

Abstract
Systemic response to DNA damage and other stresses is a complex process that includes changes in the regulation and activity of nearly all stages of gene expression. One gene regulatory mechanism used by eukaryotes is selection among alternative transcript isoforms that differ in polyadenylation [poly(A)] sites, resulting in changes either to the coding sequence or to portions of the 3′ UTR that govern translation, stability, and localization. To determine the extent to which this means of regulation is used in response to DNA damage, we conducted a global analysis of poly(A) site usage in Saccharomyces cerevisiae after exposure to the UV mimetic, 4-nitroquinoline 1-oxide (4NQO). Two thousand thirty-one genes were found to have significant variation in poly(A) site distributions following 4NQO treatment, with a strong bias toward loss of short transcripts, including many with poly(A) sites located within the protein coding sequence (CDS). We further explored one possible mechanism that could contribute to the widespread differences in mRNA isoforms. The change in poly(A) site profile was associated with an inhibition of cleavage and polyadenylation in cell extract and a decrease in the levels of several key subunits in the mRNA 3′-end processing complex. Sequence analysis identified differences in the cis-acting elements that flank putatively suppressed and enhanced poly(A) sites, suggesting a mechanism that could discriminate between variable and constitutive poly(A) sites. Our analysis indicates that variation in mRNA length is an important part of the regulatory response to DNA damage.
The necessity of a poly(A) tail at the 3′-ends of mRNAs for stability, export, and translation of the mature transcript has been well documented (Iglesias and Stutz 2008; Zhang et al. 2010; Eckmann et al. 2011). The point at which the RNA is cleaved and polyadenylated can also influence the amount and type of protein derived from that mRNA. Selection between mRNA isoforms with different poly(A) sites can alter the amount of coding sequence in the mRNA or significantly change its stability, localization, or translation because of the inclusion of post-transcriptional regulatory elements in the final mature transcript. Genome-scale studies of essentially all eukaryotes have shown that a majority of genes utilize multiple poly(A) sites (Brockman et al. 2005; Yan and Marr 2005; Zhang et al. 2005), demonstrating the scope of such variation. The spacing between alternative poly(A) sites can be as little as a few nucleotides or up to hundreds or even thousands of nucleotides. Selection among transcript isoforms with different poly(A) sites can vary during development and differentiation, between cells with different proliferation rates, or between diseased and matched healthy samples (Ji and Tian 2009; Ji et al. 2009; Singh et al. 2009; MacDonald and McMahon 2010; Neilson and Sandberg 2010; Lutz and Moreira 2011; Lembo et al. 2012). Systematic variation in poly(A) sites has generally been referred to as “alternative polyadenylation,” even though mechanisms other than changes in 3′-end processing could be responsible for selecting between the different mRNA isoforms.
It is likely that systematic, regulatory changes in mRNA length at the 3′-end could also be employed as part of the global response of a cell to environmental conditions. To assess this possibility, we conducted a genome-wide analysis of changes in poly(A) site profiles in yeast after DNA damage. Specifically, we used a large-scale measurement of direct RNA sequence (DRS) tags (Ozsolak et al. 2010) to compare expression and poly(A) site position with and without exposure to the UV-mimetic 4-nitroquinoline 1-oxide (4NQO), which introduces bulky chemical adducts into DNA. Genomic alignment and subsequent assignment of DRS tags to protein coding genes revealed significant variation in the isoform distribution in over one-third of the expressed genes, characterized by a general trend toward lengthening of mRNAs and a focusing to a reduced number of poly(A) sites in the transcripts of many genes. This surprising finding of a widespread shift in mRNA lengths suggests a novel way in which the cell can cope with DNA damage.
Results
Genome-wide measurement of poly(A) sites shows widespread trends toward transcript elongation after exposure to 4NQO
For genome-scale assessment of poly(A) site usage, DRS analysis was performed on yeast samples grown under standard conditions in the presence or absence of 4NQO. DRS generates 30- to 40-nt-long strand-specific RNA sequences that are typically anchored within 1–2 nt of the transcript's poly(A) site (Ozsolak et al. 2010). For our samples, DRS yielded 4.4 million reads for untreated cells and 10.3 million for 4NQO-treated cells. Following alignment to the reference yeast genome (sacCer3), we assigned tags to known protein coding genes (for details, see Methods).
Eukaryotic poly(A) sites are frequently degenerate with respect to position (Brockman et al. 2005; Yan and Marr 2005; Zhang et al. 2005), a phenomenon that has typically been dealt with in computational analyses either by local clustering and condensation of sites or by removal of sites that are below a threshold level of supporting evidence. In contrast, we chose to work with all sites at their indicated position and create a “cumulative polyadenylation distribution” (CPD) (see Figs. 2, 3, below), which reads from 5′→3′ through the gene and represents the empirical likelihood of polyadenylation at or before each position in the putative transcript. Comparison of the identification of genes with significant 4NQO-induced variation with and without clustering revealed only minor differences in results (Supplemental Fig. 1).
Figure 2.

Cumulative polyadenylation distribution (CPD) plots for genes that shift to longer isoforms with exposure to 4NQO. CPD plots are presented for (A) RNA14, (B) DEF1, and (C) RAD26. In all panels, black and red line-plots represent the CPD without and with exposure to 4NQO, respectively. Orange bars show CDS positions, with all plots oriented such that the 5′-3′ direction of transcription is left to right. Light blue bars identify the largest block of difference between 4NQO and STD samples, and are displayed if the block was measured as significant. Gray bars show all significantly differing regions in the CPD after 4NQO exposure. Green arrows show prominent poly(A) sites. Normalized expression levels were calculated for total (black column plot), full-length (blue column plot), and optimal transcripts (red column plot).
Figure 3.

Cumulative polyadenylation distribution (CPD) plots for genes that shift to shorter isoforms with exposure to 4NQO. CPD plots are presented for (A) RAD53, (B) MIP6, and (C) HAA1. Plot elements are the same as in Figure 2.
We compared each gene's distribution of DRS tags with and without 4NQO treatment, testing the hypothesis that the distribution of isoforms, and therefore the distribution of DRS tags, would not vary with the addition of 4NQO. Statistically significant variation was identified through a permutation analysis that randomized DRS tag–to-sample assignment (Methods), controlling for multiple testing at FDR < 0.1. We found that 2031 of the ∼5800 yeast genes had significant variation in their distribution of poly(A) sites (Table 1). Of these, 1720 genes had evidence only for a shift to elongated isoforms, 258 had evidence only for a shift to truncated isoforms, and 53 had more complex patterns. The changes in genes with elongated transcripts generally affected both the CDS and 3′ UTR, whereas genes with evidence of truncation were highly biased to affect only the 3′ UTR (Supplemental Fig. 2).
Table 1.
Count of genes with significant 4NQO-induced variation in isoform, broken down by relative position and type of change
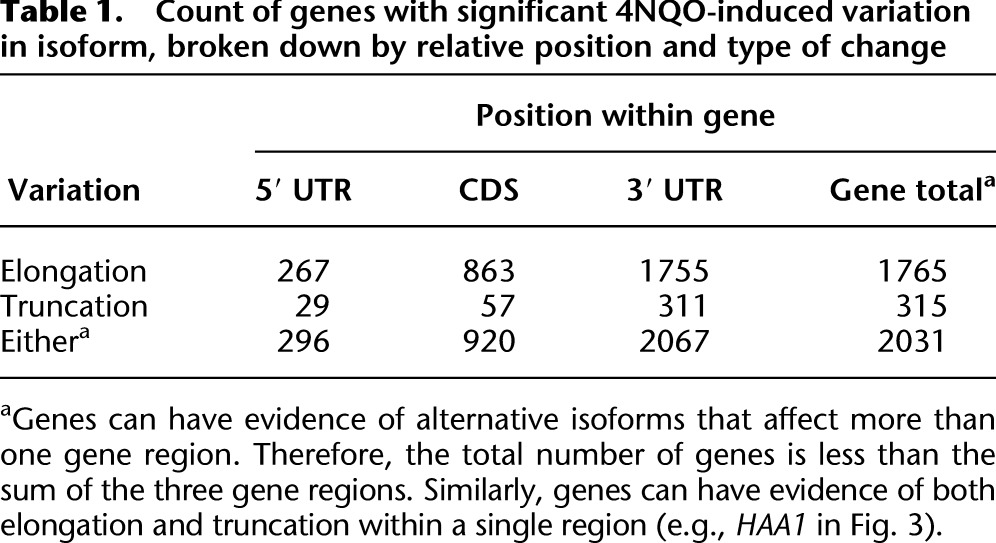
Consistent with other large-scale measurements of yeast poly(A) sites (Ozsolak et al. 2010; Yoon and Brem 2010; Johnson et al. 2011), the distribution of poly(A) sites within distinct gene regions (Fig. 1A) demonstrates that the majority of sites in both 4NQO-treated and untreated samples are in the first 250 nucleotides (nt) of the 3′ UTR. However, with 4NQO, there was a notable loss of transcripts ending at poly(A) sites within or upstream of the CDS. Comparison of the relative change in the fraction of all poly(A) sites in each gene region (Fig. 1B) shows that the largest variation is observed in the suppression of short transcripts terminating within 100 nt of the start codon. Taken transcriptome-wide (Fig. 1C), we found a distinct decrease in the fraction of the DRS tags of transcripts located within the CDS after 4NQO treatment. As noted above, over 3800 genes did not vary significantly in poly(A) site usage (examples are shown for TEF1, TEF2, CLP1, MPE1, and PTA1 in Supplemental Fig. 4).
Figure 1.

The distribution of DRS tags, indicating putative poly(A) sites, within annotated gene structures. (A) The fraction of the DRS sites identified within each gene region shows a downstream shift following exposure to 4NQO. The CDS is separated into proximal (<100 nt from the AUG start codon) and distal (>100 nt). (B) A logarithmic plot of the ratio of 4NQO to STD (untreated) counts from A. (C) The fraction of the tags in each sample assigned to distinct parts of gene structures. On the left (labeled CDS-truncated) is the fraction upstream of the stop codon, indicating a putatively truncated protein coding sequence. On the right (labeled Non-optimal) is the fraction that lies outside of the first 250 nt of the 3′ UTR.
To further assess how the expression of isoforms with alternative poly(A) sites contributes to gene regulation, the CPD plots (Figs. 2, 3) include measurements of normalized transcript expression determined in three distinct ways: (1) the total abundance, as reflected in the normalized counts assigned to each gene regardless of the location within the gene structure (black bar plots in Figs. 2, 3); (2) the abundance of transcripts that include a full-length CDS, regardless of 3′ UTR length (blue bar plots); and (3) the abundance of putatively “optimal” transcripts, defined as including the full CDS and a 3′ UTR ≤250 nt, a distance that corresponds to the ∼95th percentile of observed yeast 3′ UTRs (red bar plots) (Graber et al. 1999, 2002).
3′ RNA amplification of cDNA ends (3′ RACE) was used to validate the change in mRNA isoforms for representatives of suppressed (ACT1, NHX1, and RNA14) and enhanced poly(A) sites (RAD53) (Supplemental Fig. 3; for CPD plots, see Figs. 2A, 3A; Supplemental Fig. 4). For each of these genes, the size of the 3′ RACE products was consistent with that predicted from the DRS mapping; for ACT1 and RNA14, in agreement with the sizes of mRNAs detected by Northern blot in other studies (Mandart 1998; Sparks and Dieckmann 1998; He et al. 2003; Runner et al. 2008; Johnson et al. 2011). For NHX1 and RAD53, the upstream sites are far from any A-rich stretches, minimizing the likelihood that the 3′ RACE products would be generated by internal priming. Importantly, the amount of the 3′ RACE products associated with each gene changes in the direction predicted by the DRS data (Supplemental Fig. 3, bottom panel), with transcripts that terminate at the upstream site decreasing upon 4NQO treatment for ACT1, NHX1, and RNA14, but increasing for RAD53. In summary, the 3′ RACE analysis supports the accuracy of DRS data set in determining poly(A) site positions.
To search for functional significance in the genes that show significant changes in their CPD profiles, we extracted groups of genes with common behavior and submitted the gene lists to the GOStat tool (Beissbarth and Speed 2004), which searches for over- and underrepresented Gene Ontology (Ashburner et al. 2000) terms, including corrections for multiple hypothesis testing. Analysis of the genes whose short transcripts were suppressed revealed significant categories of both over- and underrepresented terms (Supplemental Tables 1, 2). The overrepresented GO terms fell into three general categories, focused on (1) transport and localization, (2) signal cascades in response to stimulus, and (3) catabolic processes. The underrepresented categories were primarily associated with gene transcription and translation and other biosynthetic pathways. In sharp contrast, genes that were subject to truncation upstream of the CDS had no significantly over or underrepresented terms.
CPD plots reveal gene-specific patterns for isoform variations in response to 4NQO
In this section, we present several representative examples of the different manners in which the distribution of 3′ termini can vary with exposure to 4NQO. Poly(A) sites that show the greatest suppression in terms of tag counts are those located upstream of the stop codon, compared with those in the 3′ UTR (Fig. 1A). Many genes with poly(A) sites in this category have a discrete, step-like CPD within their CDS, produced by poly(A) sites that can be further distinguished by whether the transcripts terminate distal to the CDS start codon or proximal (defined as within 100 nt) or even upstream in the 5′ UTR. An example of the first type is RNA14, the yeast homolog of the mammalian gene CSTF3 (CstF-77), which encodes a poly(A) factor with an evolutionarily conserved feedback mechanism in which polyadenylation at a site within the CDS leads to suppression of protein product (Mandart 1998; Juge et al. 2000; Pan et al. 2006). Consistent with previous studies, DRS tags indicate two distinct internal sites (Fig. 2A, positions ∼393,750 and ∼394,250) that together account for >70% of the RNA14 transcripts under standard growth conditions. With exposure to 4NQO, however, transcripts terminating at both sites are suppressed, reducing to ∼40% the fraction of transcripts terminating in the CDS. Additional examples (e.g., NHX1) can be seen in Supplemental Figure 4.
DEF1 provides an example of suppression of promoter-proximal sites (Fig. 2B). DEF1 encodes a protein that works in complex with Rad26 to ubiquitinate and degrade RNA polymerase II in response to DNA damage such as that caused by 4NQO and UV light (Woudstra et al. 2002). Under normal conditions, ∼70% of DEF1 transcripts terminate within 20 nt of the start codon (∼80% upstream of the stop codon) and produce a known short RNA (Nagalakshmi et al. 2008; Yassour et al. 2009). Under growth in 4NQO, however, the fraction drops to ∼30% (35% in the CDS), resulting in a fourfold increase in the fraction of mRNAs that include the full-length CDS. Additional examples of this type (e.g., HRP1) can be seen in Supplemental Figure 4.
We also observed genes with seemingly promiscuous polyadenylation across the CDS to give a ramp-like appearance in the CPD. An informative example is RAD26, which encodes a protein that forms a complex with Def1 and is involved in transcription-coupled repair (Woudstra et al. 2002). Under normal growth conditions, DRS tags show evidence of a large number of minor poly(A) sites spread over ∼2 kb of the CDS (Fig. 2C). After exposure to 4NQO, the transcripts are much more tightly focused to two dominant poly(A) sites (positions ∼500,000 and 500,700 in Fig. 2C), increasing the fraction of transcripts with a complete CDS from ∼45% to ∼75%. Other examples of this subclass are CFT1, CFT2, FIP1, PAP1, PCF11, PTI1, and SSU72 (Supplemental Fig. 4).
Of the 1765 genes with a 4NQO-induced shift to elongated transcripts (Table 1), 898 were affected only in their 3′ UTR (Supplemental Fig. 2). Elongation of these transcripts would presumably result in an extended 3′ UTR with additional regulatory sequences that could affect translation or stability (Kuersten and Goodwin 2003; Legendre et al. 2006; Sandberg et al. 2008; Ji and Tian 2009; Mayr and Bartel 2009). Examples of genes with 4NQO-induced elongated 3′ UTRs include ACT1, SUA7, CLP1, NPL3, PFS2, SYC1, and YTH1 (Supplemental Fig. 4).
While transcript elongation dominated our data, we also found 315 genes with increased expression of truncated transcripts. Of these, 258 did not affect the CDS but only changed the length of the 3′ UTR (Supplemental Fig. 2), generally resulting in a switch from an extended 3′ UTR to short or moderate length 3′ UTRs (<250 nt). RAD53, which encodes a protein kinase necessary for cell cycle arrest in response to DNA damage (Putnam et al. 2009), presents an interesting specific example (Fig. 3A). The DRS data indicate two major RAD53 transcripts, both including the complete CDS, and terminating ∼120 nt and ∼750 nt downstream from the stop codon, respectively. A total of 750 nt represents an extremely long 3′ UTR for yeast transcripts (Fig. 1A). 3′ RACE PCR validated expression of sequences that extended from just downstream from the upstream poly(A) site to the distal poly(A) site (Supplemental Fig. 3). Strikingly, the extended transcript is ∼2.5× more abundant than the shorter isoform in standard growth conditions, while exposure to 4NQO reversed this ratio to give 4× more of the shorter mRNA (Fig. 3A). Thus, despite a small drop in the total abundance of the transcript, the change in isoform distribution results in a significant increase in the number of transcripts with the shorter 3′ UTR (Fig. 3A).
A few genes show a shift to transcripts that terminate upstream of the stop codon, which would presumably reduce full-length protein expression relative to the transcript expression. For example, MIP6 (Fig. 3B), which encodes a putative mRNA binding protein associated with the nuclear pore, shows a significant increase in the fraction of transcripts that terminate at a promoter proximal poly(A) site, increasing from ∼15% to ∼65% with exposure to 4NQO.
Finally, a small number of genes (∼50), for example, HAA1 (Fig. 3C), show more complex changes in the poly(A) site profile. HAA1, which encodes a stress-related transcription factor (Fernandes et al. 2005), produces transcripts with four principal poly(A) sites and many additional minor sites under normal conditions (Fig. 3C). Under standard conditions, ∼50% of the transcripts are truncated within the coding sequence, and another 20% use poly(A) sites ≥450 nt from the stop codon, giving an unusually long 3′ UTR for yeast (Fig. 1A). Following 4NQO treatment, at least 80% of the transcripts utilize a poly(A) site ∼80 nt downstream from the stop codon. Comparing total transcripts (Fig. 3C), the expression increases ∼10% with exposure to 4NQO; however, the full-length and “optimal” length transcript expression levels increase by twofold and fourfold, respectively. This reduction of the number of utilized poly(A) sites in stably expressed transcripts with 4NQO treatment is reflected genome-wide, as we observed a significant shift to use of fewer poly(A) sites for many of the genes whose mRNA profiles changed (Supplemental Fig. 5).
In summary, the genome-wide assessment presented above suggests that the change in the relative expression of isoforms that differ in the position of the mRNA 3′-end provides a means of controlling expression beyond that which can be obtained by transcriptional control alone.
Comparison with a previous data set suggests both technical and biological variation
For comparative purposes, we obtained and similarly analyzed the sequence data from a previous study focused on the effects of depletion of the mRNA export factor Yra1 (Johnson et al. 2011). Our analysis of these data was largely consistent with the previous analysis, with ∼1500 genes showing significant variation between control and Yra1-depleted samples (cf. Supplemental Fig. 6 with Fig. 5 in Johnson et al. 2011). In contrast with the trend in 4NQO, depletion of Yra1 led to roughly equal numbers of genes with shifts to truncated and elongated transcripts.
A detailed comparative analysis of the data sets facilitated an assessment of technical aspects of these different methods for identifying poly(A) sites. The Yra1-depletion data were generated with a solution-based oligo-dT-VN primer followed by sequencing on an Illumina GA IIx (Johnson et al. 2011). We found that the solution-based assay was significantly more susceptible to internal priming at A- or AG-rich sequences than DRS (Supplemental Fig. 7).
Direct comparison of CPDs revealed a more striking difference between the two data sets, suggestive of systematic differences in experimental conditions. Genome-wide CPD comparison of the control samples identified 2790 genes with significant variation between experiments, compared with 2031 between 4NQO and its control and 1489 between Yra1-depletion and its control. Examples of this systematic variation can be seen in plots of the genes RNA14, DPL1, and PCF11 (Fig. 4). For these genes, the collection of poly(A) sites is largely consistent between experiments, but the relative abundance of tags at each site varies systematically between experimental groups, a phenomenon that holds true across a large fraction of surveyed genes. Consistent with this analysis, when we compared the many earlier studies that used Northern blot of RNA14 transcripts to demonstrate the effects of various mutations or growth conditions on polyadenylation in vivo, there was significant variation in the relative poly(A) site usage among the wild-type controls (Fig. 1A in Mandart 1998; Fig. 2D in He et al. 2003; Fig. 7 in Runner et al. 2008; Fig. 2 in Sparks and Dieckmann 1998).
Figure 4.

Comparison of poly(A) sets from distinct data sets reveals systematic variations in isoform abundance between experiments. CPD plots comparing the measured differences between the current DRS data and the previously measured poly(A) site profiles for standard and Yra1-depleted conditions (Johnson et al. 2011). (A) RNA14. Transcripts terminating within the RNA14 CDS are significantly suppressed with either exposure to 4NQO or Yra1 depletion; however, the baseline control (STD) plots vary significantly. Note that the putative site positioned ∼393,500 is near an A-rich block, but not close enough to be attributed to internal priming of the oligo-dT. (B) DPL1. No significant variation is measured with either 4NQO exposure or Yra1 depletion; however, significant variation is measured between the two control samples. In addition, the expression levels, shown at right are much more similar within experiments than between control samples of the two experiments (for a genome-wide analysis of expression level similarities , see Supplemental Fig. 8). (C) PCF11. Significant variation was measured in 4NQO but not with Yra1 depletion. As with DPL1, expression levels are more similar between samples in the same experiment than between controls of the distinct experiments. 4NQO, STD data from the current study. J_STD (control) and J_YRA1 (Yra1-depleted) samples from Johnson et al. (2011).
We also noted that for DPL1 and PCF11, the expression levels are much more similar within each experiment than between the two control samples (Figs. 4B,C), suggesting systematic differences in experimental conditions. A hierarchical clustering of the measured expression levels was consistent with the supposition that samples from the same experiment are more similar than the control samples (Supplemental Fig. 8). Further investigation of the relationship between expression level and variation in poly(A) site distribution intriguingly revealed that highly expressed genes are largely immune to significant changes in isoform distribution (Supplemental Fig. 9). This relationship held up in all comparisons, whether between treatment and control within an experiment, or between the control samples of different experiments. However, no correlation was found between the number of poly(A) sites and the expression level (data not shown). Finally, we compared the overlap in the genes that are subject to changes in poly(A) site distribution among the three comparisons (Supplemental Fig. 10). The overlapping categories are significantly higher than expected under the null hypothesis of independent lists, suggesting that genes that change isoforms under one set of test conditions are likely to do so under multiple conditions.
Exposure to 4NQO suppresses the mRNA 3′-end processing reaction
Suppression of polyadenylation activity at upstream sites is one possible means of reducing the relative expression of short transcripts following exposure to 4NQO. To determine the effect of 4NQO on mRNA 3′-end processing, yeast cells were exposed to 5 μg/mL of 4NQO for 2 h, and in vitro processing was examined in extracts prepared from treated and untreated cells. Measurements of a test construct containing the GAL7 poly(A) site revealed that both the cleavage and poly(A) addition steps are significantly impaired in 4NQO-treated cells (Fig. 5A). Processing was also inhibited if cells were exposed to UV light (Fig. 5B). UV exposure causes damage similar to that of 4NQO, inhibits poly(A) site cleavage in mammalian cells (Kleiman and Manley 2001), and induces a shift to longer transcripts from the yeast RPB2 gene (Yu and Volkert 2013). The 4NQO-induced inhibition of in vitro processing is unlikely to be part of the general environmental stress response as we have shown in previous studies that 3′-end formation is not inhibited by heat shock (37°C) for a similar amount of time (Cheng et al. 2004; He and Moore 2005; Ghazy et al. 2009). Furthermore, the efficiency of in vitro processing was not affected when cells were exposed to other growth-inhibitory agents, such as 10 mg/mL hydroxyurea, which causes replication stress by decreasing dNTP pools, or 20 mM caffeine, a low-affinity adenosine analog thought to inhibit enzymes such as the DNA damage checkpoint kinases ATM and ATR and the MTOR (mechanistic target of rapamycin) kinase that regulates cell growth (Fig. 5C; Reinke et al. 2006; Sabisz and Skladanowski 2008).
Figure 5.

Effects of 4NQO on mRNA 3′-end processing. (A) In vitro processing efficiency. Yeast samples were treated with 5 μg/mL 4NQO for 2 h, followed by preparation of processing extracts. Coupled cleavage and poly(A) addition was assayed using radioactive precursor containing the GAL7 poly(A) site and flanking sequences (left panel). Cleavage was assayed by replacing ATP with dATP, which blocks tail extension (right panel, first three lanes). Poly(A) addition alone was assayed using RNA substrate ending at the GAL7 poly(A) (right panel, last three lanes). Unprocessed precursor is shown in the lanes marked “Pre,” and positions of substrate and product are shown on the right. (B) Effects of UV light on processing. Extracts were made from cells with or without exposure to UV light and examined for processing as described in A. (C) Effects of other agents on processing. Extracts were made from cells treated with hydroxyurea (HU, 10 mg/mL) or caffeine (20 mM) for 2 h, and coupled cleavage and poly(A) addition reactions compared with that of extract from untreated cells. Neither treatment inhibits the efficiency of processing, but we note that poly(A) tails are longer with exposure to caffeine. (D) Protein level variation. Western blots of Rpo21, processing complex subunits, Npl3, and Sto1, with actin (Act1) as the loading control. The percentage of protein remaining after 4NQO treatment was calculated after normalizing to Act1 and is indicated next to each protein. (E) Diagram of RNA14 substrates. This figure depicts the RNA14 precursors and indicates the position of poly(A) sites used in vitro in relationship to the region mapped for the upstream and downstream sites by DRS and shows the length of sequence in each portion of the transcript. The numbers marking the regions mapped by DRS correspond to the chromosomal positions shown in the CPD plot of Figure 2A. The product indicated for pA1 refers to the reactions in F. (F) Coupled cleavage and poly(A) addition reactions using the transcripts containing the first (pA1) or third (pA3) RNA14 poly(A) sites. Reactions were performed with GAL7 substrate (lanes 1–3), RNA14 pA1 (lanes 4–6), and RNA14 pA3 (lanes 7–9) using extract from cells with (+) or without (−) 4NQO treatment. Unprocessed precursor is shown in the lanes marked “Pre.” The products from the RNA14 pA3 substrate are indicated by an asterisk.
We also examined proteins involved in mRNA 3′-end formation to see if there were any changes that might explain the decrease of polyadenylation activity in vitro. Processing of yeast poly(A) sites requires assembly of a complex of CPF, CF IA, and Hrp1 (Millevoi and Vagner 2010; Chan et al. 2011). Each factor recognizes one of the cis-acting elements that specify the poly(A) site, and the entire complex is stabilized by protein/protein interactions between the factors. To determine if these proteins were depleted or post-translationally modified in a way that significantly changed mobility by SDS-PAGE, we used antibodies against specific subunits to probe blots of 4NQO-treated and untreated extracts (Fig. 5D). As has been previously reported (Malik et al. 2008), the level of the Rpo21 (Rpb1) subunit of RNA Polymerase II is reduced following DNA damage by 4NQO. There were no significant changes in the levels of the CF IA or Hrp1 processing factors, or in the Sto1 (Cbp80) and Npl3 proteins, which are known to interfere with the interaction of CF IA and RNA (Bucheli et al. 2007; Wong et al. 2007). However, there is a striking reduction of many of the CPF subunits, including essential processing factors such as Pta1, Mpe1, Fip1, the Ysh1 nuclease, and the RNA binding proteins Cft1, Cft2, and Yth1. Mobility changes were not observed; however, post-translationally modified subunits may be present but not resolved on the gels used for Western blots. Taken together, these results suggest that loss of CPF subunits contributes to a general down-regulation of the poly(A) processing capacity of the cell after exposure to 4NQO.
In sharp contrast with the in vitro measurements of protein expression with exposure to 4NQO, the DRS data revealed that nearly all transcripts from genes involved with mRNA 3′-end formation were increased in expression (Supplemental Table 3). Only NPL3 and SYC1 transcripts were decreased in expression.
To gain further insight into the regulatory mechanisms that could produce an altered mRNA isoform profile, we focused on the RNA14 gene. As shown in Figure 2A, the two major poly(A) sites in the RNA14 coding sequence are significantly suppressed in vivo with 4NQO treatment such that ∼60% of mRNAs terminate at the third poly(A) site. Previous in vivo studies have shown that the use of the first RNA14 poly(A) site (pA1) is decreased when cleavage/polyadenylation factors are mutated (Mandart 1998; He et al. 2003), or by loss of function of Rpb4 or Yra1, both of which affect in vivo recruitment of the processing factors to the elongating transcriptional complex (Runner et al. 2008; Johnson et al. 2011). These findings suggest that RNA14 pA1 is very susceptible to the availability of the 3′-end processing machinery.
To directly test this idea, we compared the activity of RNA14 pA1 with that of the third region (pA3) in our in vitro processing assays. As can be seen from Figure 2A, these poly(A) sites span ∼70 nt for pA1 and 100 nt for pA3. Transcriptional templates were designed to give transcripts of 540 nt for pA1 and 495 nt for pA3, giving at least 190 nt of sequence on each side of the region in which most polyadenylation occurs (Fig. 5E). From our knowledge of yeast poly(A) sites (Fig. 6; Graber et al. 1999, 2002; van Helden et al. 2000), this arrangement should include the core signal elements needed for processing. Processing of RNA14 pA3 occurred as robustly as our standard test site, GAL7 (Fig. 5F), with almost all precursor being polyadenylated in 20 min. Four products were detected, and the approximate positions of the cleavage sites were determined after taking into account that the length of poly(A) tails added in vitro ranges from 70–90 adenosines. This placed the most abundant product within the region determined by DRS and two of the less abundant products just outside of it (Fig. 5E). In contrast, the RNA14 pA1 precursor was not processed (Fig. 5F, lanes 4,5), and activity on this substrate was not improved with longer times of reaction or by varying the concentration of extract, salt, or magnesium or the means of preparing the RNA substrate (data not shown). As with GAL7, the abundance of polyadenylated products from the RNA14 pA3 substrate was decreased if extract came from 4NQO-treated cells. From these experiments, we conclude that the RNA14 pA1 site is processed very poorly in vitro compared with the downstream RNA14 pA3 site or the constitutive GAL7 site. This finding is consistent with the analysis presented below, which shows that suppressed poly(A) sites have poorer matches to the consensus signal sequences.
Figure 6.

Sequence differences in the cis-elements that flank different classes of poly(A) sites. (A) Differences between enhanced and suppressed poly(A) sites plotted as the difference in the fraction of the poly(A) sites in each set with a match to the representative pentamers as a function of position relative to the poly(A) site. As indicated by the gray labels and arrows, positive (negative) values indicate more frequent occurrence in the flanks of enhanced (suppressed) sites. Asterisks mark pentamer-position pairs that passed DPWC statistical significance tests. (B) Same as A, but calculated in comparison to 4NQO-suppressed and constitutive poly(A) sites. As indicated by the gray labels and arrows, positive (negative) values indicate more frequent occurrence in the flanks of suppressed (constitutive) sites. (C) Same as in A and B, but for a comparison of 4NQO-enhanced and constitutive poly(A). As indicated by the gray labels and arrows, positive (negative) values indicate more frequent occurrence in the flanks of enhanced (constitutive) sites. The positioning of the cis-elements of the model previously developed for yeast poly(A) site prediction (Graber et al. 2002) is shown at the top for reference. (EE) Efficiency element, with optimal sequence UAUAUA; (PE) positioning element, characterized as adenine-rich; (UU) upstream uracil-rich element; (DU) downstream uracil-rich element.
Comparative sequence analysis reveals that variable poly(A) sites show differences in the sequence elements that define yeast poly(A) sites
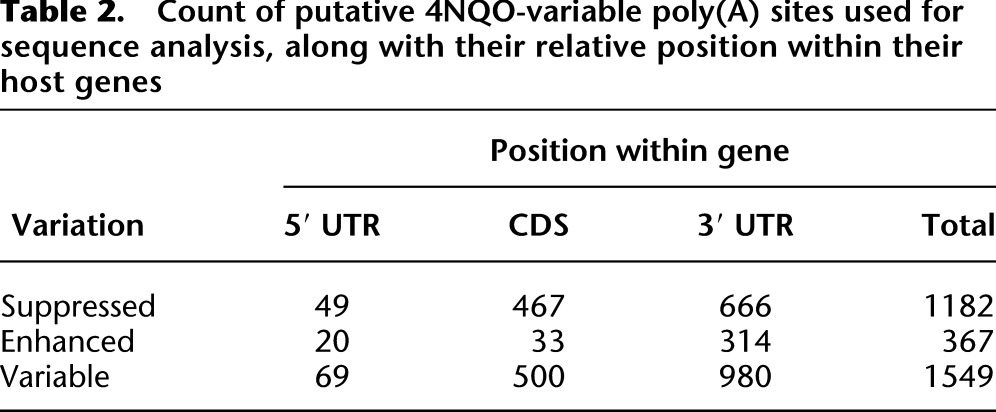
In the genes whose CPDs showed significant variation with and without 4NQO treatment, we classified poly(A) sites as “constitutive” or unchanging, enhanced (leading to transcript truncation), or suppressed (leading to transcript elongation). We hypothesized that the changes in levels of isoform could be driven at least partially by changes in the efficiency of processing at specific sites. In this case, regulated sites might show differences in the cis-acting poly(A) sequence elements. Four elements have been shown to define yeast poly(A) sites (Chen and Moore 1992; Guo and Sherman 1996a,b; Dichtl and Keller 2001). These include the efficiency element, composed of the hexamer UAUAUA or close variants and typically positioned between 30 and 80 nt upstream of the poly(A) site, the positioning element, an A-rich element typically ∼15–25 nt upstream of the poly(A) site, and two U-rich tracts that closely flank the poly(A) site (Fig. 6A; Graber et al. 1999, 2002; van Helden et al. 2000). Based on previous studies of poly(A) cis-elements (Legendre and Gautheret 2003; Hu et al. 2005; Hutchins et al. 2008), we expected that the differences could be manifested in terms of sequence content, positioning, or a combination of both. For this analysis, we extracted 400-nt-long sequences centered on putative poly(A) sites (Table 2), generating distinct sets for each class of poly(A) site and location within the associated gene (Methods). We grouped sequences by whether they were enhanced or suppressed and further classified them on the basis of their location within the gene structure as 5′ UTR, CDS, or 3′ UTR. For further comparison, we also identified a set of 484 constitutive poly(A) sites (Methods).
Table 2.
Count of putative 4NQO-variable poly(A) sites used for sequence analysis, along with their relative position within their host genes
We searched for putative changes with differential positional word counting (DPWC) (Graber et al. 2007), which identifies position-specific differences in sequence content between sets of sequences aligned on a common feature (Methods). Results are reported as pairs of pentamers and an associated positioning relative to the poly(A) and are plotted as shown in Figure 6 and Supplemental Figure 11. The plots of Figure 6 focus on representative pentamers that reflect the sequence content of the known yeast poly(A) sequence elements (e.g., UAUAU and AUAUA for the efficiency element). These pentamers consistently varied the most significantly across all comparisons made (Supplemental Fig. 11).
Comparison of suppressed versus enhanced poly(A) sites produced a significant common pattern, regardless of the specific gene region under examination (Fig. 6; Supplemental Fig. 11). A striking difference between these sets was manifested as an AAAAA or GAAAA sequence at or flanking the poly(A) site in enhanced sequences. In addition, significant differences were observed in several pentamers (e.g., UAUAU and AUAUA) at positions that suggest relative loss of the efficiency element in the suppressed sites compared to enhanced sites.
Comparison of the constitutive poly(A) sites with the suppressed (Fig. 6B) and enhanced (Fig. 6C) sites revealed common differences, including fewer matches to the efficiency element (Figs. 6B,C, pentamers UAUAU and AUAUA approximately between positions −70 and −40) and also reduced incidence of U-rich sequences both near the poly(A) site and also further upstream of the efficiency element (Fig. 6B,C, pentamer UUUUU). Since these patterns are consistent with elements of accepted models of the yeast poly(A) signal, they suggest that a threshold quality of signal is required for constitutive processing. The A-rich signal that distinguished suppressed from enhanced signals is visible in the comparison of enhanced and constitutive sites (pentamer, AAAAA in Figure 6C; Supplemental Fig. 11); however in this case, the enrichment did not pass our strict threshold for significance. A large-scale study recently identified an extended AG-rich element (GAAGAAGA) as distinctive of poly(A) sites located within coding sequences (Yoon and Brem 2010). This element did not appear in our analysis. Our results suggest that differences in the sequences that flank the poly(A) sites control the relative activity of the poly(A) sites after DNA damage.
Discussion
We have shown here for the first time that exposure to the DNA damaging agent 4NQO significantly alters the profile of poly(A) sites associated with transcripts of a large number of yeast genes. The predominant pattern of mRNA lengthening resembles that observed when mammalian cells differentiate or otherwise proliferate more slowly (Sandberg et al. 2008; Ji et al. 2009). Thus, systematic selection between mRNA isoforms that differ in poly(A) sites may represent a general mechanism by which the eukaryotic cell introduces increased regulatory potential to specific groups of mRNAs in response to external signals. In the following sections, we first discuss critical aspects of our analytical method and then mechanisms that could contribute to the changes in mRNA isoforms that we have observed.
Analysis of poly(A) site usage
The computational analysis that we have used to portray poly(A) site usage involves mapping the CPD across a gene, rather than the common practice of grouping or eliminating sites below a threshold level of supporting evidence. This technique allowed us to identify patterns of polyadenylation that might otherwise have been missed. The relevance of this novel approach is demonstrated by comparison with a recent study that described a shift to a downstream poly(A) site in the yeast RPB2 gene in response to UV-induced damage (Yu and Volkert 2013). RPB2 has one poly(A) site in the 3′ UTR close to the stop codon and a second site that is more distal, and transcripts using both sites are readily detectable in untreated cells by Northern blot analysis (Yu and Volkert 2013) and in our CPD plot (Supplemental Fig. 4). With exposure to the UV-mimetic 4NQO, the CPD plot shows a shift to RNAs with longer 3′ UTRs, similar to that seen by Yu and Volkert (2013). However, it also reveals that a significant amount of the polyadenylated RPB2 transcripts (∼38%) in undamaged cells terminate promiscuously within the coding sequence. The fraction of truncated transcripts drops to ∼15% with 4NQO treatment. The heterogeneity of this subpopulation would make these transcripts unlikely to be detected by Northern blot, yet their suppression significantly increases the proportion of mRNA that can be translated into full-length protein.
Another novel contribution of our study lies in the comparison with data from a previous study of the effects of depletion on the Yra1 mRNA export factor (Johnson et al. 2011). This comparison revealed that while the positions of major poly(A) sites were generally unchanged, their relative usage showed unexpected diversity when comparing the two putatively wild-type “control” samples. One likely explanation for this variability is that differences in experimental conditions (e.g., choice of growth media, specific strains utilized, and target density of yeast culture prior to measurement) can alter the pattern of mRNA isoforms, as well as total abundance (Supplemental Fig. 8). A possibly significant variable in this instance is that the Yra1-depletion study used synthetic complete medium (Johnson et al. 2011), while our study used the richer YPD medium. Thus, these potential sources of variability must be taken into account when comparing data sets from different studies.
Possible mechanisms for changing isoform distributions
The unexpected volatility in isoform distribution may reflect an underappreciated means of rapidly responding to a changing environment and leads to the question of what mechanisms could elicit such changes. However, mechanisms underlying genome-wide changes in polyadenylation patterns have not been well studied. Variations in the levels of different mRNA isoforms could arise from changes in mRNA stability, from changes in how well particular sites are recognized and processed by the polyadenylation machinery, or from altering the rate of transcription elongation between poly(A) sites. We discuss each potential mechanism below.
Altering mRNA stability
Previous work has shown that the general mRNA population transiently decreases after UV-induced DNA damage and is not restored until the damage is repaired (Cevher and Kleiman 2010). In mammalian cells, this decrease is manifested through regulatory changes in transcription, 3′-end processing, and mRNA degradation, yet specific transcripts can escape the general response. For example, normally unstable mRNAs needed for DNA damage response can be stabilized after DNA damage (Blattner et al. 2000).
Differences in degradation among mRNA isoforms of previously transcribed RNAs could produce the patterns we observe after 4NQO treatment, especially if these RNAs are inherently unstable. Two types of yeast mRNAs fall in this category. First, transcripts with very long 3′ UTRs are subject to destabilization through nonsense-mediated decay (NMD) (Nicholson et al. 2010; Parker 2012). In our data set, eight transcripts that are natural NMD substrates (Kebaara and Atkin 2009) were expressed highly enough to analyze. However, these did not show a consistent pattern of increase or decrease in overall expression levels (two increased, three decreased, and three showed no change). The second type of unstable mRNAs are terminated within the coding sequence and are subject to degradation by the nonstop-mediated decay (NSD) pathway, due to their lack of stop codon and 3′ UTR (Inada and Aiba 2005; Wilson et al. 2008). If transcription of genes expressing these types of RNAs was slowed after 4NQO treatment, the NSD pathway could selectively remove truncated transcripts from the general population. However, we found that in ∼200 genes, transcripts that terminate before the stop codon either increase or are unchanged in relative abundance after 4NQO treatment (Table 1; Supplemental Table 11; for specific examples, see Supplemental Fig. 4), suggesting that CDS-truncated RNAs are not universally susceptible to NSD. It is not currently known how either NMD or NSD responds to DNA damage or, whether under certain conditions, they can act on subsets of their normal targets. There are two published studies showing that stress does not change the relative stability of yeast mRNA isoforms expected to be targets of NMD or NSD, one examining CBP1 transcripts when yeast are grown on glycerol (Sparks et al. 1997) and the second looking at RPB2 transcripts in response to UV-induced DNA damage (Yu and Volkert 2013). A whole-genome analysis of changes in mRNA turnover rates will be necessary to resolve the contribution of mRNA stability to changing the ratios of different mRNA isoforms.
Altering 3′-end processing at poly(A) sites
Previously published studies of global alternative polyadenylation (e.g., Sandberg et al. 2008; Singh et al. 2009; MacDonald and McMahon 2010; Neilson and Sandberg 2010; Lembo et al. 2012) almost universally infer that the changes in mRNA lengths are due to alterations in processing of the 3′-ends. This assumption is based primarily on finding that promoter-proximal sites tend to have poorer matches to consensus sequences specifying a poly(A) site (Beaudoing et al. 2000; Jan et al. 2011; Shepard et al. 2011; Martin et al. 2012; Smibert et al. 2012; Ulitsky et al. 2012). We have found a similar poor adherence to the optimal configuration of signal elements in our yeast study. Comparison of either suppressed or enhanced sites (Fig. 6) with the constitutive sites suggests that a strong efficiency element and U-rich elements flanking the poly(A) sites are required for constitutive activity and that weakening of these elements is important if a site is to be regulated. In addition, enhanced sites tend to have an AAAAA/GAAAA motif immediately flanking the poly(A) site that is diminished in suppressed sites.
In our study, the major shift in poly(A) site usage after 4NQO treatment involves suppression of upstream sites. Our finding that variations in consensus signal sequences define such sites suggests that alterations in the general polyadenylation machinery, rather than specific regulatory proteins, could drive the large-scale increase in use of promoter-distal sites. In agreement with this idea, some studies have shown that artificial manipulation of the level of a single processing factor can create global shifts to downstream sites similar to those seen under natural conditions (Di Giammartino et al. 2011; Berg et al. 2012; Jenal et al. 2012; Martin et al. 2012; Shi 2012; Yao et al. 2012; others). However, these studies have not shown that similar changes in these factors actually happen as part of the cell's response to stress.
In the current study, we have conducted a thorough analysis of all of the proteins found in the cleavage/polyadenylation complex, as well as of known regulators of yeast polyadenylation. The only change that we detect upon treatment with 4NQO is a decrease in subunits of CPF, the factor that recognizes the U-rich elements flanking the poly(A) site and provides the enzymatic functions of cleavage and poly(A) addition (Millevoi and Vagner 2010; Chan et al. 2011). Mutations in CPF subunits cause a shift to downstream sites on common test genes such as RNA14 and ACT1 (Dichtl et al. 2002; He et al. 2003; Kyburz et al. 2003; Garas et al. 2008), similar to the changes seen for these transcripts upon exposure to 4NQO. We propose that the poorer configuration of processing signals found on suppressed promoter-proximal sites make them more susceptible to the reduced levels of CPF caused by 4NQO and thus more likely to be bypassed. However, when CPF is not limiting, as in untreated cells, preference would generally be given to the first poly(A) site transcribed.
Our data also show that exposure to 4NQO results in the increased generation of truncated transcripts for a small number of genes (Table 1). In mammalian cells, specific RNA binding proteins have been found to promote use of alternative poly(A) sites in some genes (for review, see Di Giammartino et al. 2011; Shi 2012) or to prevent the suppression of TP53 mRNA polyadenylation after DNA damage (Decorsiere et al. 2011), in spite of a global decrease in polyadenylation efficiency (Kleiman and Manley 1999; Nazeer et al. 2011). It is not known whether similar modulators will be present in yeast.
Altering transcription elongation between poly(A) sites
Another mechanism that could alter the ratios of long to short mRNAs is changes in the progression of RNA Polymerase II between poly(A) sites, such as has been reported for regulation of alternative splicing after DNA damage (Munoz et al. 2009). Slowing transcription chemically or by mutation of transcription factors can increase use of an upstream poly(A) site (Cui and Denis 2003; Pinto et al. 2011; Yu and Volkert 2013) but to our knowledge, natural examples of this mechanism have not been reported. The RNA14 gene may prove to be such an example.
We have shown that the upstream RNA14 poly(A) site is inactive in vitro, while the site in the 3′ UTR is used very efficiently (Fig. 5). However, 50% of the RNA14 mRNAs end at the upstream poly(A) site in vivo (Fig. 2A). A block to transcription downstream from this site could explain the abundance of truncated RNA14 transcripts when there is no genotoxic stress. Alternatively, it is possible that increased recruitment of processing factors to the transcriptional complex in this region of the RNA14 gene, especially when the factors are not limiting, favors the poor promoter-proximal site. Detailed molecular analyses of RNA14 and other genes with similar behavior will be necessary to clarify the mechanism and its generality.
Consequences of alternative isoform expression
We propose that regulated variation in isoforms that differ in their poly(A) sites works in conjunction with transcription and translational control to facilitate an appropriate response to environmental conditions. In the particular case that we have examined, isoform variation in response to 4NQO exposure would represent a fine-tuning mechanism for optimizing expression of proteins needed to promote repair and recovery once the damage is removed. DNA damage is known to induce a general suppression of mRNA transcription, processing and translation (Heine et al. 2008; Cevher and Kleiman 2010; Spriggs et al. 2010). The changes in length to give an mRNA optimized for translation may help the cell to produce as much full-length protein as possible from a limited amount of transcripts and translational apparatus. Differences in isoform may also contribute to the documented observation that mRNA levels in yeast do not always correspond well to protein levels (Griffin et al. 2002; Foss et al. 2007, 2011; Baek et al. 2008; Lee et al. 2010). Conversely, the near uniform divergence between transcript and protein levels in the polyadenylation complex, even with isoform distribution included (Supplemental Fig. 4; Supplemental Table 3), suggests that mechanisms regulating translation and protein stability must also play a role in maintaining the necessary balance of protein expression.
Conclusions
We have shown here that exposure of yeast to 4NQO results in systematic changes to the poly(A) site pattern that would be predicted to affect the amount and type of protein made in response to this type of DNA damage. As discussed above, multiple forces, including degradation of inherently unstable mRNAs, changes in stability of specific mRNAs, and changes in transcription elongation, could be operating to produce the final profile of mRNA isoforms observed after DNA damage. Indeed, it would be surprising if the cell limited itself to a single mechanism in its effort to enhance survival. The systematic variation in poly(A) sequence elements at regulated sites and the decreased expression of several protein components of the poly(A) complex that we report here support a role for changes in how sites are recognized by the processing machinery. Further research is needed to elucidate the extent to which the different mechanisms are used, and whether they are employed in response to other types of stress.
Methods
Direct RNA sequencing (DRS)
A 200-mL culture of W303-1a yeast cells was grown in YPD at 30°C to an OD600 of 0.8, and half of the culture was mixed with 4NQO (5 μg/mL) while the other half remained untreated. After 2 h, cells were harvested, and total RNA was extracted by the hot phenol method (Rio et al. 2011) and Phase Lock Gel (5 Prime) according to the method in the manufacturer's manual. Precipitated RNA was dissolved in 10 mM Tris-HCl and 1 mM EDTA (pH 7.6) and stored at −80°C for further analysis. Total RNA (3 μg) was submitted to Helicos for DRS as described previously (Ozsolak et al. 2010).
Alignment and assignment to gene
Alignments of DRS tags were made to the reference yeast genome (sacCer3) with the RNA Unified Mapper (RUM; Grant et al. 2011), using the –BLAT-only option, and with additional parameters –BLAT-tile-size = 10 –BLAT-step-size = 1. Following empirical investigation of error rates (Supplemental Fig. 12), we further restricted DRS tags to those that uniquely aligned to the genome with three or fewer mismatches. To protect against the probability of false-positive poly(A) sites due to sequence generation internal to the transcript, we removed any tags that were adjacent to a genome run of six or more A or G residues (Supplemental Fig. 7). Genome annotations were obtained from the Yeast Genome database project (http://www.yeastgenome.org/) and restricted to putative protein coding genes, excluding genes that are characterized as “dubious.” Tag-gene assignments were then made based on both correct orientation and relative positioning, with a restriction to implied poly(A) sites that were properly oriented and located within 100 nt upstream of the coding sequence and 1000 nt downstream from the annotated CDS. In cases where two or more genes could be assigned, an assignment was preferentially made to CDS if the tag was located between the start and stop codons, or to 3′ UTR rather than 5′ UTR for ambiguous intergenic tags, and to the closer gene, if the assignment was of the same classification. All tags not assigned in this way were discarded as beyond the scope of the present analysis, even though they may be of interest for further investigations. Nearly all genes (>5700) had at least one assigned gene for both samples after this procedure.
Expression level determination
Abundance was measured in three distinct ways: (1) all tags assigned to a gene, (2) all tags that imply a complete coding sequence, and (3) all tags with complete CDS and 3′ UTR <250 nt (95% cutoff). Since recent reports have demonstrated the dangers of normalization to total counts (Bullard et al. 2010), the tag abundances were normalized by scaling each gene's total counts such that the 75th percentile values were equal.
Identification of differences in poly(A) site distribution
Searches for changes in poly(A) site distribution were carried out as follows: poly(A) site distributions were expressed as cumulative plots extending from the furthest 5′ to furthest 3′ tags assigned to each gene across both conditions. We did not eliminate or condense low-count sites but rather used all sites to calculate a CPD, which represents the empirical probability that a poly(A) site has been reached at each point in the gene structure. We then identified the largest difference block between control and treated CPD curves, defined as a contiguous block where the sample-to-sample relationship (greater or less) is unchanging. Difference blocks were tabulated for their sequence length and sum of differences across the entire length. Genes with significant variation were identified through a permutation analysis with the null hypothesis being that the distribution of poly(A) sites would be unchanged and that differences in cumulative poly(A) usage between conditions were due to sampling. All tags assigned to the gene in question were pooled and then randomly segregated into two groups 10,000 times, preserving the size of the two groups. The number of permutations in which the largest difference block equaled or exceeded the value from the real data was used to create an empirical P-value. Multiple hypothesis testing was controlled at a false-discovery rate (FDR) < 0.05. We also arbitrarily restricted the size of the differential region to be at least 30 nt long with an average difference of at least 0.1 (representing 10% of the polyadenylation sites measured for the gene under test). The largest block for each gene is displayed as a light blue bar in the bottom of the CPD plots (Figs. 2, 3; Supplemental Fig. 4). Genes that passed significance for the distribution as a whole were subjected to further permutation analysis, in which the difference between the CPDs from each sample was measured at each position. The permutations established a null distribution that was used to calculate empirical P-values for each position in the CPD. Positions scoring <0.05 were marked as significant and are marked with gray bars in the CPD plots.
Identification of poly(A) sites
To enhance the searches and analysis of how poly(A) sites are specified for alternative processing, we focused on discrete sites where significant changes could be characterized. To identify prominent sites within a single sample, we used an edge-finding approach (diagrammed in Supplemental Fig. 13) to analyze the CPD, highlighting regions of change of at least 5% of the tags for the gene. To identify sites that varied in 4NQO, we applied the same edge-finding approach to the difference between the CPDs of the STD and 4NQO samples, restricting the identified sites to those where the implied change in poly(A) was for at least 40% of the transcripts assigned to each gene. We further restricted this analysis to “upstream” poly(A) sites within each gene, based on the rationale that regulatory control is more likely to be manifested there. We generated sets of a few tens to a few hundred sequences for each type of variation and gene region (Table 2).
For further analysis, we collected a set of “constitutive” poly(A) sites, based on the analysis of DRS data for comparison of multiple stress conditions (including growth in glycerol and growth at 37°C [JH Graber, FI Nazeer, P Yeh, JN Kuehner, S Borikar, D Hoskinson, and CL Moore, in prep.]). We used the edge-finding algorithm (Supplemental Fig. 13) on each condition-specific cumulative poly(A) distribution and specifically extracted 484 sites that represent at least 10% of the poly(A) activity for their parent genes in all of the tested conditions.
Comparative sequence analysis
Sequence analysis for elements was performed according to DPWC using the method described previously (Graber et al. 2007). Briefly, we assume that the sequence elements that specify poly(A) sites are constrained both by sequence content and relative positioning, and therefore count the occurrences of all k-mers (k = 5 in this work) across all sequences in 10-nt wide position windows surrounding the poly(A) sites. Parameters were set based on size of the sequence sets and our knowledge of the positioning of the yeast poly(A) signals. We measured the difference between two sets of sequences (e.g., suppressed vs. enhanced sites), under the null hypothesis that the k-mer positioning distributions were the same and any differences were due to sampling. Statistical significance was obtained through a permutation analysis that randomized the sequence to set assignments while preserving the number of sequences in each set. Empirical P-values obtained in this way were corrected for multiple testing controlling at FDR ≤ 0.20, with the additional requirement that the pentamer-window pair occur in at least 10% of one of the sequence sets under comparison. All of the analysis shown here is based on the fraction of sequences in each set that contain at least one copy of the pentamer in the sequence window. A similar analysis, based on the fraction of all pentamers counted in each window, gives equivalent results (Supplemental Fig. 11). Complete DPWC results are available in Supplemental Tables 4 through 10.
Gene sets were examined for enrichment of functional categories using GOStat (Beissbarth and Speed 2004).
RNA analysis
For 3′ RACE, cDNA was synthesized according to the method previously described (Rio et al. 2011). Briefly, 10 μg of total RNA was reverse transcribed using an anchor oligo(dT) adaptor (Supplemental Table 12; Borson et al. 1992) and SuperScript III Reverse Transcriptase (Invitrogen). Subsequent PCR reactions were performed using gene-specific primers and an anchor PCR primer (Supplemental Table 12). PCR products were then resolved in 0.9% Agarose gels and visualized by ethidium bromide staining. The intensity of DNA bands was quantified using Image J software.
In vitro processing reactions
Exponentially growing yeast cultures were treated with 4NQO (5 μg/mL), hydroxyurea (10 mg/mL), or caffeine (20 mM) for 2 h, followed by preparation of processing extracts. For UV treatment, cells were pelleted and resuspended in phosphate-buffered saline at a concentration of 10 OD units/mL and spread on polycarbonate plates at a depth of <2 mm. The cells were then exposed to two pulses of 200 J/m2 doses of UV using a UV crosslinker (Stratagene), pelleted, and resuspended in prewarmed YPD for 2 h in the dark before harvesting for extract preparation. Preparation of yeast cell extracts, transcription of [α-32P]UTP-labeled precursor RNA, and processing assays were described previously (Zhao et al. 1999). Reaction mixtures were assembled on ice in a volume of 12 μL containing 1 mM magnesium acetate, 75 mM potassium acetate, 2% polyethylene glycol 8000 (Fisher), 2 mM ATP, 10 nM radioactive GAL7 RNA, 1.5 μM (0.6 μg) tRNA, 1 mM DTT, 0.4 U of RNasin (Promega), and 0.1 mg/mL of bovine serum albumin, 4 μL of cell extract (∼20 μg of protein) and incubated for 20–30 min at 30°C. Reactions were stopped by addition of 2 μL of stop solution (12 mg/mL of proteinase K, 130 mM EDTA, and 6% SDS) followed by incubation for 15 min at 30°C. The mixtures were diluted to 30 μL and extracted once with phenol-chloroform-isoamyl alcohol (25:24:1), and 1/10 of the reaction mixture was separated by electrophoresis on a 5% acrylamide–8.3 M urea gel and visualized by phosphoimager. Templates for T3 polymerase transcription of RNA substrates containing the first and third RNA14 poly(A) sites were constructed by PCR using the primer pairs described in Supplemental Table 12. The primer pairs were positioned to give ∼200 nt on each side of the major cleavage site cluster. Transcripts were purified by electrophoresis on a 5% acrylamide–8.3 M urea gel according to the method described previously or using the RNeasy Mini Kit (Qiagen), following the manufacturer's instructions.
Western blot analysis
Whole-cell extracts were prepared according to the method described previously (He and Moore 2005). Proteins were resolved on a 10% SDS-PAGE gel and detected by Western blot using antibodies specific for the indicated processing complex subunits according to the method described previously (Saguez et al. 2008). Rpo21 (Rbp1) was detected using commercial 8WG16 antibody (Santa Cruz). Antibodies against Npl3 and Sto1 (Cbp80) were gifts from C. Guthrie and I. Mattaj, respectively. Protein levels before and after 4NQO treatment were quantified using BioRad Quantity One software, and the percentage of protein remaining after 4NQO treatment was calculated after normalizing for Act1 protein.
Data access
The sequences used in these analyses have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRA054029. Additional supplemental data and analysis tools can be obtained at http://harlequin.jax.org/yeast4NQO/.
Acknowledgments
We thank C. Guthrie and I. Mattaj for their generous gifts of antibodies. We thank the members of the C.L.M. and J.H.G. laboratories for stimulating discussions and Greg Carter, Kevin Mills, and Steven Munger for critical reading of the manuscript. We also thank three anonymous reviewers for their helpful critiques. This work was supported by NIH grant GM072706 (J.H.G.) and NIH grant GM041752 (C.L.M.). F.I.N. was supported by the American Cancer Society New England division, grant no. PF-12-251-01-RMC.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.144964.112.
Freely available online through the Genome Research Open Access option.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D 2000. Patterns of variant polyadenylation signal usage in human genes. Genome Res 10: 1001–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beissbarth T, Speed TP 2004. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20: 1464–1465 [DOI] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner C, Kannouche P, Litfin M, Bender K, Rahmsdorf HJ, Angulo JF, Herrlich P 2000. UV-induced stabilization of c-fos and other short-lived mRNAs. Mol Cell Biol 20: 3616–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borson ND, Salo WL, Drewes LR 1992. A lock-docking oligo(dT) primer for 5′ and 3′ RACE PCR. PCR Methods Appl 2: 144–148 [DOI] [PubMed] [Google Scholar]
- Brockman JM, Singh P, Liu D, Quinlan S, Salisbury J, Graber JH 2005. PACdb: PolyA Cleavage site and 3′-UTR database. Bioinformatics 21: 3691–3693 [DOI] [PubMed] [Google Scholar]
- Bucheli ME, He X, Kaplan CD, Moore CL, Buratowski S 2007. Polyadenylation site choice in yeast is affected by competition between Npl3 and polyadenylation factor CFI. RNA 13: 1756–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevher MA, Kleiman FE 2010. Connections between 3′-end processing and DNA damage response. Wiley Interdiscip Rev RNA 1: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y 2011. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA 2: 321–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Moore C 1992. Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol Cell Biol 12: 3470–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, He X, Moore C 2004. The essential WD repeat protein Swd2 has dual functions in RNA polymerase II transcription termination and lysine 4 methylation of histone H3. Mol Cell Biol 24: 2932–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Denis CL 2003. In vivo evidence that defects in the transcriptional elongation factors RPB2, TFIIS, and SPT5 enhance upstream poly(A) site utilization. Mol Cell Biol 23: 7887–7901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decorsiere A, Cayrel A, Vagner S, Millevoi S 2011. Essential role for the interaction between hnRNP H/F and a G quadruplex in maintaining p53 pre-mRNA 3′-end processing and function during DNA damage. Genes Dev 25: 220–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, Manley JL 2011. Mechanisms and consequences of alternative polyadenylation. Mol Cell 43: 853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Keller W 2001. Recognition of polyadenylation sites in yeast pre-mRNAs by cleavage and polyadenylation factor. EMBO J 20: 3197–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Blank D, Sadowski M, Hubner W, Weiser S, Keller W 2002. Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J 21: 4125–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann CR, Rammelt C, Wahle E 2011. Control of poly(A) tail length. Wiley Interdiscip Rev RNA 2: 348–361 [DOI] [PubMed] [Google Scholar]
- Fernandes AR, Mira NP, Vargas RC, Canelhas I, Sa-Correia I 2005. Saccharomyces cerevisiae adaptation to weak acids involves the transcription factor Haa1p and Haa1p-regulated genes. Biochem Biophys Res Commun 337: 95–103 [DOI] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, Kruglyak L 2007. Genetic basis of proteome variation in yeast. Nat Genet 39: 1369–1375 [DOI] [PubMed] [Google Scholar]
- Foss EJ, Radulovic D, Shaffer SA, Goodlett DR, Kruglyak L, Bedalov A 2011. Genetic variation shapes protein networks mainly through non-transcriptional mechanisms. PLoS Biol 9: e1001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garas M, Dichtl B, Keller W 2008. The role of the putative 3′ end processing endonuclease Ysh1p in mRNA and snoRNA synthesis. RNA 14: 2671–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazy MA, He X, Singh BN, Hampsey M, Moore C 2009. The essential N terminus of the Pta1 scaffold protein is required for snoRNA transcription termination and Ssu72 function but is dispensable for pre-mRNA 3′-end processing. Mol Cell Biol 29: 2296–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber JH, Cantor CR, Mohr SC, Smith TF 1999. Genomic detection of new yeast pre-mRNA 3′-end-processing signals. Nucleic Acids Res 27: 888–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber JH, McAllister GD, Smith TF 2002. Probabilistic prediction of Saccharomyces cerevisiae mRNA 3′-processing sites. Nucleic Acids Res 30: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber JH, Salisbury J, Hutchins LN, Blumenthal T 2007. C. elegans sequences that control trans-splicing and operon pre-mRNA processing. RNA 13: 1409–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, Stoeckert CJ, Hogenesch JB, Pierce EA 2011. Comparative analysis of RNA-seq alignment algorithms and the RNA-seq unified mapper (RUM). Bioinformatics 27: 2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin TJ, Gygi SP, Ideker T, Rist B, Eng J, Hood L, Aebersold R 2002. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics 1: 323–333 [DOI] [PubMed] [Google Scholar]
- Guo Z, Sherman F 1996a. 3′-end-forming signals of yeast mRNA. Trends Biochem Sci 21: 477–481 [DOI] [PubMed] [Google Scholar]
- Guo Z, Sherman F 1996b. Signals sufficient for 3′-end formation of yeast mRNA. Mol Cell Biol 16: 2772–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Moore C 2005. Regulation of yeast mRNA 3′ end processing by phosphorylation. Mol Cell 19: 619–629 [DOI] [PubMed] [Google Scholar]
- He X, Khan AU, Cheng H, Pappas DL Jr, Hampsey M, Moore CL 2003. Functional interactions between the transcription and mRNA 3′ end processing machineries mediated by Ssu72 and Sub1. Genes Dev 17: 1030–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine GF, Horwitz AA, Parvin JD 2008. Multiple mechanisms contribute to inhibit transcription in response to DNA damage. J Biol Chem 283: 9555–9561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Lutz CS, Wilusz J, Tian B 2005. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11: 1485–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins LN, Murphy SM, Singh P, Graber JH 2008. Position-dependent motif characterization using non-negative matrix factorization. Bioinformatics 24: 2684–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N, Stutz F 2008. Regulation of mRNP dynamics along the export pathway. FEBS Lett 582: 1987–1996 [DOI] [PubMed] [Google Scholar]
- Inada T, Aiba H 2005. Translation of aberrant mRNAs lacking a termination codon or with a shortened 3′-UTR is repressed after initiation in yeast. EMBO J 24: 1584–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, Vrielink JA, Bos AJ, Drost J, Rooijers K, et al. 2012. The poly(a)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149: 538–553 [DOI] [PubMed] [Google Scholar]
- Ji Z, Tian B 2009. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 4: e8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B 2009. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci 106: 7028–7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Kim H, Erickson B, Bentley DL 2011. The export factor Yra1 modulates mRNA 3′ end processing. Nat Struct Mol Biol 18: 1164–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge F, Audibert A, Benoit B, Simonelig M 2000. Tissue-specific autoregulation of Drosophila suppressor of forked by alternative poly(A) site utilization leads to accumulation of the suppressor of forked protein in mitotically active cells. RNA 6: 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebaara BW, Atkin AL 2009. Long 3′-UTRs target wild-type mRNAs for nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Nucleic Acids Res 37: 2771–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman FE, Manley JL 1999. Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 285: 1576–1579 [DOI] [PubMed] [Google Scholar]
- Kleiman FE, Manley JL 2001. The BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage and tumor suppression. Cell 104: 743–753 [DOI] [PubMed] [Google Scholar]
- Kuersten S, Goodwin EB 2003. The power of the 3′ UTR: Translational control and development. Nat Rev Genet 4: 626–637 [DOI] [PubMed] [Google Scholar]
- Kyburz A, Sadowski M, Dichtl B, Keller W 2003. The role of the yeast cleavage and polyadenylation factor subunit Ydh1p/Cft2p in pre-mRNA 3′-end formation. Nucleic Acids Res 31: 3936–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, et al. 2010. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Gautheret D 2003. Sequence determinants in human polyadenylation site selection. BMC Genomics 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Ritchie W, Lopez F, Gautheret D 2006. Differential repression of alternative transcripts: A screen for miRNA targets. PLoS Comput Biol 2: e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo A, Di Cunto F, Provero P 2012. Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PLoS ONE 7: e31129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz CS, Moreira A 2011. Alternative mRNA polyadenylation in eukaryotes: An effective regulator of gene expression. Wiley Interdiscip Rev RNA 2: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald CC, McMahon KW 2010. Tissue-specific mechanisms of alternative polyadenylation: Testis, brain, and beyond. Wiley Interdiscip Rev RNA 1: 494–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Bagla S, Chaurasia P, Duan Z, Bhaumik SR 2008. Elongating RNA polymerase II is disassembled through specific degradation of its largest but not other subunits in response to DNA damage in vivo. J Biol Chem 283: 6897–6905 [DOI] [PubMed] [Google Scholar]
- Mandart E 1998. Effects of mutations in the Saccharomyces cerevisiae RNA14 gene on the abundance and polyadenylation of its transcripts. Mol Gen Genet 258: 16–25 [DOI] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M 2012. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep 1: 753–763 [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Vagner S 2010. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res 38: 2757–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz MJ, Perez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. 2009. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 137: 708–720 [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazeer FI, Devany E, Mohammed S, Fonseca D, Akukwe B, Taveras C, Kleiman FE 2011. p53 inhibits mRNA 3′ processing through its interaction with the CstF/BARD1 complex. Oncogene 30: 3073–3083 [DOI] [PubMed] [Google Scholar]
- Neilson JR, Sandberg R 2010. Heterogeneity in mammalian RNA 3′ end formation. Exp Cell Res 316: 1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O 2010. Nonsense-mediated mRNA decay in human cells: Mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67: 677–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM 2010. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Zhang H, Hague LK, Lee JY, Lutz CS, Tian B 2006. An intronic polyadenylation site in human and mouse CstF-77 genes suggests an evolutionarily conserved regulatory mechanism. Gene 366: 325–334 [DOI] [PubMed] [Google Scholar]
- Parker R 2012. RNA degradation in Saccharomyces cerevisae. Genetics 191: 671–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto PA, Henriques T, Freitas MO, Martins T, Domingues RG, Wyrzykowska PS, Coelho PA, Carmo AM, Sunkel CE, Proudfoot NJ, et al. 2011. RNA polymerase II kinetics in polo polyadenylation signal selection. EMBO J 30: 2431–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Jaehnig EJ, Kolodner RD 2009. Perspectives on the DNA damage and replication checkpoint responses in Saccharomyces cerevisiae. DNA Repair (Amst) 8: 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke A, Chen JC, Aronova S, Powers T 2006. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. J Biol Chem 281: 31616–31626 [DOI] [PubMed] [Google Scholar]
- Rio DC, Ares JM, Hannon GJ, Nilsen TW 2011. Purification of RNA from natural sources. In RNA: A laboratory manual, pp. 180–185. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Runner VM, Podolny V, Buratowski S 2008. The Rpb4 subunit of RNA polymerase II contributes to cotranscriptional recruitment of 3′ processing factors. Mol Cell Biol 28: 1883–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabisz M, Skladanowski A 2008. Modulation of cellular response to anticancer treatment by caffeine: Inhibition of cell cycle checkpoints, DNA repair and more. Curr Pharm Biotechnol 9: 325–336 [DOI] [PubMed] [Google Scholar]
- Saguez C, Schmid M, Olesen JR, Ghazy MA, Qu X, Poulsen MB, Nasser T, Moore C, Jensen TH 2008. Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol Cell 31: 91–103 [DOI] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y 2011. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-seq. RNA 17: 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y 2012. Alternative polyadenylation: New insights from global analyses. RNA 18: 2105–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Alley TL, Wright SM, Kamdar S, Schott W, Wilpan RY, Mills KD, Graber JH 2009. Global changes in processing of mRNA 3′ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res 69: 9422–9430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert P, Miura P, Westholm JO, Shenker S, May G, Duff MO, Zhang D, Eads BD, Carlson J, Brown JB, et al. 2012. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep 1: 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks KA, Dieckmann CL 1998. Regulation of poly(A) site choice of several yeast mRNAs. Nucleic Acids Res 26: 4676–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks KA, Mayer SA, Dieckmann CL 1997. Premature 3′-end formation of CBP1 mRNA results in the downregulation of cytochrome b mRNA during the induction of respiration in Saccharomyces cerevisiae. Mol Cell Biol 17: 4199–4207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, Willis AE 2010. Translational regulation of gene expression during conditions of cell stress. Mol Cell 40: 228–237 [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP 2012. Extensive alternative polyadenylation during zebrafish development. Genome Res 22: 2054–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J, del Olmo M, Perez-Ortin JE 2000. Statistical analysis of yeast genomic downstream sequences reveals putative polyadenylation signals. Nucleic Acids Res 28: 1000–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Meaux S, van Hoof A 2008. Diverse aberrancies target yeast mRNAs to cytoplasmic mRNA surveillance pathways. Biochim Biophys Acta 1779: 550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CM, Qiu H, Hu C, Dong J, Hinnebusch AG 2007. Yeast cap binding complex impedes recruitment of cleavage factor IA to weak termination sites. Mol Cell Biol 27: 6520–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woudstra EC, Gilbert C, Fellows J, Jansen L, Brouwer J, Erdjument-Bromage H, Tempst P, Svejstrup JQ 2002. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415: 929–933 [DOI] [PubMed] [Google Scholar]
- Yan J, Marr TG 2005. Computational analysis of 3′-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Res 15: 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y 2012. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci 109: 18773–18778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, et al. 2009. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci 106: 3264–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon OK, Brem RB 2010. Noncanonical transcript forms in yeast and their regulation during environmental stress. RNA 16: 1256–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Volkert MR 2013. UV damage regulates alternative polyadenylation of the RPB2 gene in yeast. Nucleic Acids Res 41: 3104–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Hu J, Recce M, Tian B 2005. PolyA_DB: A database for mammalian mRNA polyadenylation. Nucleic Acids Res 33: D116–D120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Virtanen A, Kleiman FE 2010. To polyadenylate or to deadenylate: That is the question. Cell Cycle 9: 4437–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Kessler M, Helmling S, O'Connor JP, Moore C 1999. Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Mol Cell Biol 19: 7733–7740 [DOI] [PMC free article] [PubMed] [Google Scholar]