.Picard N, Levoir L, Lamoureux F, Yee SW, Giacomini KM, Marquet P. Interaction of sirolimus and everolimus with hepatic and intestinal organic anion-transporting polypeptide transporters. Xenobiotica. 2011;41:752–757. doi: 10.3109/00498254.2011.573882. [DOI] [PubMed] [Google Scholar]
- 44.König J, Glaeser H, Keiser M, Mandery K, Klotz U, Fromm MF. Role of organic anion-transporting polypeptides for cellular mesalazine (5-aminosalicylic acid) uptake. Drug Metab Dispos. 2011;39:1097–1102. doi: 10.1124/dmd.110.034991. [DOI] [PubMed] [Google Scholar]
- 45.Obaidat A, Roth M, Hagenbuch B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol. 2012;52:135–151. doi: 10.1146/annurev-pharmtox-010510-100556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20:452–477. doi: 10.2133/dmpk.20.452. [DOI] [PubMed] [Google Scholar]