

Abstract
Despite the fact that a significant percentage of the population is unable to swallow tablets and capsules, these dosage forms continue to be the default standard. These oral formulations fail many patients, especially children, because of large tablet or capsule size, poor palatability, and lack of correct dosage strength. The clinical result is often lack of adherence and therapeutic failure. The American Association of Pharmaceutical Scientists formed a Pediatric Formulations Task Force, consisting of members with various areas of expertise including pediatrics, formulation development, clinical pharmacology, and regulatory science, in order to identify pediatric, manufacturing, and regulatory issues and areas of needed research and regulatory guidance. Dosage form and palatability standards for all pediatric ages, relative bioavailability requirements, and small batch manufacturing capabilities and creation of a viable economic model were identified as particular needs. This assessment is considered an important first step for a task force seeking creative approaches to providing more appropriate oral formulations for children.
KEY WORDS: children, drugs, formulations, manufacturing, palatability, pediatrics, regulatory
INTRODUCTION
Children require different oral dosage forms from adults due to differences in swallowing abilities, taste preferences, and dosage requirements. Most medications are produced for adults as capsules and tablets, which are often not suitable for children. The pediatric formulations available in the USA are generally liquids or powders for reconstitution. These require purified water and refrigeration, both of which are not guaranteed in the developing world nor readily available in the case of natural disasters. Extemporaneous formulations are a common work-around for the lack of commercially available preparations, but concerns regarding lack of dose accuracy, stability, and consistency in preparation present difficulties for both practitioners and caregivers (1). From the pediatricians' perspective, the availability of easy to swallow and palatable formulations can mean the difference between treatment success and failure (2). The age at which children are able to swallow tablets or capsules varies widely, but is generally expected at approximately age 7 and varies with tablet and capsule size. Children commonly refuse to take medication if it tastes bad. Prednisone for status asthmaticus (3) and clindamycin for skin and soft tissue infections (4) are obvious examples; both are extremely bitter compounds which are difficult to mask with foods or liquids and commonly meet with a great deal of resistance and refusal from children. The result of lack of adherence can and does lead to hospital admissions for intravenous therapy. Children with acute lymphoblastic leukemia receive a 2-year course of daily oral 6-mercaptopurine maintenance chemotherapy, for which the only licensed product is a tablet; a quick perusal of the web shows blogs of parents describing how they grind up the tablets in their kitchens on a daily basis. In this case, not only is the sick child potentially receiving an inaccurate dose, but the entire family is exposed to the chemotherapy.
There are many reasons for the dearth of oral pediatric formulations, with no one single factor predominating. Children represent a small proportion of the sick population. Although this is fortunate, it necessarily follows that the market for these formulations is small. In addition, the pediatric population as a whole (0–17 years) is heterogeneous, with differing formulation requirements depending on the age and developmental and clinical state of the patient. A variety of technical problems are encountered when attempting to create a product that can be swallowed easily by young children or dissolved in a small amount of food or liquid and that is suitably taste masked. There exists no consistent guidance on dosage form standards for pediatric age, taste preference standards, or acceptable excipients for use in pediatric formulations, which can lead to costly delays while toxicology studies are performed. Finally, there are regulatory issues regarding need for bioequivalence which may require additional discussion between regulatory and industry stakeholders.
It should be noted that swallowing difficulties are not solely a pediatric issue. Other patient subpopulations such as the elderly, or those debilitated by stroke might also benefit from formulations specifically designed for children. In this paper, we identify and explore various issues associated with production of suitable oral pediatric dosage forms. We also propose a workflow method which could be incorporated into pediatric drug development and potential areas of future research.
CHALLENGES IN THE DEVELOPMENT OF PEDIATRIC DOSAGE FORMS FROM THE INDUSTRIAL PERSPECTIVE
The pediatric population spans a diverse range of physical size and developmental capabilities. This diversity drives the need for different formulations, a wide range of dosage strengths within each formulation, or titratable formulations. Clinical testing of prototype dosage forms in the pediatric population is limited for ethical reasons, and so, these bioequivalence studies are performed in adults.
Design requirements for oral formulations are primarily based on the patient age, body size, and swallowing capability of the target population. Establishing the design requirements is generally complicated when the age range of the target population spans from birth to 8 or 10 years of age, as one specific type of dosage form is not ideal to cover this wide range. Information exists in the literature and from the European Medicines Agency (EMA) regarding possible acceptable dosage forms for various ages of patients (5). For patients below 2 years, liquid dosage forms are widely acceptable. In some cases, orally disintegrating or film strip-type formulations may also be acceptable; their safety profile with widespread use is, however, not known. Between the ages of 2 and 6 years, the ability of a child to swallow a small tablet or capsule is highly variable and many times based on the child's past experience with a particular drug or dosage form. A 2011 EMA guideline (6) provides a guide on tablet size for various pediatric age groups; for example, tablets should be no larger than 5 mm for patients less than 6 years of age. Even so, this size can still be a challenge to swallow for many patients (7), so a liquid or orally disintegrating dosage form should be considered. When patients are over the age of 6 years, there is better acceptance of small to medium tablets intended for swallowing, but there is a significant percentage of the population that still has difficulty swallowing tablets or capsules. Most children 12 years and older can swallow a tablet or capsule of reasonable size, but what constitutes “reasonable” will vary from patient to patient. In addition to the dosage form itself, the number of strengths required is an important design issue. When the age or weight range of the treated population is wide, more flexibility in dosage strengths may be necessary. Liquid dosage forms are considered the most flexible in this regard, but liquid formulations carry some important limitations. Liquids must be accurately measured by the caregiver. If the liquid is a suspension, the bottle must be well shaken to suspend the drug and distribute it evenly throughout the liquid. Large multiple-use bottles are inconvenient to transport, and an accurate measuring device must be carried along with the bottle. Volume must be taken into consideration: too small, and the dose may be inaccurate; too large, and adherence will become problematic. Liquids also require preservatives, which may lead to excipient safety concerns. One significant liability associated with liquids is the potential for taste issues and the need for taste masking, as described later in this review. This problem is not restricted to liquids. Many solid oral dosage forms (including film coated tablets, e.g., amoxicillin, and capsules, e.g., clindamycin) can have taste problems due to the very bitter taste of the active ingredient. When solid oral dosage forms are developed, the dosage flexibility is only achieved through the available number of dosage strengths. The EMA does not generally recommend solid oral dosage forms that are split or crushed to achieve the target dose, because the active pharmaceutical ingredient (API) is generally not evenly dispersed throughout the tablet (8,9), unless there is validation of the process.
Some flexibility in dosage administration can be achieved with granules or multi-particulate dosage forms or by tablets that are intended to be orally disintegrating. These tablets can also be administered by dispersing the tablet in a liquid prior to administration, but this requires that the caregiver estimates the correct portion of liquid to administer. Although direct administration with food or beverages should not be the primary design for a dosage form, the potential use of this type of administration should be assessed and evaluated for stability and acceptability in patients.
When developing liquid dosage forms, the solubility and stability of the API are critical to designing an appropriate drug product. The API should be stable enough to allow for at least 18 months of shelf life for the intended commercial product. For APIs with high aqueous solubility and acceptable stability, it is generally easier to design a liquid dosage form as a solution that will have good dose uniformity. Special techniques are needed to develop liquid solutions with low aqueous solubility drugs. While an advantage of APIs with low aqueous solubility is that taste issues may be reduced, the challenge of dose uniformity when formulated as suspension increases significantly. Careful formulation development is required to ensure a suspension that can be accurately dosed with a reasonable amount of mixing.
It is seldom practical or desirable to perform relative bioavailability studies in pediatric subjects. The initial prototype dosage form that is developed must be studied in adults in order to understand the in vivo performance. This is the general position of most regulatory agencies, although the US FDA does offer a potential exception for drugs that are classified as Biopharmaceutics Classification System (BCS) Class I (10). This requirement needs to be factored into the overall development program for pediatric dosage forms. Recently, there has been discussion of whether the extrapolation of BCS data from adults to pediatric populations is appropriate (11). The BCS system is based on a fundamental model of the gastrointestinal tract for the estimation of the extent of absorption, taking into account important physicochemical–physiological parameters such as aqueous solubility, intestinal permeability, drug dose, volume of luminal contents, fluid flow rate, and intestinal surface area. Pediatric developmental changes must be taken into account, as they also play a key role in pharmacokinetics. For example, obvious maturation changes are related to the volume increase of luminal fluids, intestinal surface area, and intestinal permeability (12–15). Administered dose is also fundamentally important, and therefore, there may be a need for a more quantitative, dose-dependent approach to pediatric BCS (16,17). Wu and Benet (18) have proposed an alternative Biopharmaceutics Drug Disposition Classification System which includes the role of metabolism in classifying drugs. More research in this area, including potential development of a predictive dissolution testing method which could correlate in vitro data with in vivo product performance, would greatly simplify the development of pediatric dosage forms (19).
USE OF EXCIPIENTS IN PEDIATRIC FORMULATIONS
Many compounds in current development have low solubility and permeability and so require excipients to improve oral absorption. Excipients chosen for a pediatric formulation must be determined based on the specific API under development as well as the pediatric product profile. The safety of excipients in the pediatric population has recently been called into question for pediatric products. This may be especially critical for neonates. Adverse events cited, however, generally occur only when dosed in quantities much greater than the recommended accepted daily intake on a milligram per kilogram basis. Adverse and fatal reports linked to dosing benzyl alcohol or propylene glycol in neonates (20) show dosing in neonates ranged from 0.6 to 319.5 mg/kg/day for benzyl alcohol and a median of 204.9 mg/kg/day for propylene glycol. The ADIs for these compounds are 5 and 25 mg/kg, respectively. A conservative approach to minimize the use of excipients without compromising the quality of the product would be reasonable.
Prescription products intended for children have been marketed for decades. Strickley (21) summarized the excipients present in marketed commercial formulations used clinically in children for acute or chronic conditions. A global database akin to the US FDA CDER's Inactive Ingredient Guide (22), tailored specifically to define acceptable excipients and amounts in products intended for acute or chronic dosing in children, is sorely needed.
Recently, the European Pediatric Formulation Initiative (23) in cooperation with NICHD and the US Pediatric Formulation Initiative launched the Safety and Toxicity of Excipients in Pediatrics database. This is intended to become a useful tool to find information related to the safety and toxicity of excipients used in pediatric medications. Further information can be found at http://www.eupfi.org/gpage11.html.
In summary, selection of suitable excipients, including age-related safety profiles of the chosen excipients for the youngest neonates and children, and palatability and stability need to be taken into consideration when designing both adult and pediatric formulations. This is critical as there is significant off-label use.
MANUFACTURING CONSIDERATIONS
The market for pediatric medication needs is significantly smaller than for adult indications in all but a few cases. The small volume of product, and the potentially large number of dosage strengths, needed to supply the commercial market creates further difficulties to achieve an efficient supply chain using a typical commercial product manufacturing operation. Many commercial manufacturing operations are designed to produce relatively large numbers of dosage units per batch, which could easily be ten times the requirement for an entire year's supply for the pediatric market. The more suitable manufacturing operation would be approximately 10% of a normal large-scale commercial manufacturing operation, which would be more closely aligned with the batch sizes manufactured in a clinical trial production facility. In many cases, manufacturing even one of the smallest possible commercial scale batches of product for a pediatric indication is enough to supply the market for more than a year and much of the product has the potential to expire before the product is used. This makes the economics of producing commercial pediatric pharmaceutical products very challenging for most pharmaceutical companies.
There are a few potential solutions. The first possible approach would be to leverage manufacturing operations designed for the production of clinical trial materials at smaller batch sizes. A second possibility is to utilize manufacturing operations that specialize in small volume products and typically operate with much smaller batch sizes, which might be found in the range of contract manufacturing organizations that exist in the USA, EU, and Asia. One final possibility is to leverage continuous manufacturing. This approach might be well suited to manufacture several different sized dosage forms with minimal changes in the setup of equipment, with batch size determined based on time and throughput of the equipment. This is an area that needs further exploration by the pharmaceutical industry, industry associations, regulatory agencies, and contract manufacturing organizations, in order to effectively meet the demand for pediatric products. Adults will of course benefit from the availability of these more flexible dosage forms, so the market for these new dosage forms may grossly underestimate the market.
PALATABILITY AND TASTE MASKING
It is widely accepted that pediatric medicines must be palatable to ensure dose acceptance and adherence. Palatable drug products are those in which the aversive sensory attributes have been minimized or eliminated: they are not overly bitter, produce little trigeminal irritation, have smooth mouth feel, and have no perceptible malodors. Dosage form flexibility, solubility, stability, and taste masking effectiveness are important formulation design criteria. Many drug substances are bitter or have other “negative” or averse sensory characteristics (unpleasant aromas or mouth irritation). As a consequence, the development of palatable drug products can be a daunting challenge. In addition, much of the knowledge relating to the development of palatable products is concentrated in the food industry, which enjoys much greater freedom to operate with regard to food product taste testing than drug product taste testing in the pharmaceutical industry.
The term “flavor” refers to the combination of basic tastes, aromas, feeling factors, and textures perceived when a product is consumed. “Taste” refers to those sensations perceived through the stimulation of the receptor cells in the mouth. There are five separate taste types (referred to as the “basic tastes”)—sweet, sour, salty, bitter, and savory—with distinct receptor pathways. Odors (aromas) are volatile chemical compounds perceived via the sense of smell. Feeling factors are those sensations (cooling, numbing, and biting/burning) that arise when chemical compounds directly stimulate free nerve endings in the trigeminal nerve. Textures are the tactile characteristics of a product. Notable textural attributes for oral liquid drug products include viscosity, smoothness, and mouth coating. For solid oral dosage forms (tablets, soft chews, and orally dissolvable films), important texture attributes include roughness, hardness, fracturability, and cohesiveness, depending on the specific form (24).
The distinction between taste and smell is of paramount importance in developing palatable drug products as there is a general misconception that the simple addition of a flavor (e.g., grape) can reduce a bitter, or other basic, taste. Such is simply not the case, as the modalities of taste and aroma represent fundamentally different receptor pathways and loci of perception in the brain. Although flavoring aromatics cannot mask a bitter basic taste, other basic tastes are commonly used to mitigate the perception of bitterness.
As mentioned above, Strickley et al. (21) published a review of oral pediatric drugs and described a flowchart to support decision making regarding pediatric oral formulations. The framework depicted in Fig. 1 builds on this flowchart to provide more specific guidance in answering the key questions regarding palatability and is a useful paradigm in the evaluation of palatability strategies for formulation development. The flowchart is divided into four sections. The top section labeled “taste assessment” represents the key questions that need to be answered early to support the development of age-appropriate clinical trial materials. The second section labeled “technology guidance” provides a pathway for selecting and evaluating technologies such as particle coating or adsorption that may be required to develop a palatable pediatric drug product.
Fig. 1.
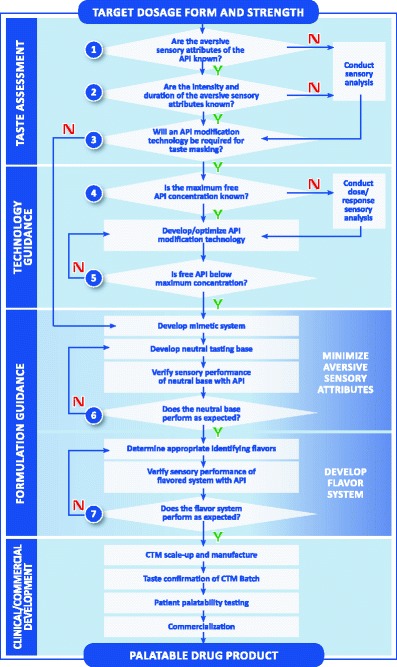
Decision framework: developing pediatric drug products [source: Senopsys LLC]
A formulation strategy for developing palatable drug products is found in the third section of the Fig. 1 flowchart labeled “formulation guidance.” The section is separated into two distinct parts. The first part is to minimize or eliminate the aversive (negative) attributes of the API. This is commonly referred to as creating a “neutral” tasting base. A neutral base exhibits balanced basic tastes, which is a foundational principle of drug palatability. Once the aversive attributes have been successfully ameliorated in the neutral base, the “desirable” (positive) attributes of the flavor system can be formulated, specifically the age-appropriate sweetness, flavoring aromatics, and feeling factors. Acidulents (sourness), sodium chloride (“salt”), and other sodium salts that produce saltiness can be beneficial in blending the basic tastes and are often included in the formulation design.
The starting point for taste optimization is a preliminary formulation containing the API and the excipients that are needed for the dosage form. The “electronic tongue” can be incorporated here in formulation development. The electronic tongue is a device which detects an electronic signal in a given formulation; the goal is to achieve an electronic signal of the formulation containing the active drug which is similar to formulation containing a placebo (25–27). A recent paper describes the use of the electronic tongue in formulating a palatable sodium phenylbutyrate preparation, an extremely difficult task (28). In certain cases, the API may have no aversive sensory attributes. Otherwise, the first step is to develop a “mimetic” system which is similar to the API, in order to reduce human exposure to unapproved drugs during development. A mimetic system uses Generally Recognized as Safe or FDA-listed excipients to replicate the aversive sensory attributes of the API. There are a number of compounds that can be used as surrogates for bitter APIs such as caffeine and quinine sulfate, sucrose octaacetate, and denatonium benzoate depending upon bitter intensity and duration.
The sweetener system is an important part of the initial formulation development. There are numerous candidate nutritive sweeteners (e.g., sucrose, fructose, or glucose) and nonnutritive sweeteners (e.g., sugar alcohols and artificial sweeteners) that differ in relative sweetness intensity, sweetness onset, duration, and chemical stability. Clinical considerations may weigh against the use of nutritive sweeteners due to their cariogenic potential and comorbidity issues (e.g., diabetes). In addition, some nonabsorbable sweeteners (e.g., sugar alcohols) may increase intestinal motility and thus may adversely affect the absorption of some medications or cause diarrhea (29).
Once candidate neutral base formulations have been developed, it is common practice to confirm that these unflavored bases perform as expected when the mimetic is replaced with the API (see Fig. 1, question 6). Based on the results, adjustments in excipient usage levels may sometimes be needed to improve the balance of the basic tastes.
When the aversive attributes have been successfully ameliorated in the neutral base formulation(s), flavors may be selected. The inclusion of identifying flavors (e.g., orange, strawberry, grape) does not play a role in the reduction of aversive basic tastes (e.g., bitterness). Appropriate flavor candidates are added to the neutral mimetic base and the formulation is evaluated for aromatic intensity, duration, and overall blend with the mimetic and excipients. Flavors of the same type share many of the same major aromatic chemicals regardless of supplier, and, therefore, multiple flavoring systems representing different flavor types (e.g., orange, strawberry, grape) are typically used. Next, the mimetic is again replaced with the API and the leading sweetened/flavored formulation is evaluated by the trained adult taste panel to confirm that it performs as expected (Fig. 1, question 7). Based on the results, final adjustments in excipient amounts are made.
Following finalization of a primary and back-up flavor system, the compatibility of formulations is assessed and then scaled up to clinical trial material batch size with requisite stability testing. The target shelf life for most drug products is typically 2 years, and the goal is to ensure the drug product remains palatable over this period. It is common practice to assess the sensory attributes of stability samples at 6-month intervals to the target expiry period.
Assessing drug palatability in children is complex and difficult. Challenges include design of the palatability study (single vs multiple dose, need for liquid dosage forms for younger children or children with swallowing problems); subject recruitment (age range, ability to communicate the complexities of palatability); ethical issues such as ability to enroll children in a nontherapeutic clinical study involving medications; logistical issues involving the family and transportation to a testing site; methodological, including baseline test preferences in various geographic and ethnic distributions, development of a test instrument and scales; and overall product acceptance. Regulatory agencies, however, are beginning to require sponsors to conduct palatability studies to ensure “acceptability” of pediatric drug products, for example as part of a pediatric investigation plan. Industry is equipped to handle challenges associated with recruitment and ethics, and many methods are available for measuring patient response to products. There is, however, no validated measure of product acceptance. There is considerable debate on the methodological aspects of affective testing in children age-appropriate study designs, test instruments, and scales. Global regulatory guidance is needed to define acceptability in children.
INNOVATIVE PEDIATRIC FORMULATIONS AND ADMINISTRATION DEVICES
Since the adoption of pediatric regulations in the USA and EU, there is a greater demand for age-appropriate medicines for children. Despite this growing demand, pediatric drug formulation science is still at an early stage, as it is complex, multiparametric, and resource and time intensive. As discussed previously, tablets and capsules cannot be swallowed by the very young, while liquid formulations may present multiple portability, stability, and dose accuracy problems (30,31).
Recently, there has been an effort to develop solid pediatric formulations that deliver the appropriate dose in a “user friendly” way, to find alternative dosage forms or drug delivery systems/approaches (e.g., mini-tablets), dosing vehicles (e.g., dairy products), and new taste masking techniques in order to improve acceptability and adherence. As the oral pathway is the most common route of drug administration, this is the area in which the greatest progress has been made. Small-sized dosage forms like mini-tablets, pellets, and sprinkles are preferred solid carriers which may be administered alone or dispersed in food. Another approach is to develop orally disintegrating drug formulations which disintegrate within few seconds in the oral cavity. Examples of these innovative dosage forms are oral lyophilisates, orally disintegrating tablets (ODTs), and orally disintegrating films (32). Combining both approaches, small-sized dosage forms and orally disintegrating formulations, have led to orally disintegrating mini-tablets (33) that may offer advantages for pediatric treatment over conventional techniques. Recently, the “pill swallowing cup” has been developed for patients who have difficulty in swallowing tablets. The cup, which contains the appropriate dose, is filled full with a beverage and then the patient drinks the liquid and drug from the cup (34). One potential disadvantage is the need for the child to swallow the full volume of the beverage, which may be problematic with young or ill children willing to drink only a sip or two at a time. Liquid dosage forms are preferred for newborn infants and young children (below 6 years old) instead of solid oral dosage forms such as capsules or tablets because of swallowing issues (7). For those forms that require administration with a measuring device, measuring spoons provide the appropriate dose, avoiding the use of inappropriate devices such as common household spoons (teaspoons and tablespoons) which can lead to inaccurate dosing (35,36). If larger volumes of medicine are required (>5 mL), then graduated measuring cups could be an alternative, although they may result in overdosing due to their restricted accuracy level. It has been found that oral syringes provide more accurate results than dosing spoons (37,38), but for the correct filling of the syringe, clear instructions should be provided to avoid air bubbles. A modified feeding bottle such as the Medibottle® has been developed, delivering the drug while the baby drinks. Dose sipping technology has been developed in order to deliver a single dose of small-sized pellets, overcoming swallowing issues (7,39). This technology incorporates small-sized pellets in a straw; when the child holds the straw in a beverage and sips, the drug is delivered in a user friendly way. An alternative drug delivery vehicle, which is familiar to infants and older children, is milk, a natural oil-in-water emulsion. Dissolving acidic drugs in alkaline solutions and then dispersing them into milk allow delivery of drugs to children in a “friendly” way (40).
BIOEQUIVALENCE AND THE CLINICAL DEVELOPMENT OF PEDIATRIC FORMULATIONS
There is no shortage of formulation technology which can be used to develop convenient, palatable, and safe formulations for children. Although liquid formulations have a number of advantages in early and late-stage clinical efficacy trials, these formulations are not always the best choice for consumer realization. The need for child-friendly formulations has been identified as a critical issue in pediatric drug development (41). One of the impediments for the pharmaceutical developer of pediatric formulations is how to bridge the market image (which could be a novel formulation such as an ODT or mini-tablet) to the standard capsule or tablet formulations(s) used in phases 2 and 3.
One possible solution is to avoid the issue entirely by using the novel formulation in the registration trials. However, this is seldom practical or cost-effective. First, early on in development, the dose may not be known for the various age groups, so a wide variety of doses would need to be developed for phase 2, which adds considerably to development timeline. Secondly, there is often a need for more than one marketed formulation—as what might be suitable for a 9-year old will not work for a 6-month old. Thus, the developer may have two (or more) formulations to develop prior to any clinical work being performed, and this could be prohibitively expensive and time consuming from a manufacturing perspective. Finally, there may be enabling studies (studies demonstrating stability and bioavailability of drug in the designated food) that would need to be performed prior to any trials being conducted for some “sprinkle”-type dosage forms that are intended to be mixed with food. These trials, although simple to perform, since they can be done using healthy adult volunteers, require quite a bit of resources to perform.
Another approach has been suggested (42) and is outlined in Fig. 2. This approach calls for enabling relative bioavailability studies of the pediatric clinical trials formulation followed by bioequivalence and food effect bridging studies upon successful completion of the pediatric phase 3 trial. Thus, a more fit-for-purpose formulation could be studied in the target pediatric population, and development of the to-be-marketed formulation would commence once there exists a reasonable chance that the drug will be safe and effective in children. Final bridging of the clinical trial formulation with the market image(s) could then occur during phase 3. On balance, this seems to be a reasonable approach, as it allows the sponsor to discharge the risk of developing a novel formulation before it is known that the compound is safe and effective in children. The needed bioequivalence studies may then be performed in adults, which are straightforward.
Fig. 2.
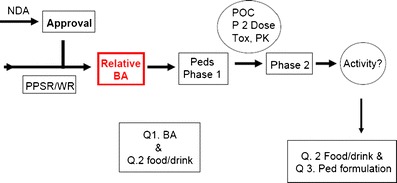
Suggested approach for pediatric formulation development (42)
To clarify the issues involving bioequivalence, some definitions may be useful. Pharmaceutical equivalence requires that the product contains the same active ingredient. Bioequivalence requires a statistical equivalence in the rate and extent of drug release available at the site of action. Drugs are considered to be therapeutically equivalent which are both pharmaceutically equivalent and bioequivalent (43).
It is important to note, however, that if there was a requirement that the market image(s) be bioequivalent to the clinical trials formulation, this could preclude the development of novel pediatric dosage forms. For example, a quickly dissolving lozenge or film, which may have many advantages when prescribed to pediatric patients, may yield a different mean Cmax than the suspension used in the phase 3 pediatric trials. Thus, the two formulations may not be equivalent with respect to Cmax. The next question is “Does it matter?” Although it is generally agreed that two formulations which are bioequivalent can be considered to have the same efficacy and safety profile, inequivalence does not automatically mean that one or the other formulation will be either ineffective or unsafe. In many cases, the efficacy of the drug depends on total exposure (AUC) and so a difference in Cmax geometric mean ratio beyond 0.8–1.25 would have no effect on efficacy or safety. A more flexible bioequivalence paradigm, taking into account the overall pharmacology and safety profile of the individual therapeutic agent (based on data provided by the sponsor before the bioequivalence study is conducted), may potentially encourage the development of novel pediatric formulations. Canada's regulations, for example, require that the geometric mean ratio fall between 0.80 and 1.250, while others require that the 90% confidence interval for the geometric mean ration fall within 0.80 and 1.250 (44). Other agencies have allowed some degree of flexibility on a case-by-case basis, but provide no specific written guidance. Raltegravir (Isentress®) is one example from the US FDA. Both peak and total exposures for the adult Isentress® tablet were markedly less than for the chewable pediatric formulation as determined by a bioequivalence study, and the chewable tablet was still approved. The labeling information reflects this difference: the dose is reduced from 400 mg twice a day for the conventional tablet to 300 mg twice a day for the chewable tablet.
It is clear that, for many therapeutic areas (e.g., analgesia), both Cmax and AUC are important, and so, these formulations would necessarily have to meet more stringent targets. Nevertheless, the option for a more fit-for-purpose bioequivalence regulatory framework for pediatrics might encourage the pharmaceutical industry to develop more innovative pediatric formulations and decrease the numbers of parents grinding up tablets every morning for their children. A clarification of this issue by FDA and other regulatory bodies would be helpful.
CLINICAL CONSIDERATIONS
Clinical development plans supporting the evaluation of pediatric formulations must address considerations for dose selection as well as study designs that permit easy interpretation of results and satisfy regulatory requirements. Intimately linked to these criteria is often the necessity to extrapolate dose requirements from either adult or preclinical (in vitro or animal) data. Typically, an equivalence approach is taken during initial pharmacokinetic and safety evaluation. Specifically, exposures that bracket and/or match adult targets are sought and dosing in pediatrics is guided by scaling methodology that compensates for size, developmental, or ontogenic considerations based on the intended pediatric population(s) (45). Pediatric formulations may present an additional hurdle as the in vitro–in vivo (IVIVC) correlation established in adults may not apply across the various targeted pediatric subpopulations. Understanding pediatric exposure requirements may be challenging for both conventional and novel formulations, especially if the pediatric indication is different from the adult clinical experience and true dose finding is required. In addition, modified release products or any delivery system providing other than immediate release oral input to achieve target in vivo exposures may require increased blood collection beyond what is allowable via normal institutional review board constraints. Satisfying these requirements requires both creative designs and candid discussions with clinical investigators regarding feasibility.
Modeling and simulation approaches, including population-based nonlinear mixed effect modeling, physiologic-based pharmacokinetic modeling, and other in silico or so-called bottom-up approaches, can facilitate the planning by providing valuable scenario testing to ensure the greatest likelihood of success (46–48). These approaches are also useful in the design of new pediatric formulations as they can be used to test assumptions regarding delivery requirements relative to the developing (particularly GI) physiology. Likewise, there is an evolving set of physiologic-based absorption models (e.g., ADAM, GITA, Grass) which are able to accommodate pediatric-specific anatomical parameters potentially improving guidance for pediatric formulations even further. Integrating in vitro drug characteristics, study design, sampling scheme, dosing requirements, and sample size into a complete trial simulation model would seem to be an optimal way of assuring adherence with both study objectives (49) and recent FDA quality standards on pediatric trial design (50).
ECONOMIC ISSUES
Innovation is driven by market forces. Although pediatric formulation needs are acute for children, their families, and health care workers, the overall pediatric market is small and hence a disincentive for research and innovation. The need for palatable formulations is not just a pediatric issue; it is estimated that approximately 50% of adults have difficulty swallowing tablets and capsules (51). The elderly, whose numbers are growing on a daily basis and those with medical conditions such as stroke which can affect swallowing, frequently cannot swallow tablet and capsule dosage forms designed for adults. This expands the intended market significantly. There is a need for discussions regarding the economic issues involved in the problem, need, and method for incentivizing production of convenient, palatable, high-quality dosage forms (52).
APPROACH TO DESIGNING THE PEDIATRIC DRUG PRODUCT
This document presents and discusses a number of the technical and economic challenges inherent to developing products for pediatric use. Given the complexity and interrelationship of these challenges, one approach to ensuring that clinical and commercial products meet the desired performance criteria is to create and document a robust product design. This approach is aligned with the concepts of quality by design, can set the foundation for defining the quality target product profile (QTPP), and places particular emphasis on the patient-centered aspects of the product design (http://www.ich.org/fileadmin/Public_Web_Site/Training/ICH_Endorsed_Training_Events/ASEAN_training_on_Q8_Q9_Q10_Guidelines/Q8_Pharma_development_JL.Robert.pdf). This documented product design, created by an interdisciplinary team, affords the opportunity for detailed discussion of the disease state, therapeutic goals, target population, and special requirements so that these challenges can be considered in conjunction with the traditional quality requirements of any drug product.
Establishing the specific pediatric population and capturing the therapeutic goals, as well as any special needs of the patient or their caregivers (e.g., physicians, parents, pharmacists), including items like comorbidities and use scenarios, disease state complications, and concomitant medications, are critical. These factors can then be integrated with more specific discussions on dose and dose flexibility needs, routes of administration, and challenges such as palatability as discussed above. It also provides an opportunity to discuss specific in vivo or pharmacokinetic performance requirements that may be different from those required in an adult population. Typically, it becomes apparent early in these discussions that the desired attributes of the drug product conflict with existing manufacturing capabilities, resulting in the need to begin to make trade-off decisions. For example, the preferred dosage form may be a solution, based upon patient population and dose flexibility needs, but technically not achievable due to a stability issue. The early identification of the tension between these factors provides opportunity to holistically optimize the dosage form and the resulting therapeutic outcome. Figure 3 outlines an approach to pediatric formulation development using elements of the QTPP approach.
Fig. 3.
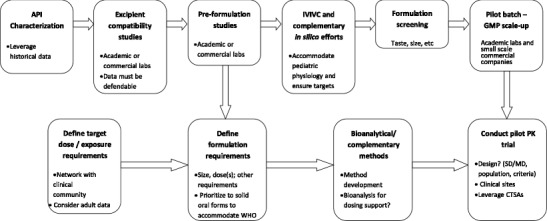
The path to pediatric formulation development: workflow for task force-led development plan. SD single dose trial, MD multiple dose trial, CTSA Clinical and Translational Science Award sites, WHO World Health Organization
Utilization of this design concept allows the team to iterate on strategic design criteria during development and make logical trade-off decisions as new information becomes available from the clinic or regulatory agencies. The design strategy also serves as a useful tool in communicating to regulatory agencies about the development strategy to be employed and therefore facilitates adherence with regulatory expectations (e.g., quality sections of a PIP).
AREAS FOR FUTURE RESEARCH
The need for improved oral pediatric dosage forms to optimize clinical care, with easy to swallow and palatable formulations in appropriate dosage increments, is overwhelming. Although this paper has focused on pediatric needs, a large number of adults would benefit from the availability of such formulations.
Areas for future research include the following:
Validation of the BCS for children of all ages or development of a modified, pediatric-specific BCS
Development of streamlined, algorithm-based approaches to formulation development, potentially based on BCS classification of the API; An interagency agreement between NICHD and FDA is exploring this possibility (http://bpca.nichd.nih.gov/collaborativeefforts/index.cfm).
Novel technologies for improving solubility and permeability with the use of pediatric-friendly and safe excipients
Scientific evaluation of pediatric excipients with long-term history of use but which have reports of anecdotal adverse events in the literature
A rational approach for the determination of pediatric dose based on adult BE studies
Pediatric dosage form preferences for specific age and developmental stage
Use of the electronic tongue and other in vitro methods of taste assessment and cross-validation with adult and pediatric taste panels
Pediatric taste preferences, validation, and cross-validation with adult taste panels
Novel technologies for taste masking with predictive evaluation methods
Research into economic models of small markets, including viable business models to reduce drug shortages and improve access to novel pediatric-friendly products
CONCLUSIONS
Clinicians, patients, and their caregivers, as well as society as a whole, place high value on pediatric clinical care. It necessarily follows that the availability of suitable pediatric dosage forms is of vital importance, as the availability of innovative, convenient, and high-quality pediatric products can spell the difference between successful treatment of a pediatric patient or failure.
The development and manufacture of innovative pediatric formulations is particularly difficult for a variety of reasons, as discussed above. By using other past challenges (e.g., orphan drug development) as a guide, the pharmaceutical industry would most likely respond to incentives, coupled with a reasonable and thoughtful regulatory framework. Consistency in requirements between regulatory agencies would facilitate and potentially expedite pediatric product development.
Any such framework must specifically consider the small market for pediatric formulations (relative to adults), the frequent necessity of developing more than one formulation, consistent guidance around excipient use and taste masking requirements, as well as consideration of a more fit-for-purpose bioequivalence strategy.
ACKNOWLEDGMENTS
The authors would like to thank Drs. Philip Mayer and David Mitchell and AAPS for facilitating this effort and to Sharon Pichon of AAPS for facilitating the calls and meetings with the co-authors
Footnotes
During the 2010 Pharmaceutical Sciences World Congress in New Orleans, 19 of the 47 partnering scientific organizations met to discuss further collaborations among all pharmaceutical scientific organizations. During that meeting, several topics were selected for further collaborations. This paper is the result of one of those topics.
DISCLAIMER
Comments and views of the authors do not necessarily represent the views of the governmental agencies or the other organizations with whom they are affiliated.
REFERENCES
- 1.Thompson KC. Extemporaneous formulations: Comparison with labeled pediatric formulations. American Pharmaceutical Review 2010 (March); 13(2).
- 2.Baguley D, Lim E, Bevan A, et al. Prescribing for children-taste and palatability affect adherence to antibiotics: a review. Arch Dis Child. 2012;97(3):293–297. doi: 10.1136/archdischild-2011-300909. [DOI] [PubMed] [Google Scholar]
- 3.Hendeles L. Selecting a systemic corticosteroid for acute asthma in children. J Peds. 2003;142:S40–S44. doi: 10.1067/mpd.2003.25. [DOI] [PubMed] [Google Scholar]
- 4.Steele RW, Russo TM, Thomas MP. Adherence issues related to the selection of antistaphylococcal or antifungal antibiotic suspensions for children. Clin Pediatr. 2006;45(3):245–250. doi: 10.1177/000992280604500306. [DOI] [PubMed] [Google Scholar]
- 5.EMEA Reflection Paper. Reflection paper formulations of choice for the paediatric population, 28 Jul 2006 EMEA/CHMP/PEG?194810/2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003782.pdf. Accessed 19 Jul 2013.
- 6.EMA Guideline on Pharmaceutical Development of Medicines for Paediatric Use. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/06/WC500107908.pdf. Accessed 19 Jul 2013.
- 7.Spomer N, Klingmann V, Stoltenberg I, et al. Acceptance of uncoated mini-tablets in young children: results from a prospective exploratory cross-over study. Arch Dis Child. 2012;97(3):283–286. doi: 10.1136/archdischild-2011-300958. [DOI] [PubMed] [Google Scholar]
- 8.Shah R, Collier J, Saveed V, Bryandt A, Habib M, Khan MA. Tablet splitting of a narrow therapeutic index drug: a case study with levothyroxine sodium. AAPS Pharm Sci Technol. 2010;11(2):818–825. doi: 10.1208/s12249-010-9414-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao N, Zidan A, Tawakkul M, Sayeed V, Khan MA. Tablet splitting: product quality assessment of metropolol succinate extended release tablets. Int J Pharm. 2010;401:25–31. doi: 10.1016/j.ijpharm.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 10.FDA Guidance to Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System, 2000. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070246.pdf. Accessed 19 July 2013.
- 11.Abdel-Rahman S, Amidon GL, Kaul A, et al. Summary of the National Institute of Child Health and Human Development-Best Pharmaceuticals for Children Act Pediatric Formulation Initiatives Workshop-Pediatric Biopharmaceutics Classification System Working Group. Clin Ther. 2012;34(11):S11–S24. doi: 10.1016/j.clinthera.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cella M, Gorter de Vries F, Burger D, Danhof M, Della Pasqua O. A model-based approach to dose selection in early pediatric development. Clin Pharmacol Ther. 2010;87(3):294–302. doi: 10.1038/clpt.2009.234. [DOI] [PubMed] [Google Scholar]
- 13.Knibbe CA, Danhof M. Individualized dosing regimens in children based on population PKPD modeling: are we ready for it? Int J Pharm. 2011;415(1–2):9–14. doi: 10.1016/j.ijpharm.2011.02.056. [DOI] [PubMed] [Google Scholar]
- 14.Cella M, Danhof M, Della Pasqua O. Adaptive trials in pediatric development: dealing with heterogeneity and uncertainty in pharmacokinetic differences in children. Br J Clin Pharmacol. 2012;74(2):346–353. doi: 10.1111/j.1365-2125.2012.04187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Cock RF, Allegaert K, Schreuder MF, Sherwin CM, de Hoog M, van den Anker JN, et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet. 2012;51(2):105–117. doi: 10.2165/11595640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Rinaki E, Valsami G, Macheras P. Quantitative biopharmaceutics classification system: the central role of dose/solubility ratio. Pharm Res. 2003;20(12):1917–1925. doi: 10.1023/B:PHAM.0000008037.57884.11. [DOI] [PubMed] [Google Scholar]
- 17.Charkoftaki G, Dokoumetzidis A, Valsami G, Macheras P. Elucidating the role of dose in the biopharmaceutics classification of drugs: the concepts of critical dose, effective in vivo solubility, and dose-dependent BCS. Pharm Res. 2012;29(11):3188–3198. doi: 10.1007/s11095-012-0815-4. [DOI] [PubMed] [Google Scholar]
- 18.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 19.Amidon G. Best Pharmaceuticals for Children Act Pediatric Formulations Initiative Workshop, Potomac, 2011, http://bpca.nichd.nih.gov/collaborativeefforts/upload/PFI_Workshop_11-1-2-11.pdf. Accessed 19 Jul 2013.
- 20.Shehab N, Lewis CL, Streetman DD, Donn SM. Exposure to the pharmaceutical excipient benzyl alcohol and propylene glycol among critically ill neonates. Pediatr Crit Care Med. 2009;10(2):256–259. doi: 10.1097/PCC.0b013e31819a383c. [DOI] [PubMed] [Google Scholar]
- 21.Strickley RG, Iwata Q, Will S, Dahl TC. Pediatric drugs—a review of commercially available oral formulations. J Pharm Sci. 2008;97(5):1731–1774. doi: 10.1002/jps.21101. [DOI] [PubMed] [Google Scholar]
- 22.Inactive Ingredients Database URL: http://www.fda.gov/Drugs/InformationOnDrugs/ucm113978.htm. Accessed 19 Jul 2013.
- 23.European Union Pediatric Formulations Initiative. http://www.eupfi.org/. Accessed 19 Jul 2013.
- 24.Meilgaard M, Civille GV, Carr BT. Sensory evaluation techniques. 3. Boca Raton: CRC; 1999. [Google Scholar]
- 25.Legin A, Rudnitskaya A, Clapham D, Seleznev B, Lord K, Vlasov Y. Electronic tongue for pharmaceutical analytics: quantification of tastes and masking effects. Anal Bioanal Chem. 2004;380(1):36–45. doi: 10.1007/s00216-004-2738-3. [DOI] [PubMed] [Google Scholar]
- 26.Lorenz JK, Reo JP, Hendl O, Worthington JH, Petrossian VD. Evaluation of a taste sensor instrument (electronic tongue) for use in formulation development. Int J Pharm. 2009;367(1–2):65–72. doi: 10.1016/j.ijpharm.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 27.Woertz K, Tissen C, Kleinebudde P, Breitkreutz J. Performance qualification of an electronic tongue based on ICH guideline Q2. J Pharm Biomed Anal. 2010;51(3):497–506. doi: 10.1016/j.jpba.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 28.Guffon N, Kibleur Y, Copalu W, Tissen C, Breitkreutz J. Developing a new formulation of sodium phenylbutyrate. Arch Dis Child. 2012;97:1081–1085. doi: 10.1136/archdischild-2012-302398. [DOI] [PubMed] [Google Scholar]
- 29.Chen ML, Straughn AB, Sadrich N, Meyer M, Faustino PJ, Ciavarella AB, et al. A modern view of excipient effects on bioequivalence: case study of sorbitol. Pharm Res. 2007;24(1):73–80. doi: 10.1007/s11095-006-9120-4. [DOI] [PubMed] [Google Scholar]
- 30.Richey RH, Craig JV, Shah UU, Ford JL, Barker CE, Peak M, et al. The manipulation of drugs to obtain the required dose: systematic review. J Adv Nurs. 2012;68(9):2103–2112. doi: 10.1111/j.1365-2648.2011.05916.x. [DOI] [PubMed] [Google Scholar]
- 31.Stegemann S, Gosch M, Breitkreutz J. Swallowing dysfunction and dysphagia is an unrecognized challenge for oral drug therapy. Int J Pharm. 2012;430:197–206. doi: 10.1016/j.ijpharm.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann EM, Breitenbach A, Breitkreutz J. Advances in orodispersible films for drug delivery. Exp Opin Drug Deliv. 2011;8:299–316. doi: 10.1517/17425247.2011.553217. [DOI] [PubMed] [Google Scholar]
- 33.Stoltenberg I, Breitkreutz J. Orally disintegrating mini-tablets (ODMTs)—a novel solid dosage form for pediatric use. Eur J Pharm Biopharm. 2011;78:462–469. doi: 10.1016/j.ejpb.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Walsh J, Bickmann D, Breitkreutz J, Chariot-Goulet M. Delivery devices for the administration of pediatric formulations: overview of current practice, challenges and recent developments. Int J Pharm. 2011;415:221–231. doi: 10.1016/j.ijpharm.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 35.Aziz MAHH, Jameela KA. How accurate are household spoons in drug administration? Med Princ Pract. 1990;2:106–109. [Google Scholar]
- 36.Madlon-Kay DJ, Mosch FS. Liquid medication dosing errors. J Fam Pract. 2000;49:741–744. [PubMed] [Google Scholar]
- 37.Griessmann K, Breitkreutz J, Schubert-Zsilavecz M, Abdel-Tawab M. Dosing accuracy of measuring devices provided with antibiotic oral suspensions. Paediatr Perinat Drug Ther. 2007;8:61–70. doi: 10.1185/146300907X178950. [DOI] [Google Scholar]
- 38.Dockhorn S, Feuersenger D, Schuenemann S, Knauf B, Duerr S, Schubert-Zsilavecz M, et al. Study of microbial contamination and dosing accuracy of oral dispensers. J Clin Pharm Ther. 2010;35:279–287. doi: 10.1111/j.1365-2710.2009.01082.x. [DOI] [PubMed] [Google Scholar]
- 39.Breitkreutz J, Boos J. Paediatric and geriatric drug delivery. Expert Opin Drug Deliv. 2007;4:37–45. doi: 10.1517/17425247.4.1.37. [DOI] [PubMed] [Google Scholar]
- 40.Charkoftaki G, Kytariolos J, Macheras P. Novel milk-based oral formulations: proof of concept. Int J Pharm. 2010;390:150–159. doi: 10.1016/j.ijpharm.2010.01.038. [DOI] [PubMed] [Google Scholar]
- 41.Rieder M. If children ruled the pharmaceutical industry: the need for pediatric formulations. Drug News Perspect. 2010;23:458–464. doi: 10.1358/dnp.2010.23.7.1458283. [DOI] [PubMed] [Google Scholar]
- 42.Booth, B. Pediatric formulations: what we want to know. Presentation to the FDA Pediatric Advisory Committee, 15 December 2009. www.fda.gov/downloads/AdvisoryCommittees/…/UCM197942.pdf. Accessed 19 Jul 2013.
- 43.Raw AS, Lionberger R, Yu LX. Pharmaceutical equivalence by design for generic drugs: modified release products. Pharm Res. 2011;28(7):1445–1453. doi: 10.1007/s11095-011-0397-6. [DOI] [PubMed] [Google Scholar]
- 44.Health Canada Guidance Document. Comparative bioavailability standards: formulations used for systemic effects. May 22, 2012. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/applic-demande/guide- ld/bio/gd_standards_ld_normes-eng.php. Accessed 19 Jul 2013.
- 45.Holford N. Dosing in children. Clin Pharmacol Ther. 2010;87:367–370. doi: 10.1038/clpt.2009.262. [DOI] [PubMed] [Google Scholar]
- 46.Barrett JS. Physiologically-based pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92(1):40–49. doi: 10.1038/clpt.2012.64. [DOI] [PubMed] [Google Scholar]
- 47.Johnson TN, Rostami-Hodjegan A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth. 2011;21:291–301. doi: 10.1111/j.1460-9592.2010.03323.x. [DOI] [PubMed] [Google Scholar]
- 48.Laer S, Barrett JS, Meibohm B. The in silico child: using simulation to guide pediatric drug development and manage pediatric pharmacotherapy. J Clin Pharmacol. 2009;49:889–904. doi: 10.1177/0091270009337513. [DOI] [PubMed] [Google Scholar]
- 49.Meibohm B, Läer S, Panetta JC, Barrett JS. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J. 2005;7:E475–E487. doi: 10.1208/aapsj070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Jadhav PR, Lala M, Gobburu JV. Clarification on precision criteria to derive sample size when designing pediatric pharmacokinetic studies. J Clin Pharmacol. 2012;52(10):1601–1606. doi: 10.1177/0091270011422812. [DOI] [PubMed] [Google Scholar]
- 51.Strachan I, Greener M. Medication-related swallowing difficulties may be more common than we realise. Pharm Pract. 2005;15:411–414. [Google Scholar]
- 52.Milne CP, Bruss JB. The economics of pediatric formulation development for off-patent drugs. Clin Ther. 2008;30(11):2133–2145. doi: 10.1016/j.clinthera.2008.11.019. [DOI] [PubMed] [Google Scholar]