

Abstract
During pregnancy, a drug’s pharmacokinetics may be altered and hence anticipation of potential systemic exposure changes is highly desirable. Physiologically based pharmacokinetics (PBPK) models have recently been used to influence clinical trial design or to facilitate regulatory interactions. Ideally, whole-body PBPK models can be used to predict a drug’s systemic exposure in pregnant women based on major physiological changes which can impact drug clearance (i.e., in the kidney and liver) and distribution (i.e., adipose and fetoplacental unit). We described a simple and readily implementable multitissue/organ whole-body PBPK model with key pregnancy-related physiological parameters to characterize the PK of reference drugs (metformin, digoxin, midazolam, and emtricitabine) in pregnant women compared with the PK in nonpregnant or postpartum (PP) women. Physiological data related to changes in maternal body weight, tissue volume, cardiac output, renal function, blood flows, and cytochrome P450 activity were collected from the literature and incorporated into the structural PBPK model that describes HV or PP women PK data. Subsequently, the changes in exposure (area under the curve (AUC) and maximum concentration (Cmax)) in pregnant women were simulated. Model-simulated PK profiles were overall in agreement with observed data. The prediction fold error for Cmax and AUC ratio (pregnant vs. nonpregnant) was less than 1.3-fold, indicating that the pregnant PBPK model is useful. The utilization of this simplified model in drug development may aid in designing clinical studies to identify potential exposure changes in pregnant women a priori for compounds which are mainly eliminated renally or metabolized by CYP3A4.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-013-9505-3) contains supplementary material, which is available to authorized users.
Key words: PBPK models, pharmacokinetics, physiological changes, pregnancy, systemic exposure
INTRODUCTION
As the use of medications during pregnancy is common (1–3), efforts continue to focus on safe and efficacious dosing regimen allowing women and their doctors to make informed decisions about continued treatments during pregnancy. While some drugs are known to show altered pharmacokinetics during pregnancy (4), anticipation of systemic exposure for medications taken by pregnant women has been difficult to determine, in part due to the lack of readily available whole-body physiologically based pharmacokinetics (PBPK) models. PBPK models have become more popular with both pharmacokinetic and formulation scientists, and advantages for the application of PBPK models as tools have been described (5,6), along with their structure, methodology, and implication in drug development (7–10). During pregnancy, apart from the development of the placenta and fetus, anatomical and physiological changes occur such as changes in blood flow, cardiac output, protein binding, renal, and hepatic functions (11,12); all of which may have significant effect on the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs. A number of recently published pregnant PBPK models (p-PBPK) incorporate comprehensive information related to anatomical, physiological, and metabolic changes which required complex and resource-intensive parameter inputs and custom programming (13,14). Although these models account for various time-variant parameter changes during gestation, key factors that influence the pharmacokinetics for pregnant women have only been partially described. Moreover, it is not always practical for clinical or pharmaceutical researchers to develop highly complex PBPK models in limited timeframes. Here, we describe an easily implementable mechanistic PBPK model that can characterize the disposition of drugs in pregnant women for four primarily renally excreted and CYP3A-metabolized compounds.
Significant changes in PK are more likely to be observed for drugs which have single elimination pathways. It is known that the CYP450 pathway involved is affected by pregnancy (15). Hence, reference drugs selected were compounds which are (a) primarily excreted via kidney, (b) known substrates of CYP450 isoenzymes and which undergo phase II metabolic pathways (e.g., N-acetyltransferase and glucoronidation, etc. (16)). Due to the regulations of the Food and Drug Administration, Clinical PK trials typically exclude both pregnant women and those of reproductive age because of ethical and practical considerations (17). For this reason, often only limited data are available for altered systemic PK parameters such as volume of distribution (Vd) and clearance (CL) change during pregnancy (17). Metformin, digoxin, midazolam, and emtricitabine were chosen as study drugs as published data were available (18–20). For metformin, digoxin, and emtricitabine, the major route of elimination is renal excretion. Midazolam is primarily metabolized and eliminated by CYP3A, which allows incorporating a specific-metabolizing enzyme change in the PBPK model. To our current knowledge, p-PBPK models have not been extensively described for prediction and analysis of PK changes for renally excreted compounds during pregnancy. Midazolam p-PBPK model has been established by Gaohua using Matlab Simulink. However, Cmax, area under the curve (AUC), and distribution phase were underpredicted (13). Here, we applied a simplified p-PBPK modeling strategy to predict the PK profiles in pregnant women by considering the essential physiologic and metabolic changes that occur during pregnancy for metformin, digoxin, midazolam, and emtricitabine. Furthermore, the simulated PK profiles were compared against published PK profiles from clinical outcomes (examples from literature) for model verification.
MATERIALS AND METHODS
Computer Hardware and Software
GastroPlus (version 8.0, Simulations Plus, Inc, CA, USA) embedded with the Advanced Compartmental Absorption Transit (ACAT) model and PBPKPlus™ module was run on a Lenovo computer with Intel® Core™ i5 processor. The ACAT model, originally established by Yu and Amidon (21), enables the prediction of rate and extent of oral drug absorption. The PBPKPlus™ module incorporates a whole-body PBPK model which defines mathematical compartments for all major tissues using a group of differential equations (22). This module can further predict the amount of drug that distributes to each tissue as a function of time as well as the PK profiles using the input parameters of physicochemical properties (e.g., solubility, permeability, LogP, pKa, and particle sizes) and disposition data (e.g., tissue-to-plasma partition coefficients (Kp), CL, and first-pass extraction ratio (FPE)).
Development and Verification of PBPK Model in Pregnant Women
General Strategy
The present work simulated the PK of four model drugs in nonpregnant or pregnant women using PBPK models comprising of 14 tissue/organ compartments (Fig. 1). Transport of drug from systemic circulation into tissues was assumed to be perfusion rate limited. Within the default GastroPlus PBPKPlus module, the reproductive organ, renamed as fetoplacental units, was used to represent lumped intra-uterine components, including the fetus, placenta, amniotic fluid, and uterus. The compartment of the fetoplacental units was also assumed to be perfusion rate limited. The p-PBPK model integrated the generic physiological parameters, physiochemical properties, ADME data, and information related to clinical trial design. The PK data of model drugs (metformin, digoxin, midazolam, and emtricitabine) for women during postpartum (PP) and pregnant periods are obtained and digitized from literature (18–20). In these clinical studies, most of subjects received treatment in the third trimester (>28 weeks) and PP (>6 weeks), except for metformin, in which some subjects also received drug during early stages (10–14 weeks) and mid stages (20–26 weeks) of pregnancy. We assumed that the physiology and pharmacokinetics in PP subjects are representative of healthy nonpregnant or prepregnant women because the physiological, anatomical, and biochemical conditions are generally considered to return to the basal level after a normal delivery. The general workflow for pregnant PBPK model development and validation is illustrated in Fig. 2. Briefly, the PBPK model was initially developed using the physicochemical, biopharmaceutical, and pharmacokinetic parameters obtained from literatures or in silico prediction tools (e.g., ADMET predictor) in nonpregnant women population, and the model was further validated by comparing the simulated PK data with the observed clinical results. Population-dependent physiological parameters in human PBPK models were obtained using the Population Estimates for Age-Related Physiology™ module in GastroPlus v8, where the generic demographic and physiological parameters (e.g., age, tissue volume, and perfusion rate) for a normal American female were based on the National Health and Nutrition Examination Survey as described previously (22). If predicted PK profile and parameters were significantly deviant from the observed data, the model was refined by parameter optimization by fitting against the nonpregnant clinical data, which is a generic refinement strategy especially for drugs in development. In GastroPlus, an optimization module was generally used to estimate parameters of interest after successful data fitting. The model refinement strategy may not be limited to this and may be case-dependent (e.g., applying scaling factor to match the observed parameter values). As illustrated in Fig. 2, a simplified p-PBPK model (model 1) was initially developed with limited physiological (e.g., body weight (BW), cardiac output (CO), glomerular filtration rate (GFR), and renal secretion) and metabolic (e.g., CL changes contributed by CYP450 enzymes) changes. Alternatively, a comprehensive pregnant model (model 2) based on model 1 was extended by further considering the physiological changes (e.g., volume and perfusion rate) in fetoplacental unit, adipose tissue, and body fluid (e.g., volume of blood). The volume and perfusion rate for each tissue were automatically calculated using direct model balancing or manually defined using the regression equations reported by Abduljalil et al. (14) for models 1 or 2, respectively. In addition, the gut physiology was assumed to be the same between nonpregnant and pregnant populations. The predicted mean PK parameters (Cmax, AUC, and peak concentration time (tmax)) for PP and pregnant women as well as the ratios of the mean PK parameters (pregnant vs. PP) were then obtained based on the simulations.
Fig. 1.
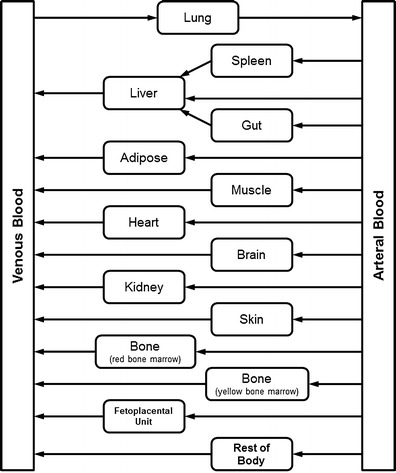
Structure of pregnancy physiologically based pharmacokinetic (p-PBPK) model
Fig. 2.
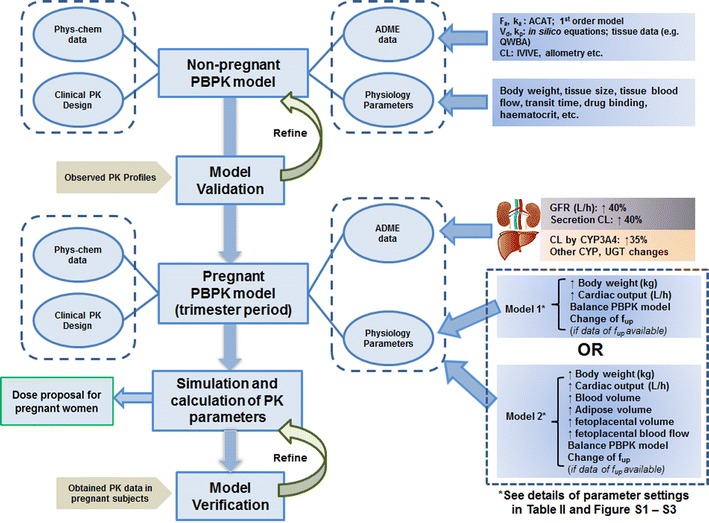
Schematic representation of the workflow of pregnancy PBPK model development and validation for compounds that are mainly renally eliminated or metabolized by CYP3A4
Development and Validation of PBPK Model in Nonpregnant Subjects
Physicochemical parameters were either obtained from literature or predicted by the GastroPlus built-in ADMET predictor. Key ADME-related parameters (e.g., solubility, effective permeability (Peff), CL, and Vd) were obtained from the literature (see details in Electronic Supplementary Material (ESM) Table S1). The ACAT model was used to quantitatively describe multiple drug absorption processes, including release, dissolution, precipitation, and passage across the GI membrane. The default gastrointestinal physiology parameters (e.g., pH, volume, transit time, bile salt concentration, etc.) under fasted and fed conditions within the ACAT model were based on the average values obtained from published population data. Rapid partitioning equilibrium of drugs was assumed between a homogenous tissue and plasma/blood, and thus perfusion-limited distribution kinetics was used for all the tissues in the PBPK model, Kp values were initially calculated using the tissue composition (TC) equations, according to the relationship between physiological data (lipids, plasma proteins and lipoproteins, and water etc.) and compound-specific determinants of distribution like lipophilicity (Log P), ionization (pKa), and plasma protein binding (fup;23). Transporter-mediated tissue distribution was not considered in the current model. The steady state Vd (Vss) was then estimated using Eq. (1)
| 1 |
where Vtissue and Vplasma is the physiological volume for each tissue and plasma. Drug elimination was assumed to occur only in liver and/or kidney and was described using reported hepatic CL (CLH), renal CL (CLR), and GFR. If measured GFR values were not available from clinical studies, a generic recent value (114 mL/min) from the literature was used (14). The renal filtration clearance (CLfiltration) and net secretion clearance (CLsecretion(net)) were calculated using Eqs. (2) and (3), respectively.
| 2 |
| 3 |
The initial model was verified by comparing the simulated PK profiles with the observed data for nonpregnant population. The model was then refined, if needed, with the following approaches, depending on different scenarios. As described, the GastroPlus optimization module was utilized to estimate parameters that significantly influence PK profiles. Common deviations in the PBPK models were observed in that the predicted Vd using in silico TC equation was significantly different from the reported values. In general, simultaneous optimization of Kp for multiple tissues to fit against observed data are not feasible unless rich tissue distribution data are available. Here, we use two approaches to refine the PBPK model for better prediction. We adjusted one or multiple Kp values using experimental data obtained for nonpregnant population from the literature. Alternatively, if no experimental Kp values were available, the in silico-predicted Kp values of all tissues were uniformly multiplied by a scaling factor. The scaling factor was manually optimized until the Vd predicted by the adjusted model matches the observed values—such an optimization could be done with clinical data from First-In-Human or other nonpregnant studies. When the predicted absorption kinetics significantly deviated from observed kinetics, the reference solubility and Peff were optimized using GastroPlus build-in optimization module™ to capture the absorption behavior. For the drug elimination, the input CL values were directly obtained from nonpregnant PK study (if CL is reported) or the mean (or median) values of the data range reported in literature. The detailed input parameters for four model compounds are were summarized in ESM Table S1, and the Kp values used in the whole-body PBPK model have been summarized in Table I.
Table I.
Tissue-to-Plasma Partition Coefficients (K p) of the Model Drugs
| Tissue | Metformina | Digoxinb | Midazolamb | Emtricitabinea | ||
|---|---|---|---|---|---|---|
| PP | Pregnancy | PP | Pregnancy | |||
| Lung | 1.53 | 9.36 | 8.95 | 0.31 | 0.33 | 3.95 |
| Adipose | 0.25 | 1.02 | 0.98 | 1.88 | 2.35 | 0.95 |
| Muscle | 1.73 | 35.7 | 35.7 | 0.59 | 0.72 | 3.75 |
| Liver | 1.64 | 10.83 | 10.34 | 0.95 | 1.16 | 3.8 |
| Spleen | 1.67 | 7.88 | 7.56 | 0.62 | 0.75 | 3.9 |
| Heart | 1.45 | 5.75 | 5.53 | 0.73 | 0.87 | 3.95 |
| Brain | 1.69 | 1.92 | 1.94 | 1.44 | 1.78 | 4 |
| Kidney | 1.55 | 11.78 | 11.24 | 0.64 | 0.77 | 3.9 |
| Skin | 1.23 | 3.61 | 3.50 | 0.9 | 1.06 | 3.6 |
| Reproductive organ (fetoplacental unit) | 1.55 | 11.78 | 11.24 | 0.66 | 0.76 | 3.9 |
| Red bone marrow | 0.95 | 2.0 | 2.0 | 1.66 | 2.03 | 2.25 |
| Yellow bone marrow | 0.25 | 1.02 | 0.98 | 1.88 | 2.35 | 0.95 |
| Rest of body | 1.67 | 7.88 | 7.56 | 0.65 | 0.78 | 3.75 |
aThe f up of metformin and emtricitabine in plasma protein is assumed constant during pregnancy and PP since there are no data available to provide evidence that the f up values change during pregnancy. Therefore, K p values are not adjusted by f up during pregnancy
bThe f up values of digoxin and midazolam in plasma during pregnancy and PP (19) are available and thereby K p values are adjusted by f up during pregnancy
Physiological Changes Considered in the Pregnant PBPK Model
The disposition of many medications is altered during pregnancy due to considerable changes in the physiology, anatomy, and biochemistry. These changes alter the exposure (absorption, distribution, and elimination) of xenobiotics during and after pregnancy in women who receive similar doses. Recently, Abduljalil et al. summarized various mathematic algorithms to describe the dynamic changes of individual parameter values during pregnancy (14). These quantitative regression equations were also applied to adjust parameter input (e.g., BW, tissue volume etc.) for the current PBPK modeling work (Table II).
Table II.
Main Anatomical, Physiological, and Biological Parameters Needed for p-PBPK Model and Equations Used to Calculate Parameter Values During Different Gestation Ages (GA) (14)
| Parameter | Unit | Equation |
|---|---|---|
| Today body weight (kg) | kg | TBW = 61.1 + 0.2409 GA + 0.0038 GA2 |
| Cardiac output (CO) | L/h | CO = 301 + 5.916 GA − 0.088 GA2 |
| Total body fat mass (TFM) | kg | TFM = 17.14 + 0.1305 GA + 0.0008 GA2 |
| Weight of the uterus | g | Weight of the uterus = 80 + 8.2931 GA + 0.3546 GA2 |
| Fetal volume | mL | Fetal volume = 0.01 exp(13.604(1 − exp(−0.0702GA))) |
| Placental volume | mL | Placenta volume = 0.0 − 0.0716 + 0.9146 GA2 − 0.0122 GA2 |
| Amniotic fluid | mL | Aminotic fluid volume = 0 + 1.9648 GA − 1.2056 GA2 + 0.2064GA3 − 0.0061 GA4 + 0.00005 GA5 |
| Volume of fetoplacental unit | mL | Fetoplacental volume = Uterus weight + Placenta volume + Fetal volume + Amniotic fluid volume |
| Blood flow of uterine | L/h | Uterine blood flow = 1.71 + 0.2068 GA + 0.0841 GA2 − 0.0015 GA3 |
| Plasma volume | L | Plasma volume = 2.5 − 0.0223 GA + 0.0042 GA2 − 0.00007 GA3 |
| Red blood cell (RBC) volume | L | RBC volume = 1.49 + 0.0098 GA |
| Total blood volume | L | Total blood volume = plasma volume + RBC volume |
| Glomerular filtration rate (GFR) | mL/min | GFR = 114 + 3.2367 GA − 0.0572 GA2 |
Drug absorption may be altered as nausea and vomiting during early stage of pregnancy may result in delayed gastric emptying and reduced small intestine motility (16,24). Prolonged GI transit time can potentially increase the tmax but reduce the Cmax (16). In addition, results have shown that there are no significant differences in basal gastric pH during pregnancy, compared with nonpregnancy values (14). However, gestation-induced alteration in absorption is clinically insignificant in most of the known case studies (25–27). Therefore, this was not considered in the current model. In the current PBPK model, the GastroPlus ACAT model (with default ASF optimized LogD v6.1) was used to describe drug absorption for nonpregnant and pregnant states.
For changes of volume of distribution, with the simplified pregnant PBPK model (model 1), drug distribution during pregnancy was assumed to be mainly impacted by changes of body weight and plasma protein levels. The average maximum weight gain during the pregnancy period is about 12.5 kg (28), which reflects an increase of total body water (∼8 L) and adipose tissue (∼4 kg) (15,16). Plasma protein levels, including albumin and α-acid glycoprotein, generally decrease during pregnancy relative to prepregnancy values. For example, plasma protein level gradually decreases since second trimester and eventually reach ∼70 to 80% of the normal level at time of delivery (14,15,29). The fraction of fup may increase due to decreased albumin or α-glycoprotein levels, which may increase drug distribution for drugs with high protein binding. However, the quantitative correlation between plasma level and fup is not well understood. Therefore, fup changes during pregnancy cannot be directly predicted based on plasma protein levels, and this is not considered in most of pregnant PBPK models (13,30). Here, fup was assumed unaltered during pregnancy unless fup was measured in pregnant women during the clinical trials (19). If needed, the Kp values were recalculated by the TC equations when plasma protein binding data were available. In the comprehensive pregnant PBPK model (model 2), more gestation-related changes and parameters were considered. A lumped fetal and placenta compartment (fetoplacental unit) was used to represent uterus, fetus, placenta, and amniotic fluid. Because substantial morphological and physiological changes in fetoplacental unit occur during pregnancy, the mass or volume of uterus, placenta, amniotic fluid, and fetus undergoes dramatic changes, which comprises about 35% of the total gestational weight gain during pregnancy (31). Increase of body fat may account for about 55% of total weight gain during pregnancy (32). During the course of pregnancy, plasma cell volume may increase 34–70% while red blood cell volume increases to a lesser extent along with the plasma volume (14). Overall, the apparent Vd may increase during pregnancy, further resulting in lower systemic drug concentration when the unadjusted dose strength (dose used in drug labeling) is used in pregnant subjects. The change in the volume of these tissues during pregnancy can be described by using the equation summarized in Table II.
The change of metabolic enzyme activities, GFR, and renal blood flow rate can significantly alter the drug elimination during pregnancy. The metabolic activities of CYP3A4, CYP2D6, UGT1A1, and UGT1A4 are known to be induced by elevated estrogen/progesterone levels while the activities of CYP1A2 and CYP2C19 are known to be inhibited by progesterone and estradiol (12,16,33). For CYP3A metabolized compounds (e.g., midazolam), an average 35% increase of systemic CL and FPE in gut and liver were included in pregnant PBPK model across the gestational ages (14–40 weeks) based on the findings by Tracy et al. (34). For really excreted compounds, the GFR and net secretion CL can increase up to 40% and 50% during pregnancy, respectively, resulting in enhanced renal excretion of unchanged drugs (16). Based on available data, GFR gradually increases during first 20–26 weeks of gestation and then reaches a plateau or slightly declines in the late stages of pregnancy (14,35). The increase in GFR in a particular gestation week was quantitatively estimated for the current pregnant PBPK model by equations reported by Abduljalil et al. (14). The dynamic changes of net renal secretion during pregnancy have not been well characterized. The net secretion clearance significantly increases during mid and late pregnancy, while is unaltered in early pregnancy (<12 weeks) as in the case of metformin (18). This finding also explains the ∼40% increase in metformin net secretion clearance in mid and late pregnancy (>22 weeks), which was considered in current PBPK model. On the other hand, the current model does not account for the change of net secretion clearance in early pregnancy (<14 weeks). In addition, there are no detailed literature data to describe the trend of secretion clearance changes between the 14 and 22 weeks of gestation. Therefore, the current PBPK model may not be suited to predict the PK of renally eliminated compounds for pregnant women during 14–22 weeks of gestation.
Cardiac output and uterine blood flow rate can significantly increase during pregnancy. The mean maternal cardiac output has a peak of ∼7.30 L/min during the third trimester, which drops down to the baseline value during postpartum period (36,37). Uterine blood flow increases during pregnancy and peaks at late pregnancy as the uterus receives about 12% of cardiac output in comparison to 0.5% in prepregnant women. The longitudinal increase of cardiac output (models 1 and 2) and uterine blood flows (model 2 only) during pregnancy were calculated using polynomial regression equations in Table II.
Case Examples
Case Example 1: Metformin (BDDCS Class 3)
Metformin is a basic compound (pKa: 10.37 and 2.33), with molecular weight (MW) of 129.2 g/mol and high aqueous solubility (129.6 mg/mL). Metformin is mainly excreted unchanged in urine with minimal metabolism (38) and is also a substrate for organic cation transporters (OCTs) (18,39–42). Thus, metformin is categorized as a biopharmaceutical drug disposition classification system (BDDCS) class 3 compound (43). Metformin pharmacokinetics was studied in 37 pregnant subjects and 12 of the subjects studied were in ≥12 weeks PP. Both pregnant and PP subjects received 500 mg oral dose of metformin twice daily. An increase of total renal clearance, creatinine clearance, and net secretion clearance was observed in mid and late pregnancy versus PP. Significant changes of metformin exposure (Cmax and AUC) were observed during mid and late pregnancy compared to PP whereas intersubject variation in PK parameters was high (18). GFR and renal clearance during nonpregnancy was assumed to be equal to the observed values obtained from clinical trial participants during PP (18), while the corresponding renal secretion clearance was calculated by Eqs. (2) and (3). Physiological changes (as ratios of pregnancy vs. nonpregnancy) and the change of drug disposition parameter were estimated based on the modeling strategy described above during early (10–14 weeks, assuming 12 weeks in average), mid (22–26 weeks, assuming 24 weeks in average), and late pregnancy (34–38 weeks, assuming 36 weeks in average; Tables III and IV). The ratios were used to adjust the input physiological parameters during pregnancy for models 1 and 2, respectively [see ESM Fig. S1 for detailed parameter values. Early and mid pregnancy parameters were generated similarly (data not shown)]. Kp values for each tissue compartments within the PBPK model were calculated using Rodgers and Rowland’s method to estimate the Vd of metformin since Roger and Rowland’s method tends to be more appropriate to predict Kp values for moderate-to-strong bases (Table I; 44). The plasma samples were reportedly collected at steady-state. Therefore, the simulations were performed for early, mid, late pregnancy, and PP until the drug concentrations reached steady-state.
Table III.
Fold Change of Gestation Age-Dependent Parameters Used for Four Case Studies in the Developed p-PBPK Model
| Gestation ages (week) | 12a | 24a | 30b | 35.3c | 36a |
|---|---|---|---|---|---|
| Body weight | 1.06 | 1.13 | 1.17 | 1.22 | 1.22 |
| Cardiac output | 1.19 | 1.30 | 1.33 | 1.33 | 1.33 |
| Volume of fetoplacental unit | 5.60 | 25.67 | 43.57 | 61.15 | 63.43 |
| Blood flow of fetoplacental unit | 8.02 | 20.10 | 25.21 | 27.97 | 28.17 |
| Volume of total body fat | 1.09 | 1.18 | 1.23 | 1.27 | 1.27 |
| Total blood Volume | 1.08 | 1.29 | 1.38 | 1.43 | 1.43 |
| GFR (mL/min) | 1.27 | 1.39 | 1.40 | 1.38 | 1.37 |
a12, 24, and 36 weeks are assumed to be the average gestation weeks reported in metaformin case study
b30 weeks is assumed to be the average gestation average gestation week for midazolam and digoxin
cMedian gestation week reported for emtricitabine case study
Table IV.
Parameters for Model Drugs in Non-Pregnant and Pregnant PBPK Model. Parameter Values Used in the Pregnancy PBPK Model are in Italics (e.g., Nonpregnant Parameter Values → Late Pregnant parameter values)
| Parameters | Metformin | Digoxin | Midazolam | Emtricitabine |
|---|---|---|---|---|
| Basic compound/formulation Information | ||||
| Dose (mg) | 500 | 0.25 | 2 | 200 |
| Dosing regimen | Twice daily | Single dose | Single dose | Once daily |
| Main clearance pathways | Renal | Renal (∼60%) | Hepatic (CYP3A4) | Renal |
| Formulations | IR tablet | IR tableta | IR tableta | IR capsulea |
| Gut physiology | Fasted | Fasteda | Fasteda | Fasteda |
| Parameters not influenced by pregnancy (e.g., physicochemical properties) | ||||
| Acid/base | Base | Neutral | Base | Base |
| MW (g/mol) | 129.2 | 781 | 325.8 | 247.3 |
| Log D at pH 7.4 | −1.386 | −1.4 | 3.56 | −0.43 |
| pK a | 10.37 | 12.98 | 5.5 | 2.65 |
| Solubility in water (mg/mL) | 129 | 0.25 | 0.024 | 112 |
| Effective permeability (10−4 cm/s) | 0.25 | 1.02 | 6.28 | 2.00 |
| Diffusion Coefficients (cm2/s) | 1.21 | 0.439 | 0.856 | 0.75 |
| Blood-to-plasma ratio | 0.7 | 1.1 | 0.55 | 1 |
| BCS/BDDCS Class | 3 | 3 | 1 | 1 |
| Parameters influenced by pregnancy (e.g., disposition parameters) | ||||
| Fraction unbound in plasma (f up, %) | 100 → 100 | 63 → 67 | 0.61 → 0.71 | 96 → 96 |
| V ss predicted by PBPK (L) | 82.7 → 101 b | 643 → 872 b | 85.8 → 125 b | 156 → 210 b |
| 100 c | 845 c | 120 c | 191 c | |
| Gut FPE (%) | 0 | 0 | 40 → 54 | 0 |
| Hepatic CL (L/h) | 0 | 4.2 → 4.2 | 24.5 → 33.1 | 0 |
| Renal CL (L/h) | 28.5 → 40.0 | 6.9 → 10.2 | N/A | 19.1 → 26.6 |
| GFR (L/h) | 9.9 → 13.8 | 7.4 → 10.5 | N/A | 6.8 → 9.4 |
| Filtration CL (L/h) | 9.9 → 13.8 | 4.6 → 7.01 | N/A | 6.6 → 9.1 |
| Secretion CL (L/h) | 18.7 → 26.2 | 2.3 → 3.11 | N/A | 12.5 → 17.6 |
aThe corresponding information is not available and was assumed
b V ss predicted by model 1
c V ss predicted by model 2
Case Example 2: Digoxin (BDDCS Class 3)
Digoxin is a neutral compound which is predominantly excreted via the kidneys and is a known P-glycoprotein (P-gp) substrate (45,46). Digoxin is a BCS/BDDCS class 3 compound due to its moderate solubility (0.25 mg/mL at pH 7.0), low permeability of (Peff, 1.02 × 10−4 cm/s), and poor metabolism. Digoxin pharmacokinetics were studied in 13 women who received a single 0.25 mg oral dose during trimester (28–32 weeks) and PP (6-10 weeks; 19). The renal clearance and net secretion clearance during PP were obtained from (19) and the filtration clearance and GFR were then backcalculated using Eqs. (2) and (3). Hepatic metabolism of digoxin is known to play a minor role (∼30%) in drug elimination (ESM Table S1). Metabolism is reported to be mediated via multiple pathways including hydrolysis and oxidation conjugation but it is not likely dependent on CYP450 system (47). CLH of digoxin in nonpregnant women was not directly reported and was calculated as 4.2 L/h using the following equation (Eq. 4)
| 4 |
where the total oral CL is calculated by the ratio of dose and AUC0 − ∞; AUC0 − ∞ (15.74 ng × h/mL) was calculated from the mean concentration vs. time curve via noncompartmental analysis using WinNonlin Phoenix v6.2 (Pharsight Corporation, Cary, NC, USA). The average bioavailability (F) of digoxin was assumed to be 70% (60–80% dependent on formulation; 48,49). Calculated CLtotal and CLH were 11.1 and 4.2 L/h, respectively (Table IV). Currently, information regarding the activity changes of non-CYP enzymes during pregnancy is not available. Therefore, hepatic clearance of digoxin during pregnancy was assumed to be same as that during nonpregnant period. Other physiological parameter changes (as ratios of pregnancy vs. nonpregnancy, Table III) during late pregnancy (assuming average gestation age is 30 weeks) were estimated based on the modeling strategy described above. The ratios were used to adjust the input physiological parameters during pregnancy for models 1 and 2, respectively (see ESM Fig. S2 for detailed parameter values). Kp values for the tissue compartments were initially calculated using Poulin and Theil’s TC equation because this TC equation yields better results for prediction of neutral compounds (23). Kp values for muscle of digoxin was estimated to be 35.7, which was obtained from the reported tissue binding data in human skeletal muscle (50). PK simulations were performed for women during late pregnancy and PP after a single oral dose (0.25 mg).
Case Example 3: Midazolam (BDDCS Class 1)
Midazolam (MW of 325.8 g/mol) is a BCS/BDDCS class 1 compounds with high aqueous solubility and high permeability (Peff, 6.28 × 10−4 cm/s). Midazolam is a sensitive CYP3A substrate with significant gut metabolism and hepatic extraction after oral dose (19,51–53). Midazolam pharmacokinetics were studied in 13 women who received an oral dose of 2 mg immediate release formulation during trimester (28–32 weeks) and PP (6-10 weeks) (19). Drug elimination of midazolam is known to be mediated mainly by CYP3A metabolism and the hepatic CL used in the current model was estimated as the average value of the CL values obtained from multiple study reports (ESM Table S1). Kp values in different compartments were calculated by Rodgers and Rowland’s TC equation (54) and fup was reported as 0.67 during PP but decreases to 0.63 during late pregnancy (19). Due to increased enzyme activity of CYP3A during pregnancy, hepatic CL and gut FPE were increased by 35% (Table IV; 34). The changes of physiological parameters (as ratios of pregnancy vs. nonpregnancy, Table III) during late pregnancy (average 30 weeks) were estimated based on the modeling strategy described above. The ratios were used to adjust the input physiological parameters during pregnancy for models 1 and 2, respectively (see ESM Fig. S2 for detailed parameter values). PK simulations were performed for women during late pregnancy and PP.
Case Example 4: Emtricitabine (BDDCS Class 1)
Emtricitabine (MW: 247.3) is a weak basic drug with pKa of 2.65, with high permeability and high solubility. The compound is mainly excreted through kidneys and is likely an OCT-2 substrate (45,46). It is a once-daily nucleoside reverse transcriptase inhibitor that selectively and potently inhibits human immunodeficiency virus type 1 (HIV-1) replication (55). Emtricitabine is commonly used in HIV-1 infected women during pregnancy. Emtricitabine pharmacokinetics were studied in 26 women who received an oral dose of 2 mg immediate release formulation during trimester (median, 35 weeks) and PP (median, 8 weeks). Clinical PK data were digitized from literature (20) for women during third trimester and PP women after 4 days of 200 mg oral doses given once daily, and the oral CL (CL/F) and absolute oral bioavailability was observed as 20.6 L/h and 93%, respectively. Therefore, in the current model, the systemic CL of emtricitabine for healthy nonpregnant women was estimated to be 19.1 L/h. The generic GFR values (114 mL/min or 6.84 L/h) of healthy nonpregnant women were used in the model (14). The corresponding renal net secretion clearance (12.5 L/h) was calculated by Eqs. (2) and (3). Changes of physiological parameters (as ratios of pregnancy vs. nonpregnancy, Table III) during late pregnancy (average 35 weeks) were estimated based on the modeling strategy described. The ratios were used to adjust input physiological parameters during pregnancy for models 1 and 2, respectively (see ESM Fig. S3 for detailed parameter values). PK simulation for emtricitabine was performed for women during late pregnancy and PP based on the parameter input provided in Table IV.
RESULTS
Case Example 1: Metformin
The simulated plasma concentration vs. time profiles based on models 1 and 2 both captured the observed PK data during late pregnancy and PP (Fig. 3a) with regression coefficients (R2) greater than 0.91. There were no significant differences in simulated results between models 1 and 2. The predicted AUC and Cmax and the fold changes of these PK parameters (pregnant vs. PP) were within 1.1-fold of observed values (Table V). The model predicted renal clearance (CLr, 39.98 L/h) during third trimesters is in agreement with observed clearance (37.5 L/h). The predicted mean fold change of AUC (pregnant vs. nonpregnant) was 0.69 and 0.71 based on the simulation using models 1 and 2, respectively, which is very close to the observed mean fold change (0.70). Although the predicted tmax was slightly longer than the observed values, the current PBPK model accurately predicts the PK of metformin during late pregnancy.
Fig. 3.
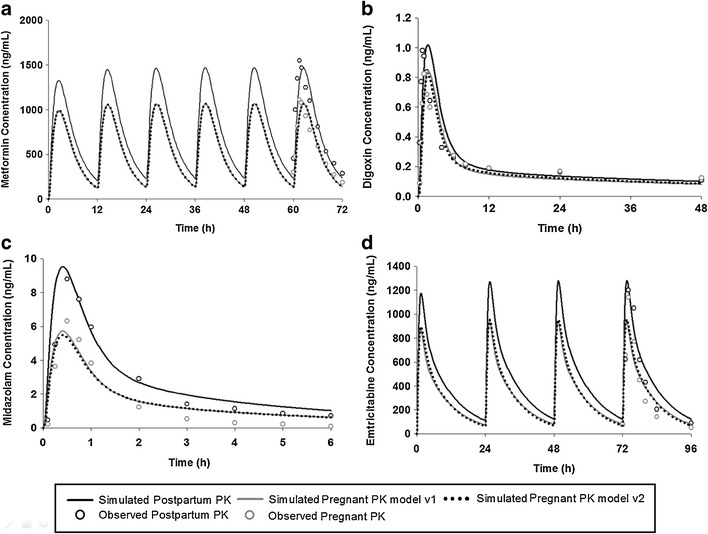
Simulated and observed mean plasma concentration in PP and late pregnant women for a metformin after 500 mg twice-daily oral dosing for 3 days, b digoxin following a single oral 0.25 mg dose, c midazolam after a single 2 mg oral dose, and d emtricitabine after once-daily 200 mg oral dose for 4 days
Table V.
Comparison Between Observed and PBPK Model Predicted Parameters
| Drug | PK Parameters | Pregnant Women (third trimester) | Non-pregnant Women (PP) | Fold Change (pregnant vs. non-pregnant) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Observeda | Predictedb | Prediction Fold errorc | Observeda | Predictedb | Prediction Fold errorc | Observeda | Predictedb | Prediction Fold errorc | ||
| Metformin | C max_ss (ng/mL) | 1,135 | 1,068, 1,070 | 1.0, 1.1 | 1,611 | 1,471 | 1.1 | 0.71 | 0.73, 0.73 | 1.0, 1.0 |
| t max (h) | 2.0 | 2.5, 2.6 | 1.2, 1.3 | 1.8 | 2.4 | 1.4 | ||||
| AUC12h_ss (ng h/mL) | 6,937 | 6,563, 6,698 | 1.0, 1.0 | 9,804 | 9,463 | 1.0 | 0.70 | 0.69, 0.71 | 1.0, 1.0 | |
| Digoxin | C max (ng/mL) | 0.8 | 0.84, 0.81 | 1.1, 1.0 | 1.1 | 1.01 | 1.1 | 0.90 | 0.83, 0.80 | 1.1, 1.1 |
| t max (h) | 1.6 | 1.7, 1.6 | 1.1, 1.0 | 1.3 | 1.6 | 1.2 | ||||
| AUC48h (ng h/mL) | 7.3 | 8.0, 8.1 | 1.1, 1.1 | 9.3 | 9.54 | 1.0 | 0.81 | 0.84, 0.85 | 1.0, 1.0 | |
| Midazolam | C max (ng/mL) | 6.4 | 5.33, 5.11 | 1.2, 1.3 | 9.3 | 9.5 | 1.1 | 0.72 | 0.56, 0.54 | 1.3, 1.3 |
| t max (h) | 0.56 | 0.4, 0.4 | 1.4, 1.4 | 0.54 | 0.4 | 1.4 | ||||
| AUCinf (ng h/mL) | 9.5 | 12.5, 12.2 | 1.3, 1.3 | 17.9 | 22.7 | 1.1 | 0.54 | 0.55, 0.54 | 1.0, 1.0 | |
| Emtricitabine | C max_ss (μg/mL) | 1.4 | 0.94, 0.96 | 1.5, 1.5 | 1.4 | 1.3 | 1.2 | 0.97 | 0.72, 0.74 | 1.3, 1.3 |
| t max (h) | 2.0 | 1.4, 1.5 | 1.4, 1.3 | 2.0 | 1.3 | 1.4 | ||||
| AUC24h_ss (μg h/mL) | 8.0 | 8.0, 8.0 | 1.0, 1.0 | 9.7 | 11.2 | 1.1 | 0.82 | 0.71, 0.71 | 1.2, 1.2 | |
aObserved data was obtained from values reported in the literatures
bPredicted PK parameters are presented as “model 1, model 2”
cFold error (FE) was calculated as FE = 10|log(Predicted/Observed)|. Calculated FE are presented as “model 1, model 2”
In addition, the simulated plasma concentration vs. time profiles based on models 1 and 2 were able to capture metformin’s elimination profiles during early and mid pregnancy (Fig. 4). The R2 of the simulation are higher than 0.78 and 0.86 for early and mid pregnancy, respectively. However, the distribution phase was slightly overpredicted in early pregnancy group whereas the Cmax was overpredicted (deviation, ∼20%) in mid pregnancy group.
Fig. 4.
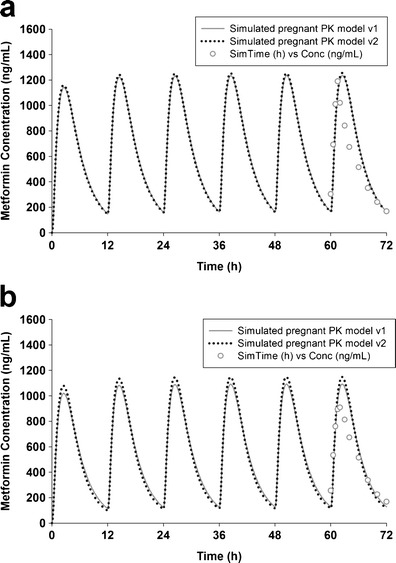
Simulated and observed mean plasma metformin concentration in early (a) and mid pregnancy (b) after 500 mg twice-daily oral dosing for 3 days
Case Example 2: Digoxin
The muscle Kp value (4.3) estimated by Poulin and Theil’ TC equation was dramatically lower than the reported Kp values (35.7), which resulted in underprediction of terminal t1/2 (predicted (11.3 h) vs. observed (46.5 h)). The model was refined with literature reported muscle Kp value and accurately simulate the terminal phase PK curve with a predicted t1/2 of 43.5 h. The simulated plasma concentrations vs. time profiles were in agreement with the observed profiles (Fig. 3b) with R2 of 0.81 and 0.90 for models 1 and 2, respectively. The predicted AUC and Cmax and the fold changes of the PK parameters (pregnant vs. PP) were within 1.2-fold of observed values (Table V). The predicted renal clearance (CLr, 10.4 L/h) for women during third trimesters was in agreement with observed data (10.9 L/h). The predicted fold change of Cmax (pregnant vs. nonpregnant) were 0.83 and 0.80 using models 1 and 2, respectively, similar to observed ratio, while the predicted fold change of AUC (model 1, 0.84; model 2, 0.85) has no large difference with the observed values (0.81).
Case Example 3: Midazolam
The ex vivo measured unbound drug fraction in plasma is 0.0071 and 0.0061 in PP and pregnant women, which were incorporated in the midazolam PBPK models. Midazolam is a compound that undergoes extensive metabolism in liver and gut. The simulated plasma concentration vs. time profile captured the observed profile well (Fig. 3c) with an R2 of 0.78 and 0.76 for models 1 and 2, respectively. The predicted AUC and Cmax were within 1.3-fold of observed values (Table V) using either models 1 or 2. The simulated ratio of Cmax (pregnant vs. PP = 0.54–0.56) was slightly underpredicted, compared to the observed values (0.72). On the contrary, the ratio of AUC was accurately predicted by the model (predicted (0.54–0.55) vs. observed (0.54)) (Fig. 4). The model successfully estimated the terminal phase half-life (predicted, 2.5 h; observed, 2.4 h). The PBPK model also predicted the short tmax and rapid absorption of midazolam. Cmax prediction for pregnant women was improved (prediction fold error, 1.2) compared with the model reported previously (13) which underpredicted Cmax by ∼40%. The prediction fold error of AUC during pregnancy using the current PBPK model was 1.3 regardless of the type of model, which was similar to the fold error of 1.35 (observed AUC of 9.5 ng h/mL vs. predicted AUC of 7.05 ng∙h/mL) predicted by a recently described midazolam p-PBPK model (13), indicating that the current PBPK model exhibits good prediction accuracy for pregnant women. In addition, there were no significant differences in simulated results between models 1 and 2.
Case Example 4: Emtricitabine
Oral Vd/F reported by the literature is about 4 L/kg (20) and the bioavailability with an IR capsule formulation is reported to be 93%, suggesting that the systemic Vss is about 3.7 L/kg. The Rodger and Rowland’s TC equation based on the input physicochemical parameters dramatically underpredicted Vss (0.60 L/kg). To our best knowledge, no observed tissue distribution data are available. Therefore, the Kp values in all tissues were multiplied by a scaling factor of 5 (based on nonpregnant data) and the simulated PK profiles fit to the observed data (Fig. 3d) with R2 of 0.80 and 0.84 for models 1 and 2, respectively. Predicted AUC and Cmax were 1.0- and 1.5-fold of observed clinical data (Table V), respectively. The predicted fold changes of AUC and Cmax (pregnant vs. nonpregnant) were within 1.3-fold of the observed ratios. The model predicted renal clearance (CLr, 26.6 L/h) during third trimesters was in agreement with observed oral clearance (25.3 L/h). Therefore, the emtricitabine pregnant PBPK model is well verified and can well predict the alternation of PK during late pregnancy. There were no significant differences in simulated results between models 1 and 2.
DISCUSSION
Ideally, the use of pharmacologic agents would be minimized for pregnant women. However, pregnant women with certain medical conditions may still require ongoing treatment. Clinical trials actively exclude pregnant women due to ethical and scientific reasons. Therefore, human PK data are seldom collected during pregnancy for appropriate dosage and frequency of administration. Even after years of marketing, product labels regarding PK and dose adjustments often provide limited information for pregnant patients. The US Food and Drug Administration and European Medicine Agency have required that application sponsors collect and maintain data on the effects of prescription medicines that are used by pregnant women with a new labeling system. Thus, regulatory agencies encourage drug companies to anticipate and predict drug exposure during pregnancy prospectively. Physiological changes during pregnancy can alter drug disposition according to clinical data reported in the literature. Currently, optimization of dosing regimen for pregnant women can be challenging, especially in the absence of clinical data. PBPK has been recently recognized as useful tool to provide valuable insights to aid clinical design. With incorporation of gestation-induced physiological changes, the pregnancy PBPK model can serve as a feasible alternative to empirical dose adjustment and guide the clinical trial design in pregnant women.
While PBPK models now have increased impact and applications in the pharmaceutical industry, the limitations in PBPK modeling need to be acknowledged and understood. In particular, Kp values can be inaccurately estimated using the in silico equations, resulting in biased prediction of Vd (56). If in vivo distribution data are not available in such situations, PBPK models cannot be readily established with high confidence. However, in some cases, if the PK profile of the compounds is characterized by a mono-exponential equation (similar to a one-compartmental model), the application of a uniform scaling factor on each Kp value likely provides good estimations for Vd and the shape of PK profiles as in the described case study of emtricitabine. The deviation in Kp predictions using in silico equations can be safely assumed to be similar across all tissues and can be corrected by applying a scaling factor to all Kp predicted by TC equations, if drug distribution can reach equilibrium rapidly (as with the assumption of a one-compartmental PK model). However, if the PK profile is described by a complicated multi-exponential equation, such assumptions may not stand because the magnitude of deviation of Kp prediction can differ between tissues (e.g., digoxin). In such cases, the PBPK modeling approach may require additional data, such as drug tissue distribution data (e.g., quantitative whole-body autoradiography; 22).
Several pregnancy PBPK models have been introduced for xenobiotic disposition in pregnant women (57–59). However, few publications focused on the investigation of maternal PK alternation caused by the effect of pregnancy. Recently, Gaohua et al. developed a first pregnancy PBPK model incorporating time-dependent activity of cytochrome P450 enzymes (e.g., CYP1A2, CYP2D6, and CYP3A4) and implementing gestational time-dependent physiological changes (13). This pregnancy PBPK model successfully described the disposition for drugs which undergoes extensive metabolism by major cytochrome P450 enzymes. Although a dynamically parameterized pregnant PBPK model can predict the PK outcome throughout the pregnancy, model development, validation, and application may be time-consuming due to its complexity. In general, most PK/PD studies in pregnant women are conducted in third trimesters, with the baseline assessment for comparison to the pregnant state during the PP period. To this end, model development efforts, especially modeling validation, mainly target the periods of late pregnancy when PK/PD endpoints are most commonly collected in clinical studies. Maternal drug clearance is often altered due to changes in metabolic activity of enzymes, GFR, and renal secretive transporters. The change in enzyme activity (induction or inhibition) plays an important role in altering PK for many drugs. Our practical and simplified PBPK model incorporates the change of essential parameters (e.g., BW, CO, GFR, CL, etc.), and successfully estimated the PK of several compounds during late pregnant and PP periods. The fetal and placenta, adipose, plasma, and blood volume have dramatic changes during pregnancy, which are likely expected to exhibit significant effects on drug distribution. Therefore, these changes are considered and incorporated in model 2. Interestingly, the predicted results were very similar for four drugs using the simple model (model 1) and the comprehensive model (model 2). This suggests that the increase of drug volume of distribution (unit, liter) during pregnancy can be explained by total body weight gain and is nearly proportional to the body weight. The individual contribution of the tissues/organs, even with substantial gestation-induced physiological changes, may be less important if body weight changes have been considered by the p-PBPK model, unless compounds distribute much more extensively into these tissues than other tissues/organs without significant alteration. For example, all four compounds are expected to have no extensive distribution into adipose tissue (Kp < 2), and thereby the proportionality between volume of distribution and body weight is not altered by fat gain, as shown by the models 1 and 2 predicted Vss values listed in Table IV. For tissue/organ gain with less composition of the total weight gain, such as fetaplacental unit which accounts for ∼35% of total weight gain (65% for adipose tissue), their volume increase may have even less influence on PK. The current p-PBPK model can be easily implemented using conventional PBPK modeling tools (e.g., GastroPlus or Simcyp), and should be sufficient to provide rational and accurate prediction for most of drugs which are mainly eliminated via the kidney or metabolized by CYP3A4 in the liver.
For renally excreted compounds, such as metformin, digoxin, and emtricitabine, the predicted fold decreases in AUC and Cmax were comparable to the observed results. The simulated results suggested that the decrease of systemic exposure in these compounds were mainly attributed to an increased renal secretion and filtration during pregnancy. The increase of GFR in pregnancy is correlated to dilation of renal vasculature caused by the systemic vasodilation possibly mediated by progesterone and relaxin (35,60). Enhanced renal secretion CL has been reported for metformin, digoxin, amoxicillin, and certain endogenous compounds during pregnancy, suggesting of pregnancy-induced enhancement of expression and/or function of renal secretion transporter regulation. Within these three modeled compounds, namely metformin, digoxin, and emtricitabine, are known as the substrates for transporters expressed on apical or basolateral (luminal) membrane of the kidney proximal tubules. For example, OCT2 plays an important role in the renal clearance of metformin in humans (61). Similarly, basolateral renal transport of emtricitabine appears to proceed via OCT2 in S2 and/or S3 segments of the proximal tubule (62). Also, digoxin is well-known as the substrate of P-gp which transports drugs across the apical membrane of renal tubular epithelium (63–65). Although magnitude of enhanced activities for these transporters during pregnancy is not well-known, the alternation of net tubular secretion during pregnancy is considered in the current PBPK model by assuming 40% increase of CLsecretion based on the previous evaluation in the pregnancy PK of metformin (18). Interestingly, the predicted and observed AUC and Cmax fold changes are less than 30% in late pregnancy, compared to PP period. Accounting for the uncertainty caused by intersubject variability, the effect of pregnancy on PK appears not be very large for drugs that are predominately eliminated via kidney.
The current PBPK modeling approach was further evaluated using midazolam which is extensively metabolized by CYP3A4. To reflect the change of CYP3A activity in third trimesters during pregnancy, the liver and gut metabolism, represented by hepatic CL and gut FPE, was increased by 30% according to previous clinical results. The prediction accuracy of the current model is similar to the PBPK model which integrates the time-dependent activity of CYP3A4. Since CYP3A4 plays the most important role in metabolism of drugs and xenobiotics, the current model can be used to anticipate the PK alteration and support drug labeling in many investigational drugs and postmarketing products for the pregnant populations. This also suggests that higher midazolam dose may be required in pregnant women to obtain the optimal pharmacological effect. In addition, alternation of activities in other metabolizing enzymes can be easily integrated in the pregnancy PBPK models using a similar approach. For example, it is known that the activities of CYP1A2 and CYP2C19 decreased by 30–65% (66) and 50% (67,68) whereas the activities of CYP2A6, CYP2D6, and UGT1A4 increased by 54% (69), 50% (70), and 200–300% (71) in late pregnant period, respectively.
The degree to which drugs are bound to the plasma proteins also influences their disposition. Albumin concentration is reported to fall by 20–30% during pregnancy, which may alter the drug concentration of albumin-bound compounds (15,29,72). Minor changes of fup have been reported for midazolam and digoxin (19). Metformin and emtricitabine are negligibly bound to plasma proteins in human. Overall, the influence of alternation of plasma protein during pregnancy has minimal effects to the PK behaviors of our modeled compounds. In addition, the increased total body water/fat creates a larger space where the drugs may distribute. Therefore, minor changes in free drug concentration are expected as a result of the compensation effects caused by more extensive distribution of unbound drugs during pregnancy. Overall, there is little practical importance of plasma protein binding in the pharmacological response in pregnant patients, although the total drug concentration may alter for some drugs such as phenytoin.
CONCLUSIONS
A simplified p-PBPK model is described, which yields good predictions for renally cleared and CYP3A4 hepatically cleared compounds evaluated (metformin, digoxin, midazolam, and emtricitabine) when compared to clinical observations. The model is readily implemented in GastroPlus and could be applied for drugs with similar clearance pathways at various stages of drug development to evaluate if altered pharmacokinetics may occur in a pregnant population based on healthy volunteer data.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
DOCX 464 kb
References
- 1.Bonati M, Bortolus R, Marchetti F, Romero M, Tognoni G. Drug use in pregnancy: an overview of epidemiological (drug utilization) studies. Eur J Clin Pharmacol. 1990;38(4):325–328. doi: 10.1007/BF00315569. [DOI] [PubMed] [Google Scholar]
- 2.De Vigan C, De Walle HE, Cordier S, Goujard J, Knill-Jones R, Ayme S, et al. Therapeutic drug use during pregnancy: a comparison in four European countries. OECM Working Group. Occupational Exposures and Congenital Anomalies. J Clin Epidemiol. 1999;52(10):977–982. doi: 10.1016/S0895-4356(99)00091-8. [DOI] [PubMed] [Google Scholar]
- 3.Lacroix I, Damase-Michel C, Lapeyre-Mestre M, Montastruc JL. Prescription of drugs during pregnancy in France. Lancet. 2000;356(9243):1735–1736. doi: 10.1016/S0140-6736(00)03209-8. [DOI] [PubMed] [Google Scholar]
- 4.Knoppert D. Safety and efficacy of drugs in pregnancy. J Popul Ther Clin Pharmacol. 2011;18(3):e506–e512. [PubMed] [Google Scholar]
- 5.Chen HS, Gross JF. Physiologically based pharmacokinetic models for anticancer drugs. Cancer Chemother Pharmacol. 1979;2(2):85–94. doi: 10.1007/BF00254079. [DOI] [PubMed] [Google Scholar]
- 6.Himmelstein KJ, Lutz RJ. A review of the applications of physiologically based pharmacokinetic modeling. J Pharmacokinet Biopharm. 1979;7(2):127–145. doi: 10.1007/BF01059734. [DOI] [PubMed] [Google Scholar]
- 7.Nestorov I. Whole body pharmacokinetic models. Clin Pharmacokinet. 2003;42(10):883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 8.Grass GM, Sinko PJ. Physiologically-based pharmacokinetic simulation modelling. Adv Drug Deliv Rev. 2002;54(3):433–451. doi: 10.1016/S0169-409X(02)00013-3. [DOI] [PubMed] [Google Scholar]
- 9.Parrott N, Lave T. Applications of physiologically based absorption models in drug discovery and development. Mol Pharm. 2008;5(5):760–775. doi: 10.1021/mp8000155. [DOI] [PubMed] [Google Scholar]
- 10.Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 11.Hill CC, Pickinpaugh J. Physiologic changes in pregnancy. Surg Clin N Am. 2008;88(2):391–401. doi: 10.1016/j.suc.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Feghali MN, Mattison DR. Clinical therapeutics in pregnancy. J Biomed Biotechnol. 2011;2011:783528. doi: 10.1155/2011/783528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol. 2012;74(5):873–885. doi: 10.1111/j.1365-2125.2012.04363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6):365–396. doi: 10.2165/11597440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989–1008. doi: 10.2165/00003088-200544100-00001. [DOI] [PubMed] [Google Scholar]
- 16.Dawes M, Chowienczyk PJ. Drugs in pregnancy. Pharmacokinetics in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2001;15(6):819–826. doi: 10.1053/beog.2001.0231. [DOI] [PubMed] [Google Scholar]
- 17.(CDER) CfDEaR. Pharmacokinetics in pregnancy—study design, data analysis, and impact on dosing and labeling. 2004. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072133.pdf. Accessed 19 Feb 2013
- 18.Eyal S, Easterling TR, Carr D, Umans JG, Miodovnik M, Hankins GD, et al. Pharmacokinetics of metformin during pregnancy. Drug Metab Dispos. 2010;38(5):833–840. doi: 10.1124/dmd.109.031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hebert MF, Easterling TR, Kirby B, Carr DB, Buchanan ML, Rutherford T, et al. Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin Pharmacol Ther. 2008;84(2):248–253. doi: 10.1038/clpt.2008.1. [DOI] [PubMed] [Google Scholar]
- 20.Stek AM, Best BM, Luo W, Capparelli E, Burchett S, Hu C, et al. Effect of pregnancy on emtricitabine pharmacokinetics. HIV Med. 2012;13(4):226–235. doi: 10.1111/j.1468-1293.2011.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu LX, Amidon GL. Characterization of small intestinal transit time distribution in humans. Int J Pharm. 1998;171(2):157–163. doi: 10.1016/S0378-5173(98)00174-4. [DOI] [Google Scholar]
- 22.Xia BF, Heimbach T, Lin TH, He HD, Wang YF, Tan E. Novel physiologically based pharmacokinetic modeling of patupilone for human pharmacokinetic predictions. Cancer Chemoth Pharm. 2012;69(6):1567–1582. doi: 10.1007/s00280-012-1863-5. [DOI] [PubMed] [Google Scholar]
- 23.Poulin P, Jones RD, Jones HM, Gibson CR, Rowland M, Chien JY, et al. PHRMA CPCDC initiative on predictive models of human pharmacokinetics, part 5: prediction of plasma concentration–time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J Pharm Sci. 2011 doi: 10.1002/jps.22550. [DOI] [PubMed] [Google Scholar]
- 24.Loebstein R, Lalkin A, Koren G. Pharmacokinetic changes during pregnancy and their clinical relevance. Clin Pharmacokinet. 1997;33(5):328–343. doi: 10.2165/00003088-199733050-00002. [DOI] [PubMed] [Google Scholar]
- 25.O'Hare MF, Leahey W, Murnaghan GA, McDevitt DG. Pharmacokinetics of sotalol during pregnancy. Eur J Clin Pharmacol. 1983;24(4):521–524. doi: 10.1007/BF00609896. [DOI] [PubMed] [Google Scholar]
- 26.Philipson A. Pharmacokinetics of ampicillin during pregnancy. J Infect Dis. 1977;136(3):370–376. doi: 10.1093/infdis/136.3.370. [DOI] [PubMed] [Google Scholar]
- 27.Philipson A, Stiernstedt G, Ehrnebo M. Comparison of the pharmacokinetics of cephradine and cefazolin in pregnant and non-pregnant women. Clin Pharmacokinet. 1987;12(2):136–144. doi: 10.2165/00003088-198712020-00004. [DOI] [PubMed] [Google Scholar]
- 28.Sachdeva P, Patel BG, Patel BK. Drug use in pregnancy; a point to ponder! Indian J Pharm Sci. 2009;71(1):1–7. doi: 10.4103/0250-474X.51941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dean M, Stock B, Patterson RJ, Levy G. Serum protein binding of drugs during and after pregnancy in humans. Clin Pharmacol Ther. 1980;28(2):253–261. doi: 10.1038/clpt.1980.158. [DOI] [PubMed] [Google Scholar]
- 30.Loccisano AE, Longnecker MP, Campbell JL, Jr, Andersen ME, Clewell HJ., 3rd Development of PBPK models for PFOA and PFOS for human pregnancy and lactation life stages. J Toxic Environ Health. 2013;76(1):25–57. doi: 10.1080/15287394.2012.722523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitkin RM. Nutritional support in obstetrics and gynecology. Clin Obstet Gynecol. 1976;19(3):489–513. doi: 10.1097/00003081-197609000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Okereke NC, Huston-Presley L, Amini SB, Kalhan S, Catalano PM. Longitudinal changes in energy expenditure and body composition in obese women with normal and impaired glucose tolerance. Am J Physiol. 2004;287(3):E472–E479. doi: 10.1152/ajpendo.00589.2003. [DOI] [PubMed] [Google Scholar]
- 33.Sato J, Sawada Y, Iga T, Hanano M. Effect of quinidine on digoxin distribution and elimination in guinea pigs. J Pharm Sci. 1983;72(10):1137–1141. doi: 10.1002/jps.2600721007. [DOI] [PubMed] [Google Scholar]
- 34.Tracy TS, Venkataramanan R, Glover DD, Caritis SN. Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A activity) during pregnancy. Am J Obstet Gynecol. 2005;192(2):633–639. doi: 10.1016/j.ajog.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 35.Davison JM, Dunlop W. Renal hemodynamics and tubular function normal human pregnancy. Kidney Int. 1980;18(2):152–161. doi: 10.1038/ki.1980.124. [DOI] [PubMed] [Google Scholar]
- 36.Robson SC, Hunter S, Boys RJ, Dunlop W. Serial study of factors influencing changes in cardiac output during human pregnancy. Am J Physiol. 1989;256(4 Pt 2):H1060–H1065. doi: 10.1152/ajpheart.1989.256.4.H1060. [DOI] [PubMed] [Google Scholar]
- 37.Flo K, Wilsgaard T, Vårtun Å, Acharya G. A longitudinal study of the relationship between maternal cardiac output measured by impedance cardiography and uterine artery blood flow in the second half of pregnancy. BJOG An Int J Obstet Gynaecol. 2010;117(7):837–844. doi: 10.1111/j.1471-0528.2010.02548.x. [DOI] [PubMed] [Google Scholar]
- 38.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 39.Wang ZJ, Yin OQ, Tomlinson B, Chow MS. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genomics. 2008;18(7):637–645. doi: 10.1097/FPC.0b013e328302cd41. [DOI] [PubMed] [Google Scholar]
- 40.Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, et al. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20(5):379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- 41.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol. 2007;74(2):359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 42.Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos. 2007;35(10):1956–1962. doi: 10.1124/dmd.107.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 44.Rodgers T, Leahy D, Rowland M. Tissue distribution of basic drugs: accounting for enantiomeric, compound and regional differences amongst beta-blocking drugs in rat. J Pharm Sci. 2005;94(6):1237–1248. doi: 10.1002/jps.20323. [DOI] [PubMed] [Google Scholar]
- 45.Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96(4):1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smit JW, Schinkel AH, Muller M, Weert B, Meijer DK. Contribution of the murine mdr1a P-glycoprotein to hepatobiliary and intestinal elimination of cationic drugs as measured in mice with an mdr1a gene disruption. Hepatology. 1998;27(4):1056–1063. doi: 10.1002/hep.510270422. [DOI] [PubMed] [Google Scholar]
- 47.Lacarelle B, Rahmani R, de Sousa G, Durand A, Placidi M, Cano JP. Metabolism of digoxin, digoxigenin digitoxosides and digoxigenin in human hepatocytes and liver microsomes. Fundam Clin Pharmacol. 1991;5(7):567–582. doi: 10.1111/j.1472-8206.1991.tb00746.x. [DOI] [PubMed] [Google Scholar]
- 48.Binnion PF. A comparison of the bioavailability of digoxin in capsule, tablet, and solution taken orally with intravenous digoxin. J Clin Pharmacol. 1976;16(10 Pt 1):461–467. [PubMed] [Google Scholar]
- 49.Marcus FI, Dickerson J, Pippin S, Stafford M, Bressler R. Digoxin bioavailability: formulations and rates of infusions. Clin Pharmacol Ther. 1976;20(3):253–259. doi: 10.1002/cpt1976203253. [DOI] [PubMed] [Google Scholar]
- 50.Jogestrand T, Ericsson F. Skeletal muscle digoxin binding in patients with renal failure. Brit J Clin Pharmacol. 1983;16(1):109–111. doi: 10.1111/j.1365-2125.1983.tb02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol. 1994;47(9):1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 52.Thummel KE, Shen DD, Podoll TD, Kunze KL, Trager WF, Bacchi CE, et al. Use of midazolam as a human cytochrome P450 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J Pharmacol Exp Ther. 1994;271(1):557–566. [PubMed] [Google Scholar]
- 53.Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36(1):89–96. [PubMed] [Google Scholar]
- 54.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94(6):1259–1276. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 55.Molina JM, Cox SL. Emtricitabine: a novel nucleoside reverse transcriptase inhibitor. Drugs Today (Barc) 2005;41(4):241–252. doi: 10.1358/dot.2005.41.4.900219. [DOI] [PubMed] [Google Scholar]
- 56.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35(10):1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 57.Luecke RH, Wosilait WD, Pearce BA, Young JF. A physiologically based pharmacokinetic computer model for human pregnancy. Teratology. 1994;49(2):90–103. doi: 10.1002/tera.1420490205. [DOI] [PubMed] [Google Scholar]
- 58.Luecke RH, Wosilait WD, Pearce BA, Young JF. A computer model and program for xenobiotic disposition during pregnancy. Comput Methods Programs Biomed. 1997;53(3):201–224. doi: 10.1016/S0169-2607(97)00020-5. [DOI] [PubMed] [Google Scholar]
- 59.Corley RA, Mast TJ, Carney EW, Rogers JM, Daston GP. Evaluation of physiologically based models of pregnancy and lactation for their application in children’s health risk assessments. Crit Rev Toxicol. 2003;33(2):137–211. doi: 10.1080/713611035. [DOI] [PubMed] [Google Scholar]
- 60.Davison JM. The effect of pregnancy on kidney function in renal allograft recipients. Kidney Int. 1985;27(1):74–79. doi: 10.1038/ki.1985.12. [DOI] [PubMed] [Google Scholar]
- 61.Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, et al. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther. 2008;84(5):559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 62.Nakatani-Freshwater T, Taft DR. Renal excretion of emtricitabine I: effects of organic anion, organic cation, and nucleoside transport inhibitors on emtricitabine excretion. J Pharm Sci. 2008;97(12):5401–5410. doi: 10.1002/jps.21370. [DOI] [PubMed] [Google Scholar]
- 63.Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR) Curr Drug Targets. 2006;7(7):893–909. doi: 10.2174/138945006777709520. [DOI] [PubMed] [Google Scholar]
- 64.Ito S. Transplacental treatment of fetal tachycardia: implications of drug transporting proteins in placenta. Semin Perinatol. 2001;25(3):196–201. doi: 10.1053/sper.2001.24566. [DOI] [PubMed] [Google Scholar]
- 65.Koren G. Pharmacokinetics in pregnancy; clinical significance. J Popul Ther Clin Pharmacol. 2011;18(3):e523–e527. [PubMed] [Google Scholar]
- 66.Knutti R, Rothweiler H, Schlatter C. Effect of pregnancy on the pharmacokinetics of caffeine. Eur J Clin Pharmacol. 1981;21(2):121–126. doi: 10.1007/BF00637512. [DOI] [PubMed] [Google Scholar]
- 67.McGready R, Stepniewska K, Seaton E, Cho T, Cho D, Ginsberg A, et al. Pregnancy and use of oral contraceptives reduces the biotransformation of proguanil to cycloguanil. Eur J Clin Pharmacol. 2003;59(7):553–557. doi: 10.1007/s00228-003-0651-x. [DOI] [PubMed] [Google Scholar]
- 68.McGready R, Stepniewska K, Edstein MD, Cho T, Gilveray G, Looareesuwan S, et al. The pharmacokinetics of atovaquone and proguanil in pregnant women with acute falciparum malaria. Eur J Clin Pharmacol. 2003;59(7):545–552. doi: 10.1007/s00228-003-0652-9. [DOI] [PubMed] [Google Scholar]
- 69.Dempsey D, Jacob P, 3rd, Benowitz NL. Accelerated metabolism of nicotine and cotinine in pregnant smokers. J Pharmacol Exp Ther. 2002;301(2):594–598. doi: 10.1124/jpet.301.2.594. [DOI] [PubMed] [Google Scholar]
- 70.Wadelius M, Darj E, Frenne G, Rane A. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400–407. doi: 10.1016/S0009-9236(97)90118-1. [DOI] [PubMed] [Google Scholar]
- 71.de Haan GJ, Edelbroek P, Segers J, Engelsman M, Lindhout D, Devile-Notschaele M, et al. Gestation-induced changes in lamotrigine pharmacokinetics: a monotherapy study. Neurology. 2004;63(3):571–573. doi: 10.1212/01.WNL.0000133213.10244.FD. [DOI] [PubMed] [Google Scholar]
- 72.Stock B, Dean M, Levy G. Serum protein binding of drugs during and after pregnancy in rats. J Pharmacol Exp Ther. 1980;212(2):264–268. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DOCX 464 kb