

Polyglutamine-expanded huntingtin specifically targeted to synapses binds to synapsin-1, inhibits its phosphorylation, and causes defects in neurotransmitter release and age-dependent defects in neurological function.
Abstract
Many genetic mouse models of Huntington’s disease (HD) have established that mutant huntingtin (htt) accumulates in various subcellular regions to affect a variety of cellular functions, but whether and how synaptic mutant htt directly mediates HD neuropathology remains to be determined. We generated transgenic mice that selectively express mutant htt in the presynaptic terminals. Although it was not overexpressed, synaptic mutant htt caused age-dependent neurological symptoms and early death in mice as well as defects in synaptic neurotransmitter release. Mass spectrometry analysis of synaptic fractions and immunoprecipitation of synapsin-1 from HD CAG150 knockin mouse brains revealed that mutant htt binds to synapsin-1, a protein whose phosphorylation is critical for neurotransmitter release. We found that polyglutamine-expanded exon1 htt binds to the C-terminal region of synapsin-1 to reduce synapsin-1 phosphorylation. Our findings point to a critical role for synaptic htt in the neurological symptoms of HD, providing a new therapeutic target.
Introduction
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by a polyglutamine (polyQ) expansion of >37 glutamines in the HD protein huntingtin (htt). The polyQ expansion also leads to at least eight other neurodegenerative diseases, including spinocerebellar ataxia (SCA-1, -2, -3, -6, -7, and -17), dentatorubropallidoluysian atrophy, and spinobulbar muscular atrophy (Orr and Zoghbi, 2007). In HD, degeneration occurs preferentially in striatal neurons and extends to other brain regions as the disease progresses (Vonsattel and DiFiglia, 1998). This selective degeneration in distinct brain regions is a paradox also seen in other neurological disorders, such as Alzheimer’s and Parkinson’s diseases. One possible explanation for this is that mutant proteins preferentially affect the specific structures and biological properties of neurons. Considering that neuronal cells have unique, long neuronal processes and synapses that may be vulnerable to toxic proteins and insults, many studies have focused on synaptic function, revealing that synaptic dysfunction is the common pathological event in a variety of neurodegenerative diseases (Wishart et al., 2006; Milnerwood and Raymond, 2010, Garden and La Spada, 2012); however, because of the widespread subcellular distribution of mutant htt and the wide range of its toxic effects, the contribution of mutant htt in synapses to the development of neurological symptoms in HD remains unknown.
In HD, htt aggregates are formed by N-terminal htt fragments generated by proteolysis because the aggregates in HD patient brains are only labeled by antibodies to the N-terminal region of htt (DiFiglia et al., 1997; Gutekunst et al., 1999), and N-terminal mutant htt readily forms intracellular aggregates in vitro and in animal models (Menalled and Chesselet, 2002; Heng et al., 2008). Furthermore, the toxicity of N-terminal mutant htt is indicated by the fact that transgenic mice expressing N-terminal mutant htt show more severe neuropathology and symptoms than those expressing full-length mutant htt (Li and Li, 2011; Ross and Tabrizi, 2011).
A study has shown the correlation between age-dependent reduction in the activity of the ubiquitin–proteasome system and the progressive formation of htt aggregates (Li and Li, 2011). A variety of HD mouse models also suggest that a high expression level of transgenic mutant proteins is necessary for accelerating disease progression in mice (Heng et al., 2008; Ross and Tabrizi, 2011), whose relatively short lifespan (<2–3 yr) may prevent them from developing the robust neuropathology that is caused by mutant proteins in human brains over decades. In support of this idea, transgenic mice expressing N-terminal mutant htt have proved to be valuable systems for uncovering important pathological changes in HD. However, in current HD mouse models, mutant htt accumulates in various subcellular regions, such as the nucleus, neuronal processes, and synapses, and we know that mutant htt in different subcellular regions affects multiple cellular functions, including mitochondrial function, intracellular trafficking, metabolism, and synaptic function (Li and Li, 2006; Ross and Tabrizi, 2011). Because of the widespread distribution of mutant htt, the current HD mouse models are limited for distinguishing between nuclear, cytoplasmic, and synaptic htt toxicity.
Because synapses are unique to neuronal cells, understanding how mutant htt affects synaptic function is critical for unraveling the selective neuropathology seen in HD. To this end, we targeted mutant htt specifically in the presynaptic terminals in a transgenic mouse model. Mice expressing mutant htt with 150Q in synapses show age-related neurological symptoms and impaired neurotransmitter release, phenotypes also seen in knockin (KI) mice expressing full-length mutant htt. We find that mutant htt binds to synapsin-1 and affects its phosphorylation, a function that is critical for release of neurotransmitters. Our results not only provide evidence for the direct contribution of synaptic mutant htt to neurological symptoms but also help identify a new therapeutic target for the selective neuropathology of HD.
Results
Generation of transgenic mice expressing synaptic mutant htt
Exon1 mutant htt has been found to be generated in the brains of HD mice and patients (Landles et al., 2010; Sathasivam et al., 2013) and can cause severe neuropathologic phenotypes (Davies et al., 1997). To express mutant htt specifically in synapses, we fused exon1 htt containing either 20Q or 150Q to synaptosomal-associated protein 25 (SNAP25; Fig. 1 A), a component of the SNARE complex that is selectively distributed in the presynaptic plasma membrane (Rizo and Südhof, 2002). The SNAP25-htt fusion protein is expressed under the mouse prion promoter. We transfected SNAP25-20Q/150Q into rat cultured primary striatal neurons. Double-immunofluorescent staining with an anti-SNAP25, which did not react with endogenous mouse SNAP25 but transfected SNAP25, showed that transfected SNAP25-htt is diffuse in the cell body and also in synapses as puncta (Fig. 1 B). We then used SNAP25-htt constructs for pronuclear injection of mouse embryos and obtained a total of 16 founders of SNAP25-150Q (150Q) transgenic mice and eight founders of SNAP25-20Q (20Q) transgenic mice. Of the SNAP-150Q founders, only six could pass the transgene via germline transmission, and the others could not be bred or died. As a result, we selected three 150Q mouse lines (line-2, line-8, and line-15) that could be bred to F1 generation and show the clear expression of transgene. Unlike SNAP25-150Q transgenic mice, all SNAP25-20Q founders lived normally as wild-type (WT) mice, suggesting that SNAP25-150Q transgene is toxic. Because all transgenic 20Q mice live normally and are indistinguishable, we focused on one 20Q transgenic line (line-45) for comparison with 150Q transgenic mice.
Figure 1.

Generation of transgenic mice expressing N-terminal htt. (A) DNA structure of the transgene for expression of exon1 htt (1–67 amino acids) containing an additional 20Q or 150Q, which is fused to the C terminus of SNAP25 (SNAP25-20Q or SNAP25-150Q) and expressed under the control of the mouse prion promoter. (B) Expression of transfected SNAP25-20Q or SNAP25-150Q in cultured neurons from the rat striatum. The cells were labeled by antibodies to htt (green, mEM48) and SNAP25 (red) and Hoechst to the nuclei (blue). Arrows indicate synapselike puncta. Bar, 10 µm. (C) Western blot analysis of tissue lysates from the cortex (Ctx) and striatum (Str) of wild-type (WT), SNAP25-20Q (20Q), and SNAP25-150Q (150Q; line-8) mice at the age of 4 mo. The blots were probed with antibodies to SNAP25 (left) or htt (right). SNAP25-150Q and its degraded product (double arrows), aggregated htt (bracket), and SNAP25-20Q (arrowhead) are indicated. Anti-SNAP25 also labeled a nonspecific band at 98 kD in WT, as it was not labeled by anti-htt. (D) Western blotting analysis of brain regions (cortex; striatum; Hipp, hippocampus; Cere, cerebellum) and peripheral tissues (lung, liver, and muscle) shows a restricted expression of transgenic mutant htt in the brain regions of SNAP25-150Q (150Q; line-2) mice at 2 mo of age. Anti-SNAP25 staining reveals that transgenic SNAP25-20Q is expressed at a higher level than SNAP25-150Q and that both SNAP25-20Q and SNAP25-150Q are lower than endogenous SNAP25. Anti-htt also labeled nonspecific bands above 50 kD in the liver and lung samples. Endog., endogenous.
Genomic DNA analysis showed that the 150Q transgenic mouse lines have the same CAG repeat expansion (Fig. S1 A). Western blot analysis revealed that these 150Q lines express mutant htt at a similar level; however, mutant htt is expressed at a lower level than transgenic htt in the 20Q line (Fig. S1 A, top). This is because Western blot was probed with mouse EM48 antibody that preferentially reacts with mutant htt (Wang et al., 2008) but still labeled less SNAP25-150Q than SNAP25-20Q. To compare the expression levels of transgenic SNAP25-htt and endogenous SNAP25, we probed the Western blot with anti-SNAP25. The results showed that transgenic SNAP25-htt is expressed at a much lower level than endogenous SNAP25 and that mutant htt in three transgenic lines is also lower than transgenic SNAP25-20Q (Fig. S1 B, bottom).
Western blotting of mouse brain tissue lysates demonstrated that anti-SNAP25 clearly labeled SNAP25-20Q and SNAP25-150Q (line-8) at 4 mo of age (Fig. 1 C). Examining line-2 transgenic mice at 2 mo of age also revealed that SNAP25-150Q is expressed in various brain regions, including the cortex, striatum, hippocampus, and cerebellum. However, in peripheral tissues, such as lung, liver, and muscle, SNAP25-150Q was not detected (Fig. 1 D). Although these Western blot results were obtained from different mouse lines, the results are consistent with earlier findings that the prion promoter drives the expression of transgenes predominantly in neuronal cells (Schilling et al., 1999; Tebbenkamp et al., 2011). Also, using different batches of anti-SNAP25, we found that the transgenic htt (Fig. 1, C and D, arrows) is expressed at lower levels than endogenous SNAP25.
It would be interesting to compare the expression levels of transgenic htt in SNAP25-150Q mice with mutant htt in HD CAG150 KI mice that is expressed at the endogenous level (Menalled et al., 2002). Western blotting with 1C2 antibody, which reacts with the expanded polyQ repeat, revealed full-length and multiple degraded htt fragments in the HD KI mouse cortex. The levels of SNAP25-150Q and its degraded products in different brain regions were lower than the levels of total full-length and truncated mutant htt or were comparable to some of N-terminal htt fragments in HD KI mice (Fig. 2 A). In the hippocampus and cerebellum, more aggregated htt is seen in SNAP25-150Q transgenic mice than in HD KI mice (Fig. 2 A), suggesting that transgenic htt in these two brain regions may be less stable and prone to aggregation. Collectively, transgenic mutant htt is expressed at a lower level than control SNAP25-20Q and is not overexpressed compared with endogenous mutant htt.
Figure 2.

Transgenic SNAP25-150Q htt is localized in axonal terminals. (A) Western blotting with mEM48 showing that SNAP25-150Q (150Q) is expressed at levels comparable to N-terminal htt fragments generated in HD 150Q KI mouse brain regions (cortex, striatum, hippocampus, and cerebellum). (B) mEM48 immunofluorescent staining showing that SNAP25-150Q is outside the nuclei of neuronal cells in the cortex (Ctx), striatum (Str), and cerebellum (Cere). The wild-type (WT) mouse cortex served as a control. (C) mEM48 immunofluorescent staining revealing the nuclear and neuropil distribution of exon1 mutant htt in the brain cortex of an 8-wk-old R6/2 mouse. (D) EM showing that mutant htt forms aggregates (double arrows) in the axon (left) and a presynaptic terminal that contains synaptic vesicles (arrow). The postsynaptic terminal, which lacks synaptic vesicles (right), is also indicated by an arrow. Bars: (B and C) 10 µm; (D) 0.2 µm.
Subcellular localization of transgenic mutant htt
Anti-htt (mEM48) immunocytochemical experiments reveal that small puncta are present in the brain cortex of SNAP25-150Q transgenic mice but not in WT and SNAP25-20Q mice (Fig. S2). These puncta are similar to the neuropil aggregates seen in HD patient brains and various HD mouse models (Gutekunst et al., 1999; Maat-Schieman et al., 2007). To verify that these puncta are indeed in neuronal processes, we performed immunofluorescent staining, which can better reveal the subcellular localization of proteins. SNAP25-150Q is clearly present as puncta outside the nuclei of neuronal cells in the cortex and striatum (Fig. 2 B). In contrast, R6/2 mice, which express exon1 htt with 150Q under the control of human htt promoter (Davies et al., 1997), show mutant htt predominantly in the neuronal nuclei and neuropil (Fig. 2 C). Such a striking difference in transgenic htt distribution between SNAP25-150Q and R6/2 mouse brain clearly indicates that SNAP25-150Q is enriched in neuropil or neuronal processes. To verify that mutant htt is indeed localized in the presynaptic terminals, we performed EM and observed mutant htt aggregates (Fig. 2 D, double arrows) in axon (Fig. 2 D, left) and presynaptic, but not postsynaptic, terminals (Fig. 2 D, right) that contain synaptic vesicles (Fig. 2 D).
Immunocytochemical examination of mutant htt in young (2 mo) and old (9 mo) SNAP25-150Q mice revealed that synaptic accumulation of mutant htt in the cortex was age dependent (Fig. 3 A). Quantitative real-time PCR (qRT-PCR) analysis showed that transcript levels of SNAP25-150Q in the cortex and striatum are not different between young and old ages (Fig. 3 B). This assay also revealed that the levels of SNAP25-150Q transcripts are lower than mutant htt transcripts in R6/2 and HD CAG150 KI mice, again supporting the idea that transgenic SNAP25-150Q is not overexpressed. Thus, the increased synaptic htt staining in older mice is likely caused by the age-dependent accumulation of mutant htt at the protein level.
Figure 3.

SNAP25-150Q mice show age-dependent accumulation of mutant htt in synapses. (A) EM48 immunostaining showing that the brain cortex in older SNAP25-150Q mouse (line-8; 9 mo) displayed more synaptic htt than in young mouse (line-8; 2 mo). Bar, 20 µm. (B) qRT-PCR analysis of mutant human htt transcripts in the striatum and cortex tissues from SNAP25-150Q (2 and 9 mo old), R6/2 mice (1 mo old), and HD CAG150 KI mice (6 mo old). Primers that can selectively amplify human htt were used. The levels of htt transcripts were calculated from their values relative to mouse actin with the value of WT mouse htt as 1 and obtained from three to four independent experiments. Error bars are means ± SEM. (C) EM48 immunoblotting showing that older SNAP25-150Q mouse (line-8) cortex had more cleaved htt products (arrows) than young mouse. (D) Western blotting of subcellular fractionation showing that transgenic htt and its cleaved products (arrows) are recognized by mEM48 and enriched in the presynaptic (P1) fraction that is also enriched by SNAP25. Because the P2 fraction contains insoluble proteins, it also shows some htt aggregates. T, total protein; S, cytosolic protein; P1, presynaptic protein; P2, insoluble fraction.
Western blotting analysis showed that the older SNAP25-150Q mouse brain accumulated more cleaved htt fragments (Fig. 3 C), consistent with the idea that proteolysis results in small htt fragments that form aggregates. To better assess the relative levels of transgenic htt in presynaptic terminals, we performed subcellular fractionation, which can enrich presynaptic proteins, such as SNAP25 (Fig. S3; Gu et al., 2009b). The results show that both SNAP25-20Q and SNAP25-150Q are enriched in the presynaptic fraction (Fig. 3 D). Importantly, degraded htt fragments (Fig. 3 D, arrows) are also present in the presynaptic fraction. Comparison of the relative levels of htt in the total lysates and the presynaptic fraction led to an estimate that ∼80% of mutant htt is distributed in the synapses. Because Western blotting had demonstrated that these htt fragments were not labeled by anti-SNAP25 (Fig. 1, C and D), we wanted to further verify that cleaved htt fragments are responsible for forming synaptic aggregates. In some neuronal cells that expressed a high level of SNAP25-150Q, diffuse cytoplasmic staining was seen by both double-immunofluorescent staining with anti-htt and anti-SNAP25 (Fig. 4 A). However, although anti-htt could clearly label synaptic htt as small puncta, the majority of these puncta were not labeled by anti-SNAP25 (Fig. 4 A and Fig. S4). These findings suggest that cleaved htt fragments without SNAP25 accumulate in the synapses. To verify this idea, we used an antibody to synapsin-1, a different synaptic protein, to perform double staining. The results clearly show that the majority of synaptic htt puncta were labeled by anti–synapsin-1 (Fig. 4 B and Fig. S5). Moreover, htt aggregates, which are apparently larger than synaptic puncta, could be labeled by anti–synapsin-1 (Fig. S5), suggesting that htt aggregates also contain synaptic proteins, despite their negative staining by anti-SNAP25. Thus, using immunofluorescent staining, fractionation assays, and EM, we found convincing evidence that transgenic htt is selectively accumulated in the presynaptic terminals in transgenic mice and that this accumulation is age dependent and largely mediated by cleaved htt fragments without fusion to SNAP25.
Figure 4.

Localization of transgenic SNAP25-150Q in synapses. (A) Double immunofluorescent staining of the cortex of a SNAP25-150Q mouse (line-8) at 6 mo of age showing that anti-SNAP25 could label diffuse mutant htt but not small synaptic htt puncta (arrows). (B) Anti–synapsin-1 could label small synaptic htt puncta, suggesting that mutant htt without SNAP25 accumulates in the synapses. Note an htt aggregate (arrows), which is larger than most synaptic htt puncta, was also labeled by anti–synapsin-1. Bars, 10 µm.
Age-dependent progression of neurological symptoms in SNAP25-150Q transgenic mice
Consistent with the age-dependent accumulation of mutant htt in synapses, transgenic SNAP25-150Q mice show progressive neurological phenotypes similar to other HD transgenic mice, such as reduced body weight, clasping, and hunchback appearance (Fig. 5, A and B). These symptoms occurred in old mice in an age-dependent manner and importantly, did not occur in SNAP25-20Q mice that express a higher level of SNAP25-20Q than SNAP25-150Q, indicating that polyQ expansion, rather than tagging htt with SNAP25, is responsible for the neurological symptoms. Strikingly, transgenic SNAP25-150Q mice die earlier than WT and SNAP25-20Q transgenic mice (Fig. 5 C). The 150Q-2 line died before 6 mo of age, whereas the 150Q-8 line could survive for a longer time, so we were able to perform a time course study to examine the behavior phenotypes of 150Q-8 mice. Rotarod performance assessment revealed that the 150Q-8 line displayed motor function deficits when they reached 11 mo old (Fig. 5 D). Consistent with their earlier death, 150Q-2 mice had more severe defects in rotarod performance than 150Q-8 mice at 4 mo of age (Fig. 5 E). A motor reflex response can also be measured by acoustic startle, which involves an intense loud noise stimulus to elicit an animal’s responses. 150Q-8 mice show decreased acoustic startle responses (Fig. 5 F), pointing to a sensorimotor deficit in the central nervous system. The light–dark box test, which assesses anxiety by quantifying general locomotor activity and a willingness to explore, revealed a significant reduction in the time 150Q-8 mice stayed in the light box with a decreased cross time (Fig. 5 G), suggesting that these mice may have increased anxiety. The beam-walking assay measures the time for the mouse to traverse a beam and the number of paw slips that occur in the process. Although the time for 150Q-8 mice to traverse the beam was not significantly different from controls (WT and SNAP25-20Q mice), the number of slips as 150Q-8 mice traverse the beam is significantly higher than controls (Fig. 5 H). Together, our findings show that SNAP25-150Q mice recapitulate the neurological symptoms of other established HD transgenic mice.
Figure 5.
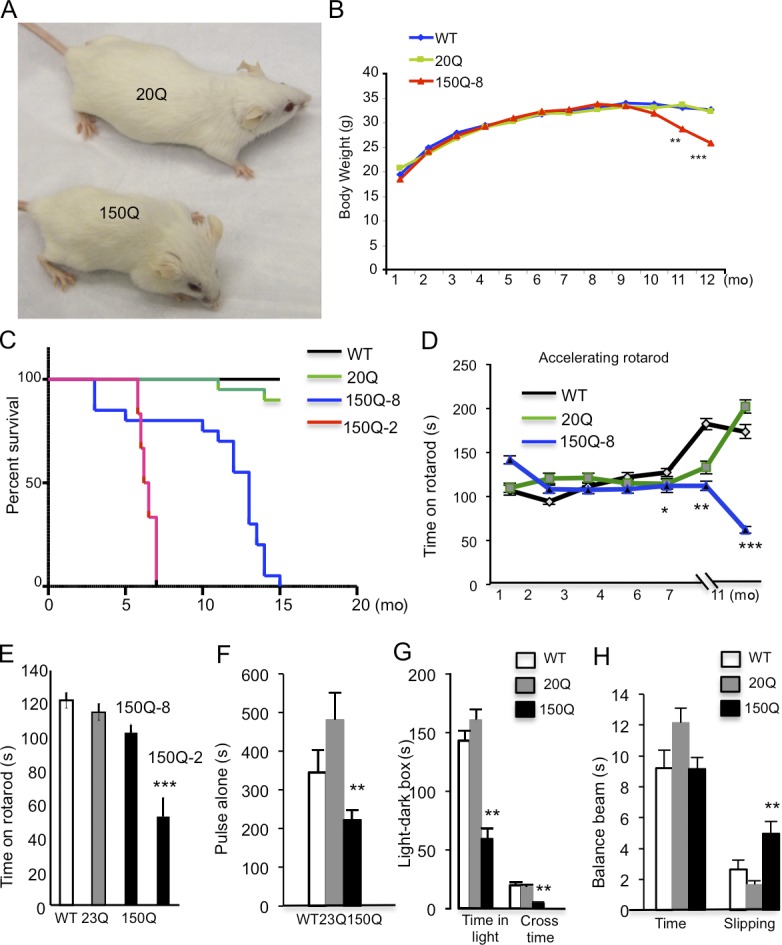
SNAP25-150Q mice show age-dependent neurological symptoms. (A) Appearance of old SNAP25-150Q mouse (150Q-8 line) and control littermate at the age of 13 mo. (B) Age-dependent decrease in the body weight of the 150Q-8 line (n = 20 each group). (C) Shorter lifespan of 150Q-8 and 150Q-2 lines compared with wild-type (WT) and SNAP25-20Q (20Q) mice (n = 20 each group). (D) Rotarod performance test showing an age-dependent decrease in motor function of SNAP25-150Q mice (line-8; n = 20 in each group). (E) Comparison of rotarod performance of transgenic mice shows that the 150Q-2 line has a more severe motor deficit than the 150Q-8 line. Mice at the age of 4 mo were examined (n = 20 each group). (F) Startle test of mice at the age of 8 mo (n = 17 each group). (G) Light–dark box test of mice at the age of 8 mo (n = 20 each group). (H) Balance beam test of time to cross the beam and slipping numbers in mice at the age of 7 mo (n = 20 in each group). In F–H, SNAP25-150Q mice (line-8) were compared with WT and SNAP25-20Q mice (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars are means ± SEM.
Synaptic mutant htt causes neuropathology and inhibits neurotransmitter release
Reactive gliosis is a nonspecific reactive change of glial cells in response to neuronal injury (Pekny and Nilsson, 2005). Using an antibody against glial fibrillary acidic protein (GFAP), an astrocytic marker protein, we found there was an obvious increase in GFAP staining in SNAP25-150Q transgenic mouse brain regions, including the cortex, striatum, and cerebellum (Fig. 6 A). Western blotting and quantitation of the ratio of GFAP to GAPDH also verified the increased astrocytic gliosis in the SNAP25-150Q mouse striatum versus WT and SNAP25-20Q transgenic mouse brains (Fig. 6 B). These results suggest that mutant htt in presynaptic terminals can mediate neuronal injury.
Figure 6.

Neuropathology in SNAP25-150Q transgenic mouse brain. (A) GFAP immunocytochemical staining shows an increase in the density of GFAP-positive glial cells in the striatum, white matter (corpus callosum), and cerebellum of SNAP25-150Q mice (line-8) at 1 yr of age compared with wild-type (WT) and SNAP25-20Q mice. Bar, 100 µm. (B) Western blotting also shows increased GFAP immunoblotting signal in the striatal tissue from the SNAP25-150Q (150Q) mouse. The ratios of GFAP to GAPDH signals on the blot were obtained from three experiments. ***, P < 0.001. (C) Paired-pulse facilitation (PPF) was examined at an interpulse interval from 20 to 500 ms. The amplitude of PPF induced by <100-ms intervals was significantly lower in SNAP25-150Q slices compared with WT (*, P < 0.05; **, P < 0.01) and SNAP25-20Q (**, P < 0.01 compared with 150Q; n = 8). (D) LTP of fEPSPs was induced (arrow) in the sensorimotor cortex. The LTP magnitude was markedly suppressed in SNAP25-150Q mice compared with WT mice (**, P < 0.01 compared with 150Q). n = 6 in each group. (E) Reduced [3H]glutamate release in the corticostriatal brain slices of SNAP25-150Q mice. No significant change was seen in [3H]GABA release between transgenic and WT mouse brain slices (n = 3 mice each group; **, P < 0.01; ***, P < 0.001). Error bars are means ± SEM.
Electrophysiological experiments of corticostriatal slices from HD YAC128 mice have revealed an age-dependent decrease of glutamate release (Joshi et al., 2009). Given the presynaptic localization of transgenic mutant htt in SNAP25-150Q mice, it is important to examine whether mutant htt affects synaptic neurotransmitter release. Using an electrophysiological approach similar to the one described in our recent study (Mohamad et al., 2013), we evaluated the strength of synaptic transmission by measuring the field excitatory postsynaptic potentials (fEPSPs) in the sensorimotor cortex of the mouse brain slices. The amplitude of paired-pulse facilitation (PPF) induced by <100-ms intervals was significantly lower in SNAP25-150Q slices (Fig. 6 C). The long-term potentiation (LTP) of fEPSPs was also significantly lower in SNAP25-150Q slices compared with WT (Fig. 6 D). These results suggest that the presynaptic function, especially the capacity for glutamate release, is impaired in SNAP25-150Q mice. In the brain slices from SNAP25-20Q, a slight reduction in PPF was also seen as compared with WT slices. Given that SNAP25-20Q is expressed at a higher level than SNAP25-150Q in the mouse brain (Fig. 1 C), expression of the higher level of SNAP25-20Q in the presynaptic terminals could cause these slight reductions. However, SNAP25-150Q caused a more marked reduction of presynaptic transmitter release than SNAP25-20Q, indicating that the expanded glutamine repeat in SNAP25-150Q has specific toxicity.
To verify that the reduction in glutamate release occurs in the striatum, we isolated the mouse brain striatal slices and performed glutamate release assays using [3H]glutamate. This in vitro assay also allowed us to compare with [3H]GABA release in the striatum. We found that glutamate release was selectively reduced in the SNAP25-150Q slices (Fig. 6 E), and [3H]GABA release did not appear to be significantly impacted, suggesting that vesicular release of glutamate is more vulnerable to synaptic mutant htt.
Mutant htt binds C-terminal synapsin-1
Because it is selectively localized in the presynaptic terminals, transgenic mutant htt may bind synaptic vesicles to impair synaptic transmission. To test this hypothesis, we used synaptosomes isolated from WT mouse cortex and then incubated them with the lysates of transfected PC12 cells, which stably expressed exon1 htt containing either normal (20Q) or expanded polyQ (150Q) repeats (Li et al., 1999). We found that more mutant htt (150Q) than normal htt (20Q) binds synaptosomes (Fig. 7 A). To explore how mutant htt binds synaptosomes, we isolated synaptosomes from SNAP25-150Q mouse brains and then immunoprecipitated synaptic proteins with an antibody to htt (mEM48). The precipitated proteins were subjected to mass spectrometry (MS), which identified 578 proteins and 1,732 peptides. This experiment revealed that synapsin-1, a presynaptic vesicle protein that plays an important role in neurotransmitter release from vesicles (Fornasiero et al., 2010; Shupliakov et al., 2011), is a good candidate to associate with mutant htt, as it was increased in SNAP25-150Q immunoprecipitates (Fig. 7 B).
Figure 7.

Mutant htt associates with synapsin-1. (A) Synaptosomes isolated from WT mouse brain cortex were incubated with lysates of PC12 cells expressing exon1 htt containing 20Q or 150Q. The precipitated synaptosomes were then analyzed via Western blotting. More 150Q bound to the synaptosomes. The blot was also probed with antibodies to synapsin-1 and synaptophysin. (B) The presynaptic fractions (P1) of WT, SNAP25-20Q, or SNAP25-150Q mice were immunoprecipitated by anti-htt (mEM48). Immunoprecipitates were subjected to MS analysis. Synapsin-1 was found to be more abundant in 150Q immunoprecipitates. (C) The interaction of synapsin-1 with mutant htt was verified by mEM48 immunoprecipitation of presynaptic fraction of SNAP25-150Q mouse brain cortex. Synapsin-1 was precipitated with mutant htt (150Q). The ratios of precipitated synapsin-1 to input are presented as the relative precipitated amounts (n = 3). (D) The presynaptic fractions of wild-type (WT) and full-length htt CAG150 knockin (KI) mice were precipitated with antibody mEM48 or mouse IgG. Synapsin-1 was also precipitated with mutant htt. (E) Reduced [3H]glutamate and [3H]GABA release was seen in the corticostriatal slices of HD KI mice compared with WT mice. *, P < 0.05; **, P < 0.01. Error bars are means ± SEM. IP, immunoprecipitation.
To verify that mutant htt associates with synapsin-1 in vivo, we performed htt immunoprecipitation using brain tissues from two HD mouse models. First, we used our HD model that expresses mutant htt in synapses and found that more synapsin-1 was coprecipitated with SNAP25-150Q (Fig. 7 C). To investigate whether synapsin-1 also associates with mutant htt in HD CAG150 KI mouse brains, we performed mEM48 immunoprecipitation of striatal lysates of these mice. Western blotting of the immunoprecipitates also revealed the association of synapsin-1 with mutant htt (Fig. 7 D). We then used brain slices from HD KI mouse brains to measure [3H]glutamate and [3H]GABA release. The results showed that glutamate release was diminished in HD KI mouse brain slices (Fig. 7 E); however, unlike the transgenic SNAP25-150Q mouse brain, HD KI mouse brain slices also showed a reduction in [3H]GABA, suggesting that this reduction is protein context dependent and can be caused by full-length mutant htt or its degraded products.
Synapsin-1 consists of two isoforms, 1A and 1B, which contain several phosphorylation sites and a proline-rich domain at their C-terminal region (Fig. 8 A). Because the proline-rich domain can serve as a binding region for vesicle trafficking (Ren and Hurley, 2011), we generated a truncated synapsin-1 (tSynapsin) by deleting the C-terminal proline-rich region and expressed it with exon1-20Q or exon1-150Q htt in transfected HEK293 cells. Immunoprecipitation of htt revealed that, although both synapsin-1A and -1B could associate with htt, depletion of the proline domain eliminated the association of synapsin-1 with htt (Fig. 8 B).
Figure 8.

Mutant htt binds to the proline-rich domain of synapsin-1. (A) The structures of synapsin-1A, synapsin-1B, and truncated synapsin-1 (tSynapsin). (B) Synapsin-1A (1A), synapsin-1B (1B), and tSynapsin were cotransfected with exon1 htt containing 20Q (20Q) or 150Q (150Q) in HEK293 cells. Cell lysates were immunoprecipitated by anti-htt (mEM48). Note that tSynapsin lacking the proline-rich domain is unable to interact with htt and that more 1A and 1B were precipitated with 150Q than 20Q. C, nontransfected cells. (C) HEK293 cells were transfected with synapsin-1B and GFP or GFP fusion proteins containing N17 (N17-GFP), exon1 htt with 97Q (97Q-GFP), or exon1 mutant htt lacking N17 (DN17-97Q-GFP). Synapsin-1 immunoprecipitates were detected by anti-GFP (top) and anti–synapsin-1 (bottom). Note that DN17-97Q-GFP and 97Q-GFP, but not GFP and N17-GFP, were coprecipitated with synapsin-1B. (D) In PC12 cells stably expressing exon1 htt containing 20Q or 150Q, more 150Q was immunoprecipitated with endogenous synapsin-1. (E) [3H-dopamine release in htt stably transfected PC12 cells. The cells were stimulated by 25 mM KCl. The released [3H]dopamine is expressed as the percentage of total cellular [3H]dopamine (n = 5 each group). **, P < 0.01 compared with wild-type (WT) PC12 cells. Error bars are means ± SEM. IP, immunoprecipitation.
The first 17 amino acids (N17) in htt have been found to modulate the subcellular localization of htt via targeting to various proteins (Gu et al., 2009a; Kim et al., 2009; Thompson et al., 2009). To examine whether N17 is involved in the association of mutant htt with synapsin-1, GFP fusion proteins containing N17 peptides (N17-GFP), exon1 htt with 97Q (97Q-GFP), or exon1 mutant htt lacking N17 (DN17-97Q-GFP) were coexpressed with synapsin-1B in HEK293 cells. GFP immunoprecipitation assay showed that synapsin-1 was selectively coprecipitated with 97Q-GFP and DN17-97Q-GFP but not GFP and N17-GFP (Fig. 8 C), suggesting that the site in htt for binding synapsin-1 exists in the exon1 htt but not N17.
PC12 cells provide a cellular model to measure vesicular dopamine release that is dependent on synapsin-1 phosphorylation (Schweitzer et al., 1995; Steiner et al., 1996; Iwata et al., 1997). We then used htt stably transfected PC12 cells to investigate whether mutant htt can affect this release. Mutant htt also bound synapsin-1 in PC12 cells in synapsin-1 immunoprecipitation (Fig. 8 D). We then measured [3H]dopamine release from these PC12 cells and found that mutant htt indeed inhibited dopamine release (Fig. 8 E).
Synaptic mutant htt inhibits synapsin-1 phosphorylation
Phosphorylation of synapsin-1 critically regulates the function of synapsin-1, and S9 phosphorylation in the N-terminal region of synapsin-1 is important for synaptic vesicle neurotransmitter release (Fornasiero et al., 2010; Shupliakov et al., 2011). Using HEK293 cells expressing GFP fusion proteins containing either N17 (N17-GFP) or exon1 mutant htt with (97Q-GFP) or without N17 (DN17-97Q-GFP), we found that GFP-97Q and DN17-97Q-GFP could also reduce S9 phosphorylation (Fig. 9 A), consistent with their interactions with synapsin-1 (Fig. 8 C). By expressing truncated synapsin-1 lacking its C-terminal region in HEK293 cells, we then tested whether mutant htt binds the C-terminal region of synapsin-1 to affect synapsin-1 phosphorylation. Comparing the phosphorylation of N-terminal synapsin-1 at serine 9 (S9) of full-length synapsin-1B and tSynapsin, we found that although S9 phosphorylation in full-length synapsin-1 is inhibited by htt-150Q, the same phosphorylation in tSynapsin is not affected by htt-150Q as compared with htt-20Q (Fig. 9 B). Thus, the inhibition of synapsin-1 phosphorylation by mutant htt depends on its interaction with C-terminal region of synapsin-1.
Figure 9.

Mutant htt reduces synapsin-1 phosphorylation via its binding to synapsin-1. (A) Western blot analysis of transfected cells showing that GFP fusion proteins containing exon1 htt (97Q-GFP and DN17-97Q-GFP) reduce the phosphorylation of synapsin-1. The ratios of phosphorylated synapsin-1 at S9 to total synapsin-1 are shown on the right (**, P < 0.01 compared with GFP; n = 3). (B) In HEK293 cells transfected with truncated synapsin-1 (tSynapsin) that lacks the C-terminal region for binding htt, mutant exon1 htt (150Q) does not affect the N-terminal S9 phosphorylation of synapsin-1 compared with cells transfected with full-length synapsin-1B. Exon1 htt containing 20Q also served as a control. **, P < 0.01. (C) Antibodies to phosphorylated synapsin-1 at serine 9 (S9), 549 (S549), and 603 (S603) were used to examine the influence of mutant htt on endogenous synapsin-1 phosphorylation in htt stably transfected PC12 cells. Representative Western blots are presented. (D) The brain cortex tissues of wild-type (WT), SNAP25-20Q, and SNAP25-150Q mice were examined via Western blotting and antibodies to phosphorylated synapsin-1. (E) Quantitation of the relative levels of phosphorylated synapsin-1 via Western blotting. Western blotting was performed in three experiments, and the ratios of phosphorylated syanpsin-1 (S9, S549, and S603) were obtained. *, P < 0.05; **, P < 0.01 compared with the control 20Q. (F) The proposed mechanism by which mutant htt in synapses affects synapsin-1 function. N-terminal mutant htt accumulates in synapses in aged neurons and interacts with the proline-rich domain of synapsin-1, reducing the phosphorylation of synapsin-1 and leading to defective release of neurotransmitters from synaptic vesicles. P, phosphorylation sites; D, proline-rich domain. Error bars are means ± SEM.
To investigate whether mutant htt can affect the phosphorylation of endogenous synapsin-1, we performed Western blotting with antibodies to different phosphorylated synapsin-1 and found a reduction in S9, S549, and S603 phosphorylation in synapsin-1 in PC12 cells expressing exon1-150Q htt (Fig. 9 C). Examination of SNAP25-150Q transgenic mouse brain tissues also revealed that these phosphorylations were reduced in the HD mouse brains (Fig. 9 D left). Quantifying the ratios of phosphorylated synapsin-1 to the total level of synapsin-1 on the same Western blots verified the specific effect of transgenic mutant htt on synapsin-1 phosphorylation (Fig. 9 E, right). Because the binding of htt to synapsin-1 is dependent on the C-terminal proline-rich domain, we propose that in HD neuronal cells, mutant htt accumulates in synapses and binds the proline-rich domain of synapsin-1 to reduce its phosphorylation, leading to impaired neurotransmitter release and contributing to neurological symptoms (Fig. 9 F).
Discussion
Mutant htt is distributed widely in various subcellular regions, and its proteolytic products form aggregates in the nucleus, neuronal processes, and nerve terminals, resulting in multiple pathological pathways. For example, mutant htt in the nucleus can affect the transcription of a number of genes, whereas mutant htt in the cytoplasm can cause intracellular trafficking defects (Ross and Tabrizi, 2011). Although synaptic dysfunction in HD mouse models has been well documented and is also an early pathological change in HD (Smith et al., 2005; Milnerwood and Raymond, 2010), the contribution of synaptic mutant htt to HD pathology and neurological symptoms remains unknown.
In our SNAP25-htt transgenic mouse model, via immunofluorescent staining, synaptic fractionation, and EM, we found that mutant htt is targeted to the presynaptic terminals in transgenic mice. Importantly, transgenic mutant htt is not overexpressed in our transgenic mice and forms aggregates without the SNAP25 epitope. First, transgenic SNAP25-htt expression is at a much lower level than endogenous SNAP25, which is evident from Western blots probed with anti-SNAP25. Second, the level of transgenic mutant htt is comparable to htt fragments in HD 150Q KI mice that express mutant htt at the endogenous level. Third, the expression level of mutant htt is lower than transgenic normal htt containing 20Q, as revealed by antibodies (EM48 and 1C2) that can preferentially react with polyQ-expanded htt but still labeled less mutant htt than control 20Q htt.
Although quantitative PCR and immunocytochemistry consistently show the lower expression level of transgenic SNAP25-150Q in our mouse model than mutant htt in R6/2 mice, SNAP25-150Q is more enriched in synapses and could produce more impact to affect synaptic function. Indeed, synaptic mutant htt causes age-dependent neurological symptoms in SNAP25-150Q mice. Such symptoms are seen in independent mouse lines and did not occur in SNAP25-20Q mice that express a higher level of transgenic htt. Thus, the phenotypes seen are caused by polyQ expansion-related toxicity, rather than the expression of SNAP25 fusion proteins or tagging htt with SNAP25. However, because mutant htt is fused to SNAP25, this fusion may modify htt toxicity in synapses by altering the half-life, posttranslational modifications, and interactions of exon1 htt with other proteins. Although our experiments cannot rule out these possibilities, several findings in our experiments support the idea that neuronal toxicity is primarily caused by cleaved htt fragments in our transgenic mice. First, there is the correlation of accumulation of mutant htt fragments in synapses with progressive neurological phenotypes. Consistently, the accumulation of cleaved htt fragments leads to the formation of synaptic aggregates that are not labeled by anti-SNAP25 in immunocytochemistry. Also, Western blotting results show that anti-SNAP25 is unable to detect htt fragments. Second, using transfected cells, we proved that exon1 mutant htt interacts with synapsin-1 and affects neurotransmitter release. Third, in HD CAG150 KI mice, we also identified cleaved htt fragments and the interactions of mutant htt with synapsin-1. It is likely that cleaved N-terminal htt fragments with expanded polyQ repeats become stable in aged neurons and accumulate in different subcellular regions, such as the nucleus and synapses, to cause age-dependent neurological phenotypes.
To our surprise, even though mutant htt is targeted to the presynaptic terminals in the mouse brain, it can cause more severe neurological phenotypes, including early death, than full-length mutant htt in YAC128 (Slow et al., 2003), bacterial artificial chromosome HD (Gray et al., 2008), and HD KI (Menalled et al., 2002) mice. These HD mice expressing full-length mutant htt apparently live longer than SNAP25-150Q mice. Thus, the neurological phenotypes in our HD mouse model underscore the synaptic toxicity of N-terminal mutant htt fragments.
Whether full-length mutant htt or N-terminal mutant htt plays an important role in synaptic dysfunction remains to be fully investigated and merits discussion here. Many studies have used full-length and N-terminal transgenic mice to identify synaptic dysfunction (Suopanki et al., 2006; Cummings et al., 2010). Because full-length mutant htt is constantly processed via proteolysis to generate N-terminal htt fragments (Zhou et al., 2003; Landles et al., 2010), studies of full-length mutant htt transgenic mice are limited for distinguishing the effects caused by full-length or N-terminal mutant htt. Although HD mice, such as R6/2 (Davies et al., 1997) and N171-82Q mice (Schilling et al., 1999), which express N-terminal mutant htt, develop more robust and progressive phenotypes than our transgenic mice and full-length htt transgenic mice, N-terminal mutant htt in R6/2 and N171-82Q mice is distributed in both the nucleus and neuronal processes and can cause both nuclear and synaptic defects. Nevertheless, the neuropathology of R6/2 mice that express exon1 htt is recapitulated by HD KI mice at the age of 22 mo (Woodman et al., 2007), suggesting that the age-dependent accumulation of small N-terminal htt fragments leads to HD pathology. Furthermore, aberrant splicing caused by a large CAG repeat in the mutant htt gene has been recently found to generate exon1 htt products in the HD mouse and patient brains (Sathasivam et al., 2013), underscoring the importance in investigating the in vivo toxicity of exon1 mutant htt. Targeting N-terminal mutant htt to the synapse is expected to reveal selective pathological events caused by synaptic mutant htt. In support of this idea, we found that synaptic mutant htt can impair presynaptic neurotransmitter release and interacts with synapsin-1. Such defects were also verified in HD KI mouse brains, in which proteolysis of full-length mutant htt can generate N-terminal mutant htt fragments.
Although presynaptic htt aggregates are present in our new transgenic mouse brains, the role of these aggregates in synaptic dysfunction remains to be investigated. However, these aggregates result from the accumulation of N-terminal mutant htt. Our experiments provide evidence for the toxicity of soluble mutant htt in the synapse because soluble mutant htt can coprecipitate synapsin-1. Mutant htt is also found to interact with other synaptic proteins (Borrell-Pagès et al., 2006; Suopanki et al., 2006; Truant et al., 2006; Shirasaki et al., 2012). Using synaptic fraction and htt immunoprecipitation, we discovered that exon1 mutant htt binds synapsin-1, identifying synapsin-1 as a new target of N-terminal mutant htt. We also expressed exon1 htt in transfected cells and found that htt binds the proline-rich domain of synapsin-1 and reduces the phosphorylation of syanspsin-1. Other studies that used MS analysis also suggested that synapsin-1 may associate with htt (Kaltenbach et al., 2007; Shirasaki et al., 2012).
In R6/2 mice, phosphorylation of site-6 (S549) in synapsin-1 was also found to be decreased, which is consistent with our observation in SNAP25-150Q mice. However, site-1 (S9) phosphorylation was unchanged, and site-3 (S603) phosphorylation was increased in R6/2 mouse brain (Liévens et al., 2002). The authors suggested that an imbalance between kinase and phosphatase activities accounts for the abnormal synapsin-1 phosphorylation in R6/2 mice. This imbalance is likely caused by the effects of mutant htt on gene transcription in R6/2 mice in which mutant htt is abundantly accumulated in the nucleus. In fact, altered expression of various kinases and phosphatases has been reported in R6/2 mice (Bibb et al., 2000; Deckel et al., 2001; Giralt et al., 2011; Fusco et al., 2012). In our HD mouse model, however, mutant htt is selectively accumulated in the presynaptic terminals and can have more direct effect on synaptic proteins. Thus, studies of this new HD mouse model may provide new mechanistic insight into synaptic dysfunction in HD. We propose that binding of mutant htt to syanpsin-1 may alter the conformation of synapsin-1, thereby impairing its phosphorylation and function in releasing neurotransmitters from synaptic vesicles.
Given the fact that synaptic dysfunction is an early pathological event, identification of synapsin-1 as a target opens up a new avenue for reversing synaptic dysfunction. This is because syanpsin-1 phosphorylation is affected by mutant htt and can be altered by drug treatment, and improving synaptic function may more effectively delay or prevent the progressive development of HD neurological symptoms.
Materials and methods
Antibodies
Rabbit polyclonal antibody (EM48) and mouse monoclonal antibodies (mEM48) against the N-terminal region (amino acids 1–256) of human htt (Li et al., 2002) were used in this study. Other mouse antibodies were GFAP (astrocyte-specific marker; EMD Millipore), PSD95 (Thermo Fisher Scientific), synaptophysin (Sigma-Aldrich), CaMkII-β (Thermo Fisher Scientific), Complexin-1, SNAP25 (BD), antibodies against polyQ (1C2; EMD Millipore), γ-tubulin (Sigma-Aldrich), and GAPDH (EMD Millipore). Rabbit antibodies were synapsin-1 (Cell Signaling Technology), phosphorylated synapsin-1 (S9 [Cell Signaling Technology]; S549 and S603 [Thermo Fisher Scientific]), syntaxin-7 (Sigma-Aldrich), GABA-A (EMD Millipore), and EAAT-2 (Cell Signaling Technology), and the goat antibody is synapsin-1 (Santa Cruz Biotechnology, Inc.).
Plasmid construction and mutagenesis
Exon1 htt plasmids encoding either normal (20Q) or expanded polyQ (150Q) repeats (htt-20Q or htt-150Q) in pRK5 vector were generated in our previous studies (Li et al., 2002; Wang et al., 2008). Exon1 htt plasmids (htt-GFP and DN17-htt-GFP) encoding 97Q with or without the first 17 amino acids constructed in pcDNA 3.1 vector (Invitrogen) were provided by W. Yang at the University of California, Los Angeles (Los Angeles, CA). Full-length synapsin-1A and synapsin-1B were generated by PCR from mouse cDNA. Primers used were as follows: forward, 5′-ACTCTAGACCACCATGAACTACCTGC-3′, and reverse, 5′-AGTGTCGACTCAGTCGGAGAAGAGGCTG-3‘. The mutant 1,447-bp synapsin-1 fragment without the C-terminal polyproline domain was engineered by PCR, with full-length synapsin-1A as the template. Primers used were as follows: forward, 5′-ACTCTAGACCACCATGAACTACCTGC-3′, and reverse, 5′-GTTAGTCGACTCAACCTGCAGGCTGCTG-3‘. The PCR products were digested with restriction enzyme Xba1 and Sal1 and linked to the pRK5 vector.
Generation of mice and genotyping
To generate SNAP25-htt transgenic mice, we fused exon1 htt containing normal (20Q) or 150Q to the C terminus of SNAP25 in a vector that expresses transgene under the control of the mouse prion promoter. Microinjection of linearized construct was conducted by the Emory University Transgenic Mouse Core Facility. Founder mice (FVB strain) were obtained and maintained in the animal facility at Emory University under specific pathogen-free conditions in accordance with institutional guidelines of The Animal Care and Use Committee at Emory University. Genomic DNA was isolated from mouse tails, and PCR genotyping was used for screening transgenic mice. Primers with sequences flanking the polyQ repeat were used for PCR. The sequences of the forward and reverse primers were as follows: forward, 5′-ATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3′, and reverse, 5′-AAACTCACGGTCGGTGCAGCGGCTCCTCAG-3′. We obtained a total of 16 founders for SNAP25-150Q (150Q) transgenic mice and eight founders for SNAP25-20Q (20Q) transgenic mice. Three 150Q mouse lines (line-2, line-8, and line-15) could be bred to F1 generation. Unlike some SNAP25-150Q founder mice that do not survive for more than 5 mo, all SNAP25-20Q founders lived normally as WT mice.
Western blotting, subcellular fractionation, and coimmunoprecipitation
For Western blots, cultured cells or brain tissues were homogenized in radioimmunoprecipitation assay buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, pH 8.0, 1 mM EGTA, pH 8.0, 0.1% SDS, 0.5% deoxycholate, and 1% Triton X-100) with protease inhibitor (P8340; Sigma-Aldrich). The cell or tissue lysates were diluted in SDS sample buffer (62.6 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, and 0.01% bromophenol blue) and sonicated for 10 s after incubation at 100°C for 5 min. The total lysates were resolved in a Tris-glycine gel (Invitrogen) and blotted to a nitrocellulose membrane. The Western blots were developed using the ECL Prime kit (GE Healthcare).
Subcellular fractionation to isolate the presynaptic fraction was performed using the method similar to that previously described (Gu et al., 2009b). The frontal cortex tissues were homogenized in ice-cold lysis buffer (10 ml/g of 15 mm Tris, pH 7.6, 0.25 m sucrose, 1 mm phenylmethylsulfonyl fluoride, 2 mm EDTA, 1 mm EGTA, 10 mm Na3VO4, 25 mm NaF, 10 mm sodium pyrophosphate, and protease inhibitors). The homogenates served as total lysis. After centrifuging the homogenates at 800 g for 5 min, the supernatant was subjected to 10,000 g centrifugation for additional 10 min to separate supernatant and pellet. This supernatant was again centrifuged at 180,000 g for 40 min to obtain the cytosolic fraction. The pellet was homogenized in lysis buffer including 1% Triton X-100 and 300 mM NaCl and centrifuged at 16,000 g for 30 min to obtain its supernatant or P1 fraction, which contains synaptosomal proteins, and the pellet or P2 fraction, which consists of membrane-associated proteins from synaptosomes.
For immunoprecipitation, protein samples were preclarified by incubation with a 50-µl volume of protein A–agarose beads (Sigma-Aldrich), and the supernatant was then incubated with antibody overnight followed by adding 30 µl protein A–agarose beads for 1 h. The immunocomplexes were washed three times in ice-cold lysis buffer and then eluted by boiling in SDS sample buffer for Western blotting.
qRT-PCR
For reverse transcription reaction, total RNA was isolated from mouse cortex and striatum tissues with the RNeasy Lipid Tissue Mini kit (QIAGEN). Reverse transcription reactions were performed with 1 µg of total RNA using the SuperScript First-Strand Synthesis System kit (Invitrogen). We performed qRT-PCR to determine transgenic htt mRNA levels in different mouse models using primers that can amplify human and mouse htt (HD1S forward, 5′-ATGGCGACCCTGGAAAAGCT-3′, and HD40A reverse, 5′-TGCTGCTGGAAGGACTTGAG-3′). The mouse actin transcript served as an internal control and was amplified with primers 452S (forward), 5′-TGAGACCTTCAACACCCCAG-3′, and Actin 669A (reverse), 5′-GTGGTGGTGAAGCTGTAGCC-3′. SYBR Select Master Mix kit (Applied Biosystems by Life Technologies) was used for qRT-PCR, which consisted of 3 µl of diluted cDNA reaction mixture, 10 µl of SYBR Select Master Mix, and 100 nM of each primer in a final volume of 20 µl. The qRT-PCR was performed with a thermal cycler (Realplex Mastercycler; Eppendorf). Relative levels of transgenic human htt were obtained by comparing with the mouse actin transcript expression levels, with the control WT mouse htt value set as 1.
Immunofluorescence, immunohistochemistry, and EM
Mice were anaesthetized with 5% chloral hydrate and perfused with 10 ml of 0.9% NaCl through the left cardiac ventricle. Mouse brains were then isolated and cut into sections with a cryostat (Leica) at −20°C. Fresh mouse brain sections were fixed in PBS/4% PFA and then examined by immunofluorescent staining at room temperature with mouse mEM48 and rabbit SNAP25 antibodies. Secondary antibodies (1:500 dilution) conjugated with DyLight 594 (Jackson ImmunoResearch Laboratories, Inc.) or Alexa Fluor 488 (Invitrogen) were used. For immunochemistry with DAB staining, anaesthetized mice were perfused with 4% PFA in 0.1 M phosphate buffer (PB), pH 7.4, through the left cardiac ventricle. Perfused brains were removed and postfixed overnight in 4% PFA at 4°C. Fixed brains were transferred into 30% sucrose at 4°C for 2 d and then sectioned to slices. Light micrographs were taken using a microscope (Axiovert 200 MOT; Carl Zeiss) and a 63× lens (LD-Achroplan 63×/0.75 NA) with a digital camera (ORCA-100; Hamamatsu Photonics). The Openlab software (PerkinElmer) was used for imaging acquisition. Images were stored in a computer and processed using Photoshop CS5 (Adobe) for proper brightness/contrast adjustments. For EM, the mice were perfused with 0.1 M sodium PB, pH 7.3, containing 4% PFA and 0.2% glutaraldehyde. The mouse brain was then postfixed in this perfusion buffer for 1 h and sectioned into 50 µm using a vibratome (CM1850; Leica). The ultrathin sections were processed for electron microscopic examination, as described in our previous publication (Li et al., 2003). In brief, all sections were osmicated in 1% OsO4 in PB and embedded in Eponate12 (Ted Pella). The cortex sections were cut into ultrathin sections (60 nm) with an ultramicrotome (Ultracut S; Leica). The ultrathin sections were examined using an electron microscope (H-7500; Hitachi).
Excitatory fEPSP recording and LTP induction
Mice were anaesthetized with isoflurane (3%) and decapitated, and the brains were removed into ice-cold artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 6.0 mM MgCl2, 26 mM NaHCO3, 2.0 mM CaCl2, and 10 mM glucose. ACSF was saturated with 95% O2 and 5% CO2, at pH 7.4. The brain was cut on an ice-cold base plate into forebrain coronal slices of 400 µm thick using a vibratome (Vibratome 1000 Plus Sectioning System; The Vibratome Company). Slices were collected with a soft brush and incubated at room temperature (23–24°C) in ACSF for 60–90 min before recording.
Coronal brain slices were transferred to a recording chamber (RC-22C; Warner Instruments) on a microscope (CX31; Olympus) and superfused with normal ACSF at the rate of 3 ml /min at 23–24°C. The fEPSPs from the sensorimotor cortex in the same column were used to evaluate the strength of synaptic transmission. A glass microelectrode filled with ACSF containing 5 mM bicuculline methiodide with tip resistance of 3–4 MΩ was placed in the layer 2/3 cortex, and a 0.1-MΩ tungsten monopolar stimulate electrode was placed in layer 4. The stimulation output (Master-8; A.M.P.I.) was controlled by the trigger function of an amplifier (EPC-9; HEKA). fEPSPs were recorded in a current-clamp mode of EPC-9. Data were filtered at 3 kHz and digitized at sampling rates of 20 kHz using the data acquisition software PULSE (HEKA). PPF was examined by applying pairs of pulses, which were separated by 20–500-ms different intervals. To induce tetanus LTP of fEPSP, the stimulus intensity was set to evoke 40% of the maximum fEPSP initial slope, and the stimulus train of 100 pulses at 100 Hz was applied three times at 30-s intervals. The magnitude of LTP was expressed as the mean percentage of baseline fEPSP initial slope.
Neurotransmitter release assay
In vitro [3H]glutamate and [3H]GABA release from brain slices were measured using established assays (Maneuf and McKnight, 2001; Patel et al., 2001). Coronal corticostriatal slices (six slices of 300 mm thick) prepared from each mouse were perfused with 37°C oxygenated (95% O2 and 5% CO2) Krebs–Ringer buffer (mM: 11.5 glucose, 25 NaHCO3, 1.2 MgCl2, 1.2 NaH2PO4, 118 NaCl, 4.8 KCl, 2.5 CaCl2, and 0.004 Na2EDTA, pH 7.4). Slices were incubated with 200 nM/liter [3H]glutamate (PerkinElmer) or 100 nM/liter [3H]GABA (PerkinElmer) for 30 min. After extensive rinsing, slices were treated with 25 mM KCl with 1 mm/liter dihydrokainic acid (Sigma-Aldrich) for 5 min to evoke the release of [3H]glutamate or treated with 25 mM KCl with 200 µm/liter nipecotic acid (Sigma-Aldrich) for 5 min to evoke the release of [3H]GABA. Fractions were collected every 5 min after adding 25 mM KCl. The net efflux of tritium in the fraction right before (basal value) and after KCl depolarization was measured in triplicate using a scintillation counter (LS6500; Beckman Coulter). For detecting dopamine release in PC12 cells, [3H]dopamine was incubated with cultured PC12 cells, and [3H]dopamine release was trigged by 25 mM KCl. The results were expressed as the percentage increase in [3H]glutamate or [3H]GABA release above the basal level using the following equation: [(K + −evoked basal release)/basal release] × 100%. Data for each group were obtained from three mice.
Behavioral studies
Rotarod testing was performed with a Rotamex 4/8 (Columbus Instruments International). Mice were trained for 10 min each day for 3 d before examination. After the training, mice were tested for additional consecutive 3 d, three trials per day, and allowed to rest for 20 min between trials. The accelerating rotarod test used the rod speed that was set at 5 rpm and increased by 0.1 rpm/s.
Chambers (SR-LAB; San Diego Instruments) were used to measure startle reactivity in mice. In each chamber, there was a platform inside a ventilated box. A clear, nonrestrictive Plexiglas cylinder was on the platform. In the chamber, a continuous background noise of 68 dB and the 120-dB startle pulse were produced by a high-frequency loud speaker, which would cause mice to move in the Plexiglas cylinder. The vibrations associated with mouse body movement were then recorded by a piezoelectric unit attached to the platform. The no-stimulus trial consisted of 68-dB background noise, which was presented for 5 min as acclimation period and continued throughout the test session. For the pulse-alone trial, 40-ms and 120-dB pulse of the broadband noise was used. During the test session, there were six presentations of the 120-dB pulse-alone trial and a mean of 15 s (range, 12–30 s) between trials. Each trial type was presented in a pseudorandom order.
The light–dark test used the apparatus (length 45 cm, width 24 cm, and height 21 cm) consisting of two equal acrylic compartments, one dark and one light, which were separated by a divider with a 5 × 7–cm opening at floor level. The light area was illuminated by a 60-W bulb lamp. A mouse was placed in the center of the light area and allowed to explore the novel environment for 5 min. During the next 5 min, the numbers of transfers between two compartments and the time staying in the light area were recorded.
On the balance beam, mice were trained for 2 d to walk the entire length of a 0.6-cm (∼1 inch)-wide × 80-cm-long wooden beam that was suspended 50 cm above the floor. On each trial the mouse was released onto the end of the beam and required to run down the entire beam and into the dark box. Each mouse was tested, and each session was the mean of three trials. The time it took to cross to the end and the number of slips were recorded.
MS analysis
The mouse presynaptic fraction (P1) was immunoprecipitated with anti-htt (mMEM48). The immunoprecipitates were analyzed by reverse phase liquid chromatography–MS at the Emory core facility. All analyses were performed on an ion trap mass spectrometer (LTQ Orbitrap; Thermo Fisher Scientific). Acquired MS/MS spectra were searched against the mouse reference database of the National Center for Biotechnology Information and identified 578 proteins and 1,732 peptides in our MS analysis.
Statistical analysis
Unless stated otherwise, values in the figures and text are presented as means ± SEM. Statistical analysis used Excel (Microsoft) using Student’s t test (two tailed) for comparing two different groups. For behavioral data analysis, we used one-way analysis of variance, which was followed by Tukey’s honestly significant difference post hoc test. A value of P < 0.05 was considered to be statistically significant.
Online supplemental material
Fig. S1 shows the expression of transgenic htt in transgenic mice. Fig. S2 shows that EM48 selectively labels mutant htt in the brain cortex of SNAP25-150Q, but not WT or SNAP25-20Q, mice. Fig. S3 shows Western blot analysis of synaptic fractions of mouse brains. Fig. S4 shows double staining of SNAP25-150Q mouse cortex with anti-htt (EM48) and anti-SNAP25. Fig. S5 shows colocalization of transgenic mutant htt with synpasin-1 in synapses in the cortex of the SNAP25-150Q mouse at 6 mo of age. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201303146/DC1.
Supplementary Material
Acknowledgments
We thank Duc M. Duong and Nicholas T. Seyfried at ENNCF (Emory Neuroscience National Institute of Neurological Disorders and Stroke [NINDS] Core Facility) proteomics core at Emory University for MS analysis, Heju Zhang at the Transgenic Mouse/Gene Targeting Core Facility at Emory University for generating transgenic mice, Dr. William Yang for providing the plasmid for expressing N-terminal htt, and Cheryl Strauss for critical reading of this manuscript.
The work was supported by National Institutes of Health grants (AG19206 and NS041449 to X.-J. Li, AG031153 and NS0405016 to S. Li, and NS057255 to S.P. Yu), and Q.Q. Xu was supported in part by the graduate program at Tongji Medical College, Huazhong University of Science and Technology. This research project was supported in part by the Proteomics Core of the Emory Neuroscience NINDS Core Facilities grant (P30NS055077) and the State Key Laboratory of Molecular Developmental Biology, China.
Footnotes
Abbreviations used in this paper:
- ACSF
- artificial cerebrospinal fluid
- fEPSP
- field excitatory postsynaptic potential
- GFAP
- glial fibrillary acidic protein
- HD
- Huntington’s disease
- htt
- huntingtin
- KI
- knockin
- LTP
- long-term potentiation
- MS
- mass spectrometry
- PB
- phosphate buffer
- PPF
- paired-pulse facilitation
- qRT-PCR
- quantitative real-time PCR
- SNAP25
- synaptosomal-associated protein 25
- WT
- wild type
References
- Bibb J.A., Yan Z., Svenningsson P., Snyder G.L., Pieribone V.A., Horiuchi A., Nairn A.C., Messer A., Greengard P. 2000. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. USA. 97:6809–6814 10.1073/pnas.120166397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell-Pagès M., Zala D., Humbert S., Saudou F. 2006. Huntington’s disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 63:2642–2660 10.1007/s00018-006-6242-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings D.M., Cepeda C., Levine M.S. 2010. Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington’s disease. ASN Neuro. 2:147–156 10.1042/AN20100007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. 1997. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90:537–548 10.1016/S0092-8674(00)80513-9 [DOI] [PubMed] [Google Scholar]
- Deckel A.W., Gordinier A., Nuttal D., Tang V., Kuwada C., Freitas R., Gary K.A. 2001. Reduced activity and protein expression of NOS in R6/2 HD transgenic mice: effects of L-NAME on symptom progression. Brain Res. 919:70–81 10.1016/S0006-8993(01)03000-1 [DOI] [PubMed] [Google Scholar]
- DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. 1997. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 277:1990–1993 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- Fornasiero E.F., Bonanomi D., Benfenati F., Valtorta F. 2010. The role of synapsins in neuronal development. Cell. Mol. Life Sci. 67:1383–1396 10.1007/s00018-009-0227-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco F.R., Anzilotti S., Giampà C., Dato C., Laurenti D., Leuti A., Colucci D’Amato L., Perrone L., Bernardi G., Melone M.A. 2012. Changes in the expression of extracellular regulated kinase (ERK 1/2) in the R6/2 mouse model of Huntington’s disease after phosphodiesterase IV inhibition. Neurobiol. Dis. 46:225–233 10.1016/j.nbd.2012.01.011 [DOI] [PubMed] [Google Scholar]
- Garden G.A., La Spada A.R. 2012. Intercellular (mis)communication in neurodegenerative disease. Neuron. 73:886–901 10.1016/j.neuron.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A., Saavedra A., Carretón O., Xifró X., Alberch J., Pérez-Navarro E. 2011. Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington’s disease. Hum. Mol. Genet. 20:4232–4247 10.1093/hmg/ddr351 [DOI] [PubMed] [Google Scholar]
- Gray M., Shirasaki D.I., Cepeda C., André V.M., Wilburn B., Lu X.H., Tao J., Yamazaki I., Li S.H., Sun Y.E., et al. 2008. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 28:6182–6195 10.1523/JNEUROSCI.0857-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X., Greiner E.R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J.S., Thompson L.M., Wetzel R., Yang X.W. 2009a. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 64:828–840 10.1016/j.neuron.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Liu W., Yan Z. 2009b. beta-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 284:10639–10649 10.1074/jbc.M806508200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst C.A., Li S.H., Yi H., Mulroy J.S., Kuemmerle S., Jones R., Rye D., Ferrante R.J., Hersch S.M., Li X.J. 1999. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 19:2522–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng M.Y., Detloff P.J., Albin R.L. 2008. Rodent genetic models of Huntington disease. Neurobiol. Dis. 32:1–9 10.1016/j.nbd.2008.06.005 [DOI] [PubMed] [Google Scholar]
- Iwata S.I., Hewlett G.H., Ferrell S.T., Kantor L., Gnegy M.E. 1997. Enhanced dopamine release and phosphorylation of synapsin I and neuromodulin in striatal synaptosomes after repeated amphetamine. J. Pharmacol. Exp. Ther. 283:1445–1452 [PubMed] [Google Scholar]
- Joshi P.R., Wu N.P., André V.M., Cummings D.M., Cepeda C., Joyce J.A., Carroll J.B., Leavitt B.R., Hayden M.R., Levine M.S., Bamford N.S. 2009. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 29:2414–2427 10.1523/JNEUROSCI.5687-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach L.S., Romero E., Becklin R.R., Chettier R., Bell R., Phansalkar A., Strand A., Torcassi C., Savage J., Hurlburt A., et al. 2007. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 3:e82 10.1371/journal.pgen.0030082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.W., Chelliah Y., Kim S.W., Otwinowski Z., Bezprozvanny I. 2009. Secondary structure of Huntingtin amino-terminal region. Structure. 17:1205–1212 10.1016/j.str.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landles C., Sathasivam K., Weiss A., Woodman B., Moffitt H., Finkbeiner S., Sun B., Gafni J., Ellerby L.M., Trottier Y., et al. 2010. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 285:8808–8823 10.1074/jbc.M109.075028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Wyman T., Yu Z.X., Li S.H., Li X.J. 2003. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum. Mol. Genet. 12:2021–2030 10.1093/hmg/ddg218 [DOI] [PubMed] [Google Scholar]
- Li S., Li X.J. 2006. Multiple pathways contribute to the pathogenesis of Huntington disease. Mol. Neurodegener. 1:19 10.1186/1750-1326-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.H., Cheng A.L., Li H., Li X.J. 1999. Cellular defects and altered gene expression in PC12 cells stably expressing mutant huntingtin. J. Neurosci. 19:5159–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.H., Cheng A.L., Zhou H., Lam S., Rao M., Li H., Li X.J. 2002. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 22:1277–1287 10.1128/MCB.22.5.1277-1287.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.J., Li S. 2011. Proteasomal dysfunction in aging and Huntington disease. Neurobiol. Dis. 43:4–8 10.1016/j.nbd.2010.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévens J.C., Woodman B., Mahal A., Bates G.P. 2002. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington’s disease transgenic mice. Mol. Cell. Neurosci. 20:638–648 10.1006/mcne.2002.1152 [DOI] [PubMed] [Google Scholar]
- Maat-Schieman M., Roos R., Losekoot M., Dorsman J., Welling-Graafland C., Hegeman-Kleinn I., Broeyer F., Breuning M., van Duinen S. 2007. Neuronal intranuclear and neuropil inclusions for pathological assessment of Huntington’s disease. Brain Pathol. 17:31–37 10.1111/j.1750-3639.2006.00040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneuf Y.P., McKnight A.T. 2001. Calcitonin gene-related peptide-mediated increase in K(+)-induced [(3)H]-dopamine release from rat caudal striatal slices. Neurosci. Lett. 310:73–76 10.1016/S0304-3940(01)02056-0 [DOI] [PubMed] [Google Scholar]
- Menalled L.B., Chesselet M.F. 2002. Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 23:32–39 10.1016/S0165-6147(00)01884-8 [DOI] [PubMed] [Google Scholar]
- Menalled L.B., Sison J.D., Wu Y., Olivieri M., Li X.J., Li H., Zeitlin S., Chesselet M.F. 2002. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J. Neurosci. 22:8266–8276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood A.J., Raymond L.A. 2010. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 33:513–523 10.1016/j.tins.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Mohamad O., Song M., Wei L., Yu S.P. 2013. Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J. Physiol. 591:149–168 10.1113/jphysiol.2012.239251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr H.T., Zoghbi H.Y. 2007. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30:575–621 10.1146/annurev.neuro.29.051605.113042 [DOI] [PubMed] [Google Scholar]
- Patel D.R., Young A.M., Croucher M.J. 2001. Presynaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor-mediated stimulation of glutamate and GABA release in the rat striatum in vivo: a dual-label microdialysis study. Neuroscience. 102:101–111 10.1016/S0306-4522(00)00463-2 [DOI] [PubMed] [Google Scholar]
- Pekny M., Nilsson M. 2005. Astrocyte activation and reactive gliosis. Glia. 50:427–434 10.1002/glia.20207 [DOI] [PubMed] [Google Scholar]
- Ren X., Hurley J.H. 2011. Proline-rich regions and motifs in trafficking: from ESCRT interaction to viral exploitation. Traffic. 12:1282–1290 10.1111/j.1600-0854.2011.01208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J., Südhof T.C. 2002. Snares and Munc18 in synaptic vesicle fusion. Nat. Rev. Neurosci. 3:641–653 [DOI] [PubMed] [Google Scholar]
- Ross C.A., Tabrizi S.J. 2011. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10:83–98 10.1016/S1474-4422(10)70245-3 [DOI] [PubMed] [Google Scholar]
- Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D., et al. 2013. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA. 110:2366–2370 10.1073/pnas.1221891110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. 1999. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8:397–407 10.1093/hmg/8.3.397 [DOI] [PubMed] [Google Scholar]
- Schweitzer E.S., Sanderson M.J., Wasterlain C.G. 1995. Inhibition of regulated catecholamine secretion from PC12 cells by the Ca2+/calmodulin kinase II inhibitor KN-62. J. Cell Sci. 108:2619–2628 [DOI] [PubMed] [Google Scholar]
- Shirasaki D.I., Greiner E.R., Al-Ramahi I., Gray M., Boontheung P., Geschwind D.H., Botas J., Coppola G., Horvath S., Loo J.A., Yang X.W. 2012. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 75:41–57 10.1016/j.neuron.2012.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O., Haucke V., Pechstein A. 2011. How synapsin I may cluster synaptic vesicles. Semin. Cell Dev. Biol. 22:393–399 10.1016/j.semcdb.2011.07.006 [DOI] [PubMed] [Google Scholar]
- Slow E.J., van Raamsdonk J., Rogers D., Coleman S.H., Graham R.K., Deng Y., Oh R., Bissada N., Hossain S.M., Yang Y.Z., et al. 2003. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12:1555–1567 10.1093/hmg/ddg169 [DOI] [PubMed] [Google Scholar]
- Smith R., Brundin P., Li J.Y. 2005. Synaptic dysfunction in Huntington’s disease: a new perspective. Cell. Mol. Life Sci. 62:1901–1912 10.1007/s00018-005-5084-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J.P., Dawson T.M., Fotuhi M., Snyder S.H. 1996. Immunophilin regulation of neurotransmitter release. Mol. Med. 2:325–333 [PMC free article] [PubMed] [Google Scholar]
- Suopanki J., Götz C., Lutsch G., Schiller J., Harjes P., Herrmann A., Wanker E.E. 2006. Interaction of huntingtin fragments with brain membranes—clues to early dysfunction in Huntington’s disease. J. Neurochem. 96:870–884 10.1111/j.1471-4159.2005.03620.x [DOI] [PubMed] [Google Scholar]
- Tebbenkamp A.T., Swing D., Tessarollo L., Borchelt D.R. 2011. Premature death and neurologic abnormalities in transgenic mice expressing a mutant huntingtin exon-2 fragment. Hum. Mol. Genet. 20:1633–1642 10.1093/hmg/ddr040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L.M., Aiken C.T., Kaltenbach L.S., Agrawal N., Illes K., Khoshnan A., Martinez-Vincente M., Arrasate M., O’Rourke J.G., Khashwji H., et al. 2009. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187:1083–1099 10.1083/jcb.200909067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R., Atwal R., Burtnik A. 2006. Hypothesis: Huntingtin may function in membrane association and vesicular trafficking. Biochem. Cell Biol. 84:912–917 10.1139/o06-181 [DOI] [PubMed] [Google Scholar]
- Vonsattel J.P., DiFiglia M. 1998. Huntington disease. J. Neuropathol. Exp. Neurol. 57:369–384 10.1097/00005072-199805000-00001 [DOI] [PubMed] [Google Scholar]
- Wang C.E., Zhou H., McGuire J.R., Cerullo V., Lee B., Li S.H., Li X.J. 2008. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J. Cell Biol. 181:803–816 10.1083/jcb.200710158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart T.M., Parson S.H., Gillingwater T.H. 2006. Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 65:733–739 10.1097/01.jnen.0000228202.35163.c4 [DOI] [PubMed] [Google Scholar]
- Woodman B., Butler R., Landles C., Lupton M.K., Tse J., Hockly E., Moffitt H., Sathasivam K., Bates G.P. 2007. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 72:83–97 10.1016/j.brainresbull.2006.11.004 [DOI] [PubMed] [Google Scholar]
- Zhou H., Cao F., Wang Z., Yu Z.X., Nguyen H.P., Evans J., Li S.H., Li X.J. 2003. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 163:109–118 10.1083/jcb.200306038 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.