

Abstract
Periventricular heterotopias is a malformation of cortical development, characterized by ectopic neuronal nodules around ventricle lining and caused by an initial migration defect during early brain development. Human mutations in the Filamin A (FLNA) and ADP-ribosylation factor guanine exchange factor 2 [ARFGEF2; encoding brefeldin-A-inhibited guanine exchange factor-2 (BIG2)] genes give rise to this disorder. Previously, we have reported that Big2 inhibition impairs neuronal migration and binds to FlnA, and its loss promotes FlnA phosphorylation. FlnA phosphorylation dictates FlnA–actin binding affinity and consequently alters focal adhesion size and number to effect neuronal migration. Here we show that FlnA loss similarly impairs migration, reciprocally enhances Big2 expression, but also alters Big2 subcellular localization in both null and conditional FlnA mice. FlnA phosphorylation promotes relocalization of Big2 from the Golgi toward the lipid ruffles, thereby activating Big2-dependent Arf1 at the cell membrane. Loss of FlnA phosphorylation or Big2 function impairs Arf1-dependent vesicle trafficking at the periphery, and Arf1 is required for maintenance of cell–cell junction connectivity and focal adhesion assembly. Loss of Arf1 activity disrupts neuronal migration and cell adhesion. Collectively, these studies demonstrate a potential mechanism whereby coordinated interactions between actin (through FlnA) and vesicle trafficking (through Big2-Arf) direct the assembly and disassembly of membrane protein complexes required for neuronal migration and neuroependymal integrity.
Introduction
Periventricular heterotopia (PH) is a congenital malformation of cortical development, characterized by reduced brain size and ectopic neuronal nodules along the ventricular neuroepithelium (Lu and Sheen, 2005). The disorder results from a defect in the initiation of neuronal migration, following disruption of the neuroependyma and impaired neuronal motility.
Human mutations in Filamin A (FLNA) and ADP-ribosylation factor guanine exchange factor 2 (ARFGEF2) genes cause PH (Fox et al., 1998; Sheen et al., 2004). The actin binding FlnA protein forms homodimers that regulate the cytoskeleton through interactions via multiple proteins, thereby promoting actin changes at the cell periphery to maintain cell shape. FlnA regulation of actin may directly affect migration and maintenance of the neuroependyma (Carreno et al., 2004; Smythe and Ayscough, 2006). ARFGEF2 encodes for brefeldin-A-inhibited guanine exchange factor-2 (BIG2; Sheen et al., 2004). BIG2 is a protein kinase A-anchoring protein, which regulates GDP–GTP conversion of the ARFs and directs vesicle trafficking.
Both human and mice genes implicated in PH formation involve FlnA overexpression or actin dynamics as a final common pathway. The mitogen-activated protein kinase kinase4 (Mekk4) interacts with FlnA, and loss of function leads to PH formation and increased FlnA expression (Sarkisian et al., 2006). Loss of Big2 also causes an increase in FlnA and phosphoFlnA-serine 2152 levels (Zhang et al., 2012). Overexpression of both FlnA and phosphoFlnA impairs migration in vivo. Cdc42 RhoGTPase loss of function causes PH, FlnA binds the Rho GTPases, and the Rho GTPases affect actin dynamics (Ohta et al., 1999; Cappello et al., 2006). Dysfunction in stem cell factor-c-kit, Spred1 (a negative regulation of Ras-MAPK-ERK) and αSnap [a protein involved in soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-mediated vesicle fusion] all lead to PH in mice, although their effects on FlnA and Big2 are not known (Clary et al., 1990; Ferland et al., 2009; Soumiya et al., 2009; Phoenix and Temple, 2010).
A fundamental question remains as to whether FlnA and its regulation of actin dynamics alone are the primary endpoints leading to PH formation. Several observations suggest that this may not be the case, as most genes associated with PH can be linked to vesicle trafficking. Big2 regulates Arf function and vesicle coat formation, αSNAP mediates SNARE-dependent vesicle fusion, and Spred1 localizes within vesicles. The RhoGTPases indirectly target actin polymerization to discrete sites on the plasma membrane and regulate vesicle movement. Finally, FlnA itself associates dynamically with Golgi membranes during trafficking of transport vesicles.
To explore the interplay between actin and vesicle trafficking in contributing to the PH phenotype, we have focused on Big2 and FlnA interactions. Here, we show that FlnA loss inhibits cell-intrinsic neuronal migration, increases Big2 expression, and impairs redistribution of Big2 from the Golgi to the membrane periphery. Loss of FlnA phosphorylation blocks Big2-dependent Arf1 activation, which disrupts both neuronal migration and cell–cell interactions through changes in cell adhesion molecules at the membrane. In this context, the precise regulation between actin and vesicles at the membrane serves to maintain the stability and turnover of cell adhesion molecules responsible for appropriate migration and neuroependymal integrity.
Materials and Methods
Mice.
FlnA knock-out mice were obtained from the Comparative and Developmental Genetics Department, MRC, Human Genetics Unit (Edinburgh, UK). This strain carries a T-to-A base-point mutation in the FlnA gene, leading to a stop codon in exon 46 and complete absence of FlnA expression (Hart et al., 2006). The previously reported mice were early embryonic lethal, thereby preventing analyses of cortical migration. FlnA-null mice were bred onto an s129/sv strain to prolong survival into late embryonic ages (Lian et al., 2012). All animal studies were conducted under guidelines set by the Institutional Animal Care and Use Committees (IACUCs) of Harvard Medical School and Beth Israel Deaconess Medical Center in accordance with The National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
To generate FlnA conditional knock-out (CKO) mice, FlnA loxp+/+ was inserted into introns 2 and 7 of the mouse FlnA gene (Feng et al., 2006) and crossbred with conditional Cre-ER mice (B6.129-Gt (ROSA)26Sortm1(cre/ERT2)Tyj/, The Jackson Laboratory). Inducible Cre strains contain a transgene that expresses a modified form of Cre recombinase, which is nonfunctional until tamoxifen induction. Tamoxifen (50 mg/kg, Sigma) mixed with progesterone (10 mg/kg, Sigma) was injected intraperitoneally at embryonic day 11.5 (E11.5) for 3 continuous days, and the phenotype and protein level were checked at E17.5. The recombination of genomic loxp with Cre enables temporal control of floxed gene expression in vivo.
Plasmid reagents.
All FlnA and Arfgef2 DNA plasmids have been previously described (Zhang et al., 2012). The FlnA short hairpin RNA interference (shRNAi; AAGGTGCTGCCTACACATGAT) was located between bases 4612 and 4632 of FlnA mRNA CDS sequence (NM_010227). The forward 5′ GGTGCTGCCTACACATGATTTCAAGAGAATCATGTGTAGGCAGCACCTTTTTT3′ and reverse 5′AATTAAAAAAGGTGCTGCCTACACATGATTCTCTTGAAATCATGTGTAGGCAGCACCGGCC-3′ oligos were synthesized (Operon company), annealed, and ligated between restriction site ApaI and EcoRI of pSilencer 1.0-mU6 Vector carrying the EGFP gene. The knock-down efficiency was verified by Western blotting (data not shown). GST-GGA3-GAT and Arf1 (HA-Arf1, HA-Arf1T31N, HA-Arf1Q71L) constructs were provided by J. Bonifacino (NIH). GFP-tagged Arf1 and dominant-negative constructs were amplified by PCR from the above DNA plasmids and inserted into pCAG-GFP vector (Addgene).
Cell culture and transfection.
HEK293, Neuro2A, polarized Madin-Darby canine kidney (MDCK) cells, and CHP100 cells were cultured in DMEM (Invitrogen) with 10% FBS and 1% penicillin-streptomycin. M2 and A7 cells were cultured in DMEM supplemented with 2% FBS, 8% newborn calf serum, and 1% penicillin-streptomycin. The G418 (final concentration, 500 μg/ml) was added to A7 cell culture to maintain FLNA expression. Neuronal progenitors were isolated from the E13.5–E14.5 mouse embryonic frontal cortex, digested, and filtrated. The dissociated single progenitors were stained for trypan blue to ensure cell viability before quantification, and were resuspended in knock-out DMEM/F12 neural progenitor culture medium (Invitrogen) with EGF and FGF supplements as previously described (Ferland et al., 2009; Zhang et al., 2012). Mouse embryonic fibroblast (MEF) cells were isolated from E14.5 embryos and cultured in DMEM with 10% FBS and 1% penicillin-streptomycin. Transfectin (Bio-Rad) and Lipofectamine 2000 (Invitrogen) were used for transfection following the manufacturer's protocols. Cells were allowed to grow in growth medium for 48 h and then were serum starved overnight (15∼24 h) for the next experiments.
Immunohistochemistry and immunocytochemistry.
Timed embryos were surgically dissected according to IACUC guidelines, fixed in 4% paraformaldehyde (PFA), and paraffin embedded or frozen sectioned, as previously described (Ferland et al., 2009; Zhang et al., 2012). Wild-type (WT) and FlnA-null neural progenitors (from E13.5 embryos) were plated on 10 μg/ml laminin-coated coverslips and cultured in neural stem cell medium (Invitrogen) at 37°C for 5 h. All cultured cells on fibronectin or laminin-coated coverslips were fixed in 4% PFA. Briefly, after blocking with 10% goat serum, the antibodies were incubated overnight against bromodeoxyuridine (BrdU; catalog #ab6326, rat anti-BrdU 1:100, Abcam), nestin (catalog #Rat-401, mouse monoclonal antibody 1:50, Developmental Studies Hybridoma Bank), RC2 (catalog #RC2 mouse anti-RC2 1:25, Developmental Studies Hybridoma Bank), zona occludens-1 (Zo-1; catalog #R26.4C, rat anti-Zo-1, 1:25, Developmental Studies Hybridoma Bank), doublecortin (DCX; catalog #sc-8066, mouse monoclonal antibody 1:100, Santa Cruz Biotechnology), FlnA (catalog #sc-58764, mouse anti-flnA 1:100, Santa Cruz Biotechnology), HA (catalog #MMS-101P, mouse antibody 1:100, Covance), Tuj1 (catalog #MMS-435P, monoclonal mouse antibody, 1:250, Covance), GFAP (catalog #600-101-215, goat antibody, 1:200, Rockland), β-catenin (catalog #MAb 519001921, 1:200, BD Transduction Laboratories), phosphoFlnA ser2152 (mouse antibody, 1:100; a gift from F. Nakamura, Brigham and Women's Hospital, Harvard Medical School, Boston, MA), paxillin (catalog #610051, mouse anti-paxillin, 1:250, BD Transduction Laboratories), FlnA (catalog #2242-1, rabbit monoclonal antibody, 1:100, Epitomics), and Big2 (rabbit polyclonal antibody, 1:50, Sigma; Lu et al., 2006; Zhang et al., 2012) for immunohistochemistry and immunocytochemistry. All appropriate secondary antibodies and phalloidin (Invitrogen) were incubated for 1 h at room temperature. Fluorescent microscopic images were prepared using an Olympus AX70 microscope with the SNAP microphotographic system. Confocal images were obtained using a Zeiss laser scanning microscope.
Detection of GTP-bound Arf1 in Neuro2A cells.
To detect the levels of active Arf1-GTP after incubation with clustered EphB2-Fc receptor, we followed a previously published GST-GGA3-GAT pull-down protocol (Lock and Stow, 2005; Tanaka et al., 2007) and instructions according to the active Arf1 pull-down assay kit (Thermo Fisher Scientific). Briefly, serum-starved Neuro2A cells overexpressing HA-Arf1 or dominant-negative Arf1T31N were treated with preclustered EphB2-Fc receptor (4 μg/ml) for 20 min. Lysates were prepared in GGA3 pull-down buffer [50 mm Tris, pH 7.5, 150 mm NaCl, 10 mm MgCl2, 10% glycerol, 1× protease inhibitor cocktail (Roche), and 1% Triton X-100] then incubated in GST-GGA3-GAT-bound glutathione-Sepharose beads for 1 h at 4°C. Subsequently, beads were washed three times in pull-down buffer and processed for Western blotting.
Bromodeoxyuridine injection and in utero electroporation.
Briefly, E13.5–E14.5 pregnant WT and FlnA-null dams were injected once intraperitoneally with BrdU (100 mg/kg) and were killed at different time points for immunohistochemistry. In utero electroporations were performed at E15.5 under anesthesia with ketamine/xylazine. The GFP or GFP-Arf1T31N constructs 1.0–3.0 μl mixed with 0.03% Fast Green were injected into either lateral ventricle using a beveled pipette, and the DNA was induced into neuronal cells by five pulses at 40 V with a 50 ms duration at 1 s intervals using Electro Square Porator, ECM 800 (BTX, Harvard Apparatus). The embryos were allowed to survive for another 72–96 h. The dissected embryonic brains were fixed in 4% PFA, sucrose protected, cryostat sectioned at 20 μm, and analyzed as previously described (Zhang et al., 2012).
Data analysis.
For quantifying BIG2 distribution, we divided M2 and A7 cells into four radial quadrants (designated by drawing three concentric circles extending radially from the center of the nucleus). BIG2 immunostaining intensity in each radial quadrant was measured using ImageJ software (NIH). To calculate the relative BIG2 distribution the intensity within in each quadrant was divided by the intensity of the total cell. The data are represented as the mean of 10 cells ± SD. Significance was determined using a paired Student's t test.
For BrdU and electroporated GFP-positive neuronal analysis, the brain structure was divided into the proliferating zone (PZ; ventricular and subventricular zones), intermediate zone (IZ), and cortical plate (CP) using nuclear staining by Hoechst. The percentages of positive GFP and BrdU cell somas found in the curtain area were calculated as a ratio to the total positive cell number (Zhang et al., 2012). Six to eight serial sections for each embryo from either sex were scored, and a total of four embryos for each group were used in this study. Significance was determined using a paired Student's t test.
For focal adhesion paxillin analysis, we quantified cells with similar shape and size in control and HA-Arf1T31N-transfected cells. The total cell and paxillin+ focal adhesion areas were traced using Photoshop. The total paxillin area combined from the many focal adhesion sites is reflected as a percentage of the total cell area.
Results
Loss of FlnA impairs neuronal migration in vivo
Prior studies have suggested that the early (E14) lethal null FlnA mouse did not exhibit neuronal migration defects at embryonic ages (Feng et al., 2006). However, FlnA inhibition through in utero electroporation did result in migration defects (Nagano et al., 2004), raising the concern for artifacts from the approach (in utero electroporation), inappropriate timing given the early mouse lethality, or compensatory effects due to other filamins in the FlnA-null mouse. To address these discordant observations, we crossbred the null FlnA mouse onto an s129/sv background, which allowed for viability to E18. We then tracked the cortical migration of cells, following the injection of BrdU (100 mg/kg) at E14.5. An increased percentage of BrdU-positive cells became situated in the ventricular, subventricular, and intermediate zones of the FlnA-null compared with WT after 72 h, consistent with a migration defect. In contrast, significantly higher proportions of BrdU-positive neurons were observed in the cortical plate of WT mice (Fig. 1A,B). There were no significant differences in the staining pattern of the radial glial markers nestin (Fig. 1C) and RC2 (Fig. 1D) or in the neuroependymal lining marker Zo-1 (Fig. 1E) between FlnA-null and WT mice at E15.5 and E18, thereby excluding a cell-extrinsic problem in causing the migration defect at this developmental age. Last, we examined the neural progenitor cell morphology since these cells undergo multipolar to bipolar morphological changes with the onset of migration from the ventricular zone to the cortical plate (Nishimura et al., 2012). Progenitors isolated from E13.5 FlnA WT and null embryos were placed on laminin-coated coverglasses, and cell morphology was monitored. WT progenitors spread out more rapidly 1 h after plating, while FlnA-null cells retained a round shape even after 3 h (Fig. 2A). Staining for actin stress fibers (fluorescein-labeled phalloidin) demonstrated a defect in filopodia formation in neural progenitors (Fig. 2B), and staining for cell-specific markers indicated that the cortical cells at E13.5 were largely progenitors (GFAP+, nestin+, and Tuj1−; Fig. 2C). The impairment in process outgrowth was seen in both neurons and neural progenitors, suggesting that process outgrowth defects could provide an underlying mechanism for disrupted migration.
Figure 1.
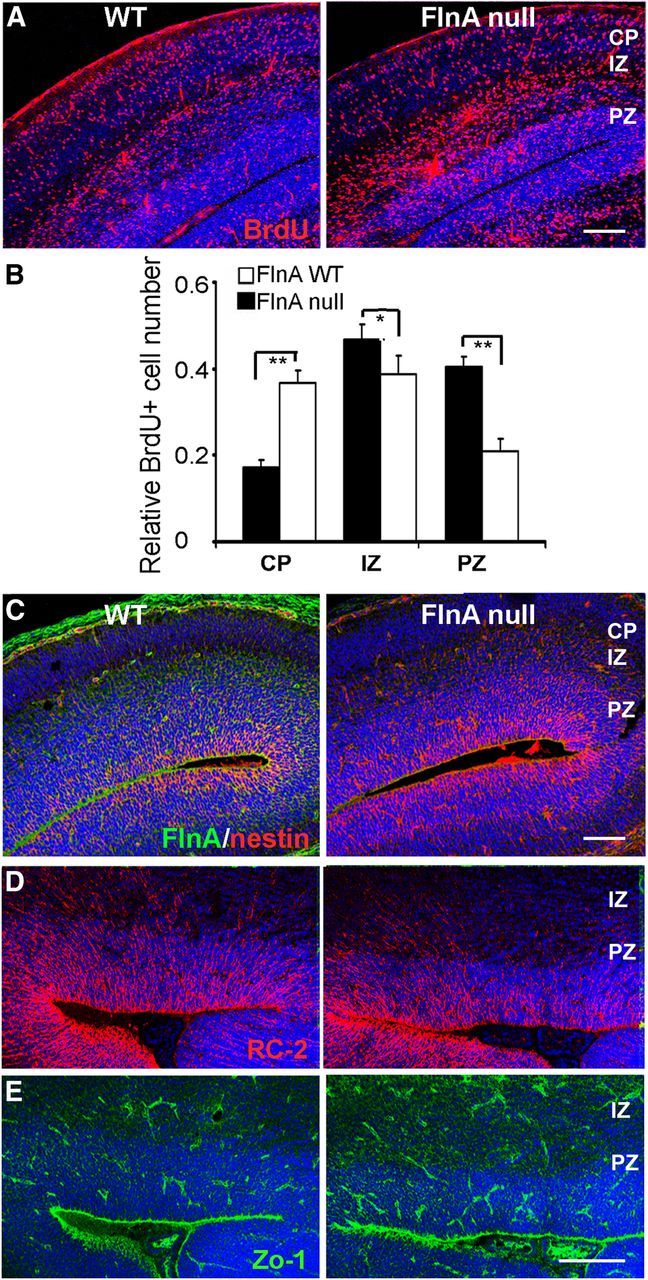
Loss of FlnA impairs neural progenitor migration. A, B, Fluorescent photomicrographs demonstrate a greater number of BrdU-positive cells residing in the ventricular and subventricular zones of the cerebral cortex of E17.5 null FlnA mice (72 h after pulsed BrdU administration) compared with WT control, indicative of an impairment in neuronal migration. Quantitative analyses of the relative number of BrdU-positive cells in the CP, IZ, and PZ for both WT and null FlnA mice are provided graphically in B (n = 3 for either sex of embryos). *p < 0.5, **p < 0.05. C–E, Fluorescent photomicrograph demonstrates no significant difference in immunostaining for radial glial markers, nestin, RC2, and Zo-1, between WT and FlnA-null brain tissues at E15.5, suggesting that the altered migration is not due to some disruption in the neuroependymal lining or radial glial scaffolding. Scale bars: A, C, top, 100 μm; D, E, 150 μm.
Figure 2.

Impairment in progenitor attachment and filopodia extension. A, Images taken under bright-field microscopy show FlnA WT neural progenitors attachment and process extension 1–3 h after plating onto laminin-coated coverslips. In contrast, null FlnA progenitors adopt a rounded morphology and more limited process extension at comparable time points (n = 3 triplicate individual experiments). B, Fluorescent photomicrographs taken under fluorescein show phalloidin-labeled actin stress fibers in both null and FlnA E13.5 neural progenitors, 1 h after plating. WT progenitors show extension of filopodia, required for neural migration, whereas progenitors lacking FlnA fail to extend appropriate projections on laminin-coated coverslips. C, E13.5 neural progenitors uniformly immunostain for the progenitor markers nestin (rhodamine) and GFAP (fluorescein), but not Tuj1, suggesting that the migratory neural population can be affected at an early stage of neuronal progenitor development. D, Summary of findings in A–C are graphically summarized from n > 3 independent experiments per variable. Scale bars: A, 100 μm; B, 20 μm; C, 40 μm.
To address whether acute loss of FlnA would similarly disrupt neuronal migration, we crossed FlnA loxp+/+ mice with a conditional Cre-ER strain, in which the Cre recombinase was activated upon tamoxifen exposure. FlnA expression was nearly undetectable in E17.5 conditional FlnA Loxp+/+/Cre+/+ mice by Western blotting after administration of tamoxifen at E11.5 for 3 d (Fig. 3A). RC2 and nestin staining did not show significant differences in radial glial and ventricular lining morphology (Fig. 3B,C). Immunostaining for DCX, a microtubule-associated protein that labels early migratory postmitotic neurons, was more widely dispersed throughout the cortical plate in the FlnA loxp+/+/Cre+/+ versus loxp+/+/Cre−/− mice (arrowheads), indicating some disruption in early neuronal migration (Fig. 3D). Similarly, BrdU labeling at E13.5 further confirmed a migration defect in the FlnA conditional mice after 48 h, with more BrdU-positive neurons found in the PZ and IZ of the loxp+/+/Cre+/+ mouse brains (Fig. 3E, arrowheads). Collectively, these studies show that loss of FlnA regardless of acute loss or gene knockout in vivo is sufficient to disrupt cell-intrinsic migration, potentially through impaired cell adhesion and/or process extension.
Figure 3.

Neuronal migration defect following acute loss of FlnA function. A, Western blot shows the near absence of FlnA protein expression in tamoxifen-treated FlnA loxp mice crossbred with tamoxifen-inducible Cre mice. Pregnant dams were administered tamoxifen at E11.5 for 3 d and killed at E17.5 (n = 3 individuals). B, C, Fluorescent photomicrographs show no significant differences by immunostaining for the radial glial markers RC2 and nestin between FlnA WT and conditional KO mice. As with the straight FlnA-null mice, there is no suggestion of disruption in the neuroependyma or radial glial scaffolding to account for any migration defect at this age. D, Fluorescent photomicrographs demonstrate more diffuse staining across the cortical width (arrowheads) for the early postmitotic neuronal marker DCX in the conditional KO vs WT mouse, consistent with a neuronal migration problem. E, Similarly, fluorescent photomicrographs demonstrate a greater proportion of BrdU-positive cells (BrdU pulse performed at E13.5) in the PZ and IZ of the conditional FlnA CKO mice (arrowheads) compared with WT embryonic brain at E15.5. F, The relative distribution of BrdU-positive cells in the WT and CKO FlnA in the CP, IZ, and PZ is graphically shown (n = 4 for either sex of embryos, six sections for each mouse were quantified; *p < 0.5, **p < 0.05). Scale bars: B, C, D, 150 μm; E, 200 μm.
FlnA regulates Big2 expression and subcellular localization
Our prior studies have shown that Big2 physically interacts with FlnA and loss of Big2 impairs neuronal migration (Zhang et al., 2012). Given the similar migratory phenotype between these two proteins, we then asked whether Big2 (202 kDa) expression was altered following loss of FlnA (280 kDa). First, Big2 protein levels were tested in FlnA-depleted M2 and repleted A7 melanoma cells (Zhang et al., 2012). We found that BIG2 expression was increased in M2 cells by Western blotting (Fig. 4A). Moreover, we detected similar increases in Big2 expression in Neuro2A cells following acute loss of FlnA by shRNAi (Fig. 4B), as well as primary FlnA-null neural progenitors (Fig. 4C). Big2 immunostaining of FlnA-null tissue showed overall increased expression within neural progenitors (Fig. 4D, asterisks), given the intensity of staining.
Figure 4.
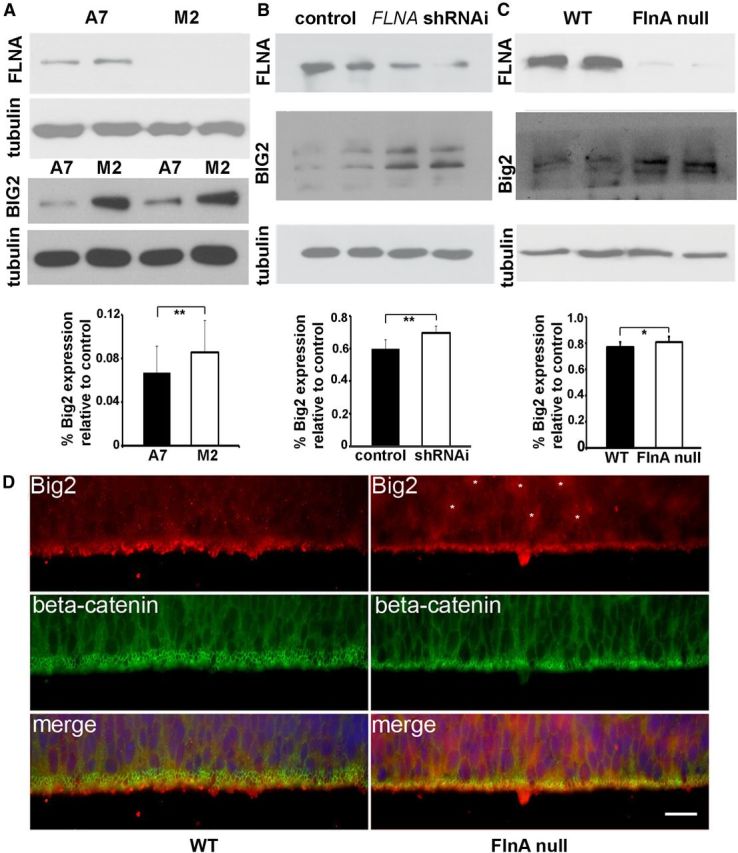
FlnA regulates Big2 expression. A, Western blot shows increased BIG2 protein levels in FLNA-deficient M2 cells compared with FLNA-replete A7 cells. B, Similarly, BIG2 levels are elevated by Western blot following FlnA knockdown by shRNAi in stably transfected Neuro2A neuroblastoma cells. C, Last, enhanced Big2 expression is found in FlnA-null progenitor cells compared with FlnA WT cells. Tubulin is used as a loading control. D, Fluorescent photomicrograph shows an overall increase in Big2 immunostaining (asterisks) in null FlnA ventricular zone. Scale bar, 20 μm.
To assess the spatial distribution of Big2 protein overexpression in the null FlnA cells, we first compared the subcellular distribution of BIG2 in serum-starved M2/A7 cells. In M2 cells, BIG2 was restricted to the perinuclear region of the cell, whereas in A7 cells BIG2 was dispersed toward the cell periphery (Fig. 5A). Quantification of BIG2 distribution revealed stronger levels of BIG2 immunostaining intensity within the peripheral cellular quadrants of A7 cells compared with M2 cells. The BIG2 perinuclear distribution pattern was also confirmed in FlnA-null progenitor cells (Fig. 5B). Previously, we had demonstrated that amino acids 441–654 for BIG2 was sufficient to physically interact with the receptor binding domain of FLNA (Zhang et al., 2012). We therefore asked whether BIG2–FLNA interactions could influence BIG2 localization. BIG2 fragments that either bind (EGFP-BIG2-NT) or do not bind (EGFP-BIG2-NT1) FLNA were transfected into HEK293 cells, and BIG2 distribution was visualized under fluorescence microscopy. Loss of the FLNA binding domain resulted in BIG2 redistribution to the perinuclear region, suggestive of localization to the Golgi apparatus. Fragments that contained the FLNA–BIG2 interacting domain, however, showed a dispersed distribution in the cytoplasm, similar to that seen with the FLNA containing A7 cells and normal neural progenitors (Fig. 5C). Finally, in the absence of FLNA binding, EGFP-BIG2-NT1 colocalized with the Golgi marker giantin, whereas the FLNA interacting EGFP-BIG2-NT2 fragment was dispersed throughout the cell and along the cell membrane in A7 cells (Fig. 5D). Overall, these studies indicate that FlnA regulates both BIG2 expression and subcellular localization.
Figure 5.

FlnA regulates Big2 subcellular localization. A, Fluorescent photomicrograph demonstrates BIG2 expression primarily resides in the cell cytoplasm in serum-starved FLNA-replete A7. In serum-starved FLNA-deficient M2 cells, BIG2 expression is generally restricted to the Golgi or perinuclear region. To quantify BIG2 distribution, M2 and A7 cell bodies are divided into four radial quadrants spanning from the nucleus to the cell periphery. The adjacent graph displays the relative BIG2 immunostaining intensity (quadrant intensity/total cellular intensity) for each quadrant in M2 and A7 cells. The relative BIG2 immunostaining intensity in A7 cells is significantly smaller than M2 cells in quadrant 1 (n = 10, *p = 0.00394) and is significantly larger than M2 cells in quadrant 3 (n = 10, **p = 0.001825), indicative of cytoplasmic versus perinuclear BIG2 localization in the presence of FLNA. B, Similarly, the fluorescent photomicrograph demonstrates Big2-dispersed localization in the cytoplasm of FlnA WT neural progenitor cells, as opposed to a perinuclear distribution in FlnA-null progenitors. C, Schematic displays of various fragments along the N-terminal region of BIG2, which either are missing BIG2-NT1 (amino acids 1–441) or contain BIG2-NT2 (amino acids 221–654) and BIG2 NT (amino acids 1–654) the FlnA binding domain (blue). Fluorescent photomicrographs show that GFP-BIG2 NT (containing the FlnA binding domain) is localized to the cytoplasm, whereas GFP-BIG2 NT1 (which lacks the FlnA binding domain) is restricted primarily to the perinuclear region in HEK293 cells. A similar spatial distribution as seen with GFP-BIG2 NT is also appreciated for GFP-BIG2 NT2. D, The expression of BIG2-NT1, but not BIG2-NT2 or BIG2-NT, is more restricted to the perinuclear Golgi (giantin immunostaining). The distribution and overlap with the Golgi is quantified graphically to the right (n = 15, *p < 0.05, **p < 0.01). Scale bars: A, 20 μm; B, D, 10 μm; C, 50 μm.
FlnA phosphorylation induces Big2 relocation to the cell membrane
Forskolin activation of transmembrane adenylyl cyclase activity increases cAMP, which phosphorylates FlnA and causes its translocation from the cytosol and Golgi to the cell membrane (Hastie et al., 1997; Sayner et al., 2011). Serine 2152 of FlnA is a PKA phosphorylation target and FlnA phosphorylation tethers signaling proteins, adhesion molecules, and the vesicle-trafficking complex to the peripheral actin rim, so as to stabilize the cortical structure and epithelial membrane (Jay et al., 2000; Sayner et al., 2011). We therefore tested whether FlnA phosphorylation dynamically redirected Big2 localization from the perinuclear region toward the membrane periphery. Forskolin treatment induced redistribution of Big2 toward the cell periphery in FLNA-replete A7 but not FLNA-absent M2 melanoma cells. Moreover, BIG2 colocalized with FLNA within the peripheral membrane ruffles in A7 cells (Fig. 6A, arrowheads). Similarly, phosphorylation of FLNA (ser2152) through overexpressing constitutively activated p-21-associated kinase 1 (PAK1; Gorlin et al., 1990) redirected BIG2 localization toward the membrane ruffles in A7 but not M2 cells, as reported previously (Fig. 6B, inset). Furthermore, Big2 redistribution to the membrane ruffles following FlnA ser2152 phosphorylation was also observed in FlnA neural progenitor cells, but not FlnA-null cells (Fig. 6C, small arrowheads). Finally, forskolin and serum can induce activation of various signaling cascades, in addition to FlnA phosphorylation. We wanted to address whether specific activation of phosphoFlnA ser2152 alone was responsible for the relocalization of Big2. EphB2-Fc signaling promoted phosphorylation of FLNA (ser2152; Fig. 6D) and produced a similar redistribution of FLNA and BIG2 to the cell periphery (Fig. 6E, small arrows). Overall, the data demonstrate that FlnA phosphorylation at ser2152 redirects Big2 subcellular localization in the Golgi and cytoplasm to the cell membrane periphery.
Figure 6.

FlnA phosphorylation directs Big2 membrane redistribution from the Golgi to the cell periphery. A, Fluorescent photomicrograph shows BIG2 localized to the membrane lamellipodial ruffles (arrowheads) after forskolin treatment in FLNA-replete A7 but not FLNA-deficient M2 cells. B, Overexpression of constitutively active PAK1 [PAK1-CA; which stimulates FLNA (ser2152) phosphorylation] induces redistribution of HA-BIG2 to the membrane ruffles in FLNA-replete A7 but not FLNA-deficient M2 cells (boxed area). C, Big2 redistribution toward the membrane ruffles (small arrowheads) is colocalized with phosphoFlnA ser2152 after forskolin-induced phosphorylation in FlnA WT progenitor cells. Higher-magnification photomicrograph is seen in bottom panel. D, Treatment of serum-starved CHP100 neuroblastoma cells with forskolin (10 μm, 10 min), serum (10% FBS, 10 min), or preclustered EphB2-Fc (4 μg/ml, 10 min) results in enhanced levels of phosphorylated FLNA (ser2152) compared with untreated serum-starved cells. E, EphB2 treatment redistributes phosphorylated FLNA (ser2152) toward the membrane ruffles with BIG2 in CHP100 cells. Scale bars: A–C, E, 10 μm.
FlnA controls Big2-dependent Arf1 activation
Changes in subcellular distribution of Big2 through FlnA activation/deactivation could significantly impact the dynamic activation of Arf1 proteins. Arfs are GTP binding proteins, associated with membranes, which function as regulators of vesicular trafficking and actin remodeling. Given that the Big2 activates Arf through its guanine exchange factor (Sec7 domain), we examined whether FlnA could regulate Big2-dependent Arf1 activation. Stimulation of serum-started Neuro2A cells with EphB2-Fc resulted in increased GTP-bound HA-Arf1 levels (Fig. 7A, top blot). Depletion of Big2 via Arfgef2 siRNA-mediated knockdown decreased GTP-bound HA-Arf1 levels in Neuro2A cells following EphB2-Fc stimulation (Fig. 7B, top blot). Furthermore, the depletion of FlnA by FlnA RNAi-mediated knockdown also led to decreased levels of GTP-bound HA-Arf1 (Fig. 7C, top blot) after EphB2-Fc treatment. Finally, we asked whether Arf1 activation levels were chronically changed with FlnA loss. In stably transfected FlnA knock-down Neuro2A cell lines. Arf1 activity was decreased in both serum-starved and serum-supplemented FlnA knock-down cells compared with control cells (Fig. 7D), and Big2 expression was upregulated with FlnA inhibition, as seen in the FlnA-null mice. Serum-induced Arf1 activation (presumably through FlnA phosphorylation) was also diminished with inhibition of FlnA (Fig. 7E). These experiments suggest that Big2 is required for the activation of Arf1 within neural cells, and FlnA modulates Big2-dependent Arf1 activation.
Figure 7.

FlnA modulates Big2-dependent Arf1 activation in response to Ephrin signaling. A, Treatment of serum-starved Neuro2A cells with preclustered EphB2-Fc (4 μg/ml, 20 min) enhances levels of GTP-bound HA-Arf1 (top blot) detected by Western blot. B, Knockdown of Big2 in serum-starved Neuro2A cells (by Arfgef2 siRNA) decreases the levels of GTP-bound HA-Arf1 (top blot) detected after treatment with preclustered EphB2-Fc (4 μg/ml, 20 min). C, FlnA acute knockdown in Neuro2A cells (by FlnA siRNA) decreases the levels of active HA-Arf1 detected after treatment with preclustered EphB2-Fc (4 μg/ml, 20 min). D, Increased Big2 expression levels, but decreased active Arf1 levels, are detected in FlnA knock-down stable Neuro2A cells line (n = 3 independent experiments for all the results). E, Statistical analysis shows that loss of FlnA leads to decreased Arf1 activity (diminished Arf1-GTP level) and FlnA phosphorylation increases Arf1 activation (n = 3, *p < 0.5, **p < 0.05).
Arf1 activation directs neuronal migration and cell adhesion
The functional state of Arf1, in general, is an important regulator of cell migration (Turner et al., 2001; Tanaka et al., 2007; Boulay et al., 2008). To investigate whether the migration defect seen in FlnA-null mice could be attributable to Arf1 activation, we inhibited Arf1 activity by targeting cortical progenitors through in utero electroporation of a GFP-tagged Arf1 dominant-negative (T31N) mutation. Injection and electroporation at E15.5 and analyses at E19 revealed that loss of Arf1 function significantly reduced the migration of cortical progenitors to the cortical plate, compared with GFP alone. The majority of the Arf1T31N-positive cells were situated around the PZ and IZ (Fig. 8A,B), implying that altered Arf1 activation in the loss of FlnA contributed to the migration defect in FlnA-null mice. At times, a disruption in the neuroependymal lining could also be appreciated in neural cells expressing the Arf1T31N construct and situated along the VZ (Fig. 8A, adjacent patent).
Figure 8.
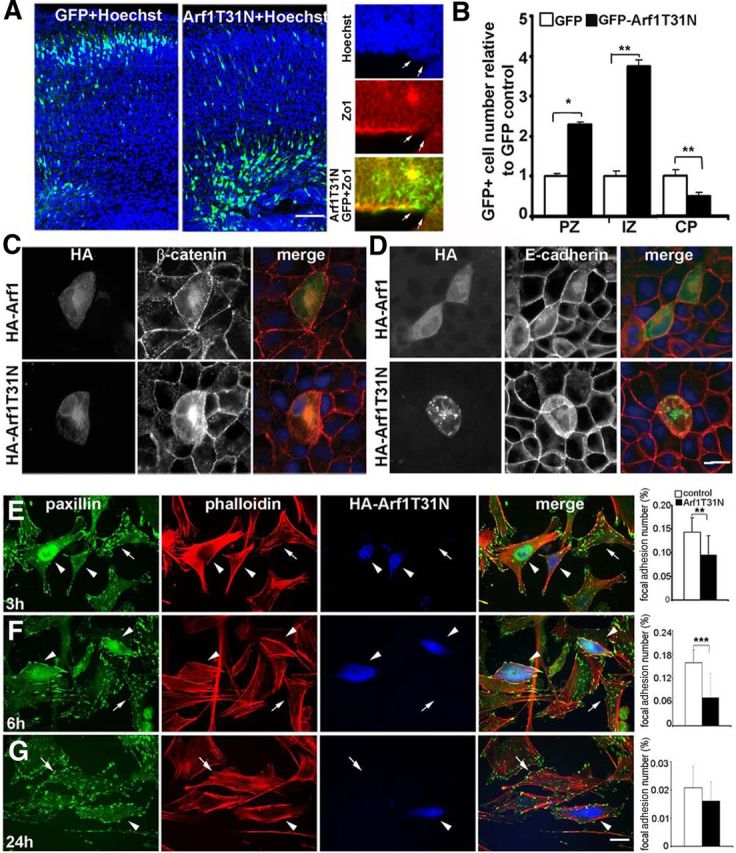
Downregulation of Arf1 activity alters cell membrane stability and impairs migration. A, Fluorescent photomicrograph of the cerebral cortices shows that electroporation of the Arf1 dominant-negative construct Arf1T31N leads to a greater number of cells situated near the ventricular lining and intermediate zones compared with GFP labeled cells, which migrate mostly to the cortical plate (right panels). At times, transfection of the Arf1T31N-GFP construct (fluorescein) led to the disruption of the neuroependymal lining (arrows) as shown through loss of Zo-1 (rhodamine) staining and protrusion of cells (Hoechst, blue) into the lateral ventricles. Electroporation of either GFP alone or GFP-tagged dominant-negative Arf1T31N were microinjected into lateral ventricles at E15.5, and mice were killed at E19 for analyses. The brain sections were stained with Hoechst to define the cortical layers (n = 4 for either sex of embryos, and 6–8 serial sections for each embryo were scored). B, The impairment in migration with loss of Arf1 activity in A is graphically summarized (*p < 0.5, **p < 0.05). C, D, In polarized MDCK cells, overexpression of the dominant-negative mutant HA-Arf1T31N causes mislocalization of β-catenin (C) and E-cadherin (D) away from the cell membrane into the cell cytoplasm. In addition, the cell membranes begin to protrude upward into the vertical axis, consistent with a disruption in the integrity of the epithelium. E, In MEF cells, overexpressing HA-Arf1T31N results in a significant decrease in the number of paxillin focal adhesions. MEF cells were transfected with HA-Arf1T31N or HA alone, incubated for 24 h, and then split and plated onto fibronectin-coated cover glasses. Cultures were fixed 3–24 h after replating, and stained for paxillin, phalloidin, and HA-Arf1T31N, as well as vinculin. No changes were seen with vinculin or control HA vector alone (data not shown). The paxillin focal adhesion number is significantly decreased compared with control by 3 h after replating (n = 15 cells and **p < 0.005). F, By 6 h after replating, MEF cells expressing HA-Arf1T31N show a more dramatic decline in the number of paxillin focal adhesions along the cell periphery (n = 15 cells and ***p < 0.001). G, By 24 h after plating, MEF cells expressing HA-Arf1T31N show a trend toward fewer paxillin-stained focal adhesions, although this was not statistically significant (n = 15 cells; arrowheads refer to HA-Arf1T31N-transfected cells, and arrows refer to control cells). Scale bars: A, 200 μm; D, E, 10 μm.
Neuronal migration and the integrity of the neuroependyma lining have both been implicated in PH formation. At the cellular level, altered recruitment of proteins to the adhesion sites at the cell periphery and their turnover via Arf-mediated vesicle trafficking could contribute to these processes (Carreno et al., 2004; Sarkisian et al., 2006; Zhang et al., 2012). We therefore inhibited Arf1 through dominant-negative transfection in polarized MDCK cells, as a model to assess for epithelial integrity. Arf loss led to the redistribution of the cell adhesion molecules, β-catenin (Fig. 8C) and E-cadherin (Fig. 8D), away from the membrane surface toward the cytoplasm. Overexpression of dominant-negative Arf1 also caused the expansion of MDCK cell volume with extrusion of the cell body vertically out of the x–y-plane, presumably as a consequence of the loss of cell–cell adhesion.
Both Big2 and FlnA have also been implicated in vesicle trafficking of receptors and adhesion molecules (i.e., paxillin, vinculin, and FAK) along the cell membrane of migrating cells (Xu et al., 2010; Zhang et al., 2012; Chen et al., 2013). Therefore, we analyzed the focal adhesion number in MEF cells 24 h after transfection of Arf1T31N, followed by dissociation of the cells and replating. We did not see dramatic changes in vinculin 3–48 h after replating, nor in HA vector control-tranfected cells compared with untransfected cells (data not shown). Paxillin number, however, showed significant reduction after the first 3–6 h compared with control cells (Fig. 8E,F). The difference was diminished by 24 h (Fig. 8G), suggesting that Arf1 activity is most important in paxillin involving new focal adhesion synthesis in motile cells. In total, the experiments suggest that Arf1 may serve as a downstream effector of both Big2 and FlnA function, and disruption of Arf1-dependent vesicle trafficking may contribute to the neuropathology seen in PH.
Discussion
Prior studies have suggested that the initial migration delay seen in PH resulted from a defect in the cytoskeleton through disruption of FlnA-dependent actin turnover. The current work extends upon this initial framework and demonstrates that actin binding proteins such as FlnA may indirectly influence the activation of vesicle trafficking proteins such as Big2 by directing their subcellular localization within the cell. Big2 redistribution to the cell membrane promotes Arf1 activation at the membrane and allows for turnover of cell adhesion molecules and receptors that guide neuronal motility and neuroependymal integrity. These findings begin to implicate a common shared pathway, namely, that of actin-dependent vesicle trafficking, in the pathogenesis of this disorder.
An inconsistency surrounding the role of FlnA in migration derives from conflicting reports which either support or refute a migration defect in cortical neurons following the loss of FlnA function. Several studies using null FlnA mice did not demonstrate a migration defect during early embryonic ages (before E15.5; Ozanne et al., 2000; Feng et al., 2006). Loss of FlnA function was early embryonic lethal in the male mice, and the heterozygous females did not show a cortical migration problem. These findings contrasted with the observed impairment in migration following in utero electroporation of FlnA siRNA constructs (Nagano et al., 2002, 2004), raising the question of whether the techniques altered FlnA function, the early lethality in the mice prevented observable defects in migration, or some compensatory role was played by other filamin proteins in the null FlnA mouse models. The current findings reconcile the differences in that the prior findings of preserved migration in null FlnA mice likely were limited by the embryonic lethality. We also find a cell-intrinsic defect in neuronal migration and that more immediate loss of FlnA (through a tamoxifen-inducible cre) is sufficient to produce the migratory defect.
Reciprocal interactions between Big2 and FlnA mediate vesicle trafficking and affect neuronal migration. Previously, the loss of function of genes associated with PH has been shown to cause impaired migration due to overexpression of FlnA. Both Big2 and Mekk4 bind FlnA and the loss of expression of either of these proteins results in increased FlnA levels (Sarkisian et al., 2006; Zhang et al., 2012). We also show that loss of FlnA impairs migration. These studies indicated that changes in FlnA could alter actin dynamics and disrupt the actin cytoskeleton, thereby changing the size and number of cell adhesion molecules involved in neuronal migration. The current findings of increased Big2 levels following loss of FlnA, however, suggest another possible explanation for the observed changes. The increases in Big2 or FlnA from loss of FlnA or Big2, respectively, could serve as compensatory feedback mechanisms. Loss of FlnA (and phosphoFlnA) would prevent sufficient Big2 from reaching the cell membrane and lead to the upregulation of Big2 to promote redistribution to the cell periphery. Conversely, in the absence of Big2 protein expression, one possible interpretation for increased FlnA expression and FlnA phosphorylation levels would be a compensatory feedback to promote Big2 redistribution to the membrane periphery. In support of this possibility, we did not find significant migration abnormalities with overexpression of Big2 after electroporation (data not shown). Thus, protein expression did not drive Big2 function alone. Rather, Big2 subcellular localization is required to direct Arf-dependent functions, and FlnA would provide the mechanism for redistribution of Big2. Finally, this homeostatic process would also provide a rationale for the observation of both increased and decreased levels of FlnA disrupting migration. Either increased or decreased FlnA levels would alter FlnA phosphorylation levels and thereby disrupt Big2 localization and consequent Arf1-dependent trafficking.
Arf1 regulation of focal adhesion turnover may serve as a final pathway in PH formation. Arf1 activation is triggered by the GDP-to-GTP conversion through the GEF family of proteins, including Big2. Overexpression of the dominant-negative Arf1T31N prevents GTP-bound Arf1 formation (activated Arf1), and consequently disrupts vesicle coat protein formation and prevents cargoing of proteins from the cytosol to the membrane and back, thereby providing a mechanism to alter cell adhesion (Bonifacino and Glick, 2004; Nie and Randazzo, 2006). Overexpression and microinjection of the Arf1 constitutively active mutant Q71L in serum-starved Swiss 3T3 fibroblasts causes stress fiber formation with increased paxillin focal adhesion formation. Conversely, overexpression or microinjection of the inactive N-terminal truncation Δ17Arf1 into cells reduces stress fiber and paxillin formation (Norman et al., 1998). In an analogous fashion, FlnA knockdown causes rapid focal adhesion disassembly represented by cleavage of paxillin and vinculin compared with WT cells (Xu et al., 2010), consistent with our observation of decreased paxillin number with the loss of Arf1 activation. The current studies provide a mechanistic link between the GEFs and filamins in regulation of Arf activation, leading to consequent changes in focal adhesion size and/or number. Arf-dependent changes in focal adhesion turnover can affect neuronal migration and neuroependymal integrity.
A defect in vesicle trafficking may help to explain other phenotypes associated with PH. While loss of Big2 and FlnA function promotes heterotopia formation, mutations in these genes have also been shown to give rise to microcephaly. Our prior studies have shown that the loss of FlnA leads to a slowing in progenitor proliferation due to prolongation through G2 to M phase of the cell cycle (Lian et al., 2012). This impairment results from decreased degradation of cyclinB/cdk1-associated proteins, suggesting that FlnA plays some role in the clearance and/or recycling of specific cytoplasmic proteins—as would be appreciated in defects involving trafficking of proteins. More broadly, FLNA mutations have been recognized to affect extra-CNS organ systems in giving rise to phenotypic heterogeneity (Parrini et al., 2006; Solé et al., 2009). Uniformly, each of the organ abnormalities can be associated with problems in cell–cell adhesion or proliferation, presumably of progenitors, similar to that seen with PH in the nervous system. Loss of FlnA leads to shortened bones and midline fusion problems (proliferative defect in chondroprogenitors; Parrini et al., 2006); cutaneous manifestations such as Ehlers-Danlos syndrome, a connective tissue disorder of epidermal progenitors (Sheen et al., 2005; Gómez-Garre et al., 2006; Sun et al., 2010); vascular anomalies from bleeding due to loss of endothelial progenitor cell–cell contacts (Feng et al., 2006); and pulmonary abnormalities with lobar emphysema, bilateral atelectasis, lung cysts, and tracheobronchomalacia due to defects in bronchial chondroprogenitors (de Wit et al., 2009; Masurel-Paulet et al., 2011). Finally, FlnA dysfunction and PH has been linked to intestinal pseudo-obstruction with shortened gut, again suggestive of a proliferative defect (Gargiulo et al., 2007; Clayton-Smith et al., 2009; Kapur et al., 2010). Each of these defects potentially reflects a similar pathophysiology (loss of cell–cell adhesion, impaired migration, and proliferation), as is seen with PH formation.
Our findings of reciprocal interactions between FlnA and Big2 in mediating neuronal migration through cell–cell adhesion proteins imply a finely regulated balance between vesicle transport and actin assembly/disassembly. RhoGTPases are known effectors of FlnA and have been implicated in membrane protrusions and vesicle trafficking (Ridley, 2006). However, the FlnA-dependent downstream effectors of the RhoGTPases, which are involved in vesicle trafficking, are not apparent. Further studies will be needed to elucidate this pathway before we can fully appreciate the role of filamins and GEFs in progenitor development across various organ systems.
Footnotes
This work was supported in part by National Institutes of Health Grant NS063997-01 to V.S. We thank Drs. T. Stossel, K. Nakayama, J. Flanagan, J. Bonifacino, J. Blenis, and F. Nakamura for providing the valuable reagents specified in the experimental procedures of this paper.
The authors declare no competing financial interests.
References
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/S0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Boulay PL, Cotton M, Melançon P, Claing A. ADP-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J Biol Chem. 2008;283:36425–36434. doi: 10.1074/jbc.M803603200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch-Bräuninger M, Eilken HM, Rieger MA, Schroeder TT, Huttner WB, Brakebusch C, Götz M. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci. 2006;9:1099–1107. doi: 10.1038/nn1744. [DOI] [PubMed] [Google Scholar]
- Carreno S, Engqvist-Goldstein AE, Zhang CX, McDonald KL, Drubin DG. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J Cell Biol. 2004;165:781–788. doi: 10.1083/jcb.200403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PW, Jian X, Yoon HY, Randazzo PA. ARAP2 signals through Arf6 and Rac1 to control focal adhesion morphology. J Biol Chem. 2013;288:5849–5860. doi: 10.1074/jbc.M112.415778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-T. [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J, Walters S, Hobson E, Burkitt-Wright E, Smith R, Toutain A, Amiel J, Lyonnet S, Mansour S, Fitzpatrick D, Ciccone R, Ricca I, Zuffardi O, Donnai D. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Hum Genet. 2009;17:434–443. doi: 10.1038/ejhg.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit MC, de Coo IF, Halley DJ, Lequin MH, Mancini GM. Movement disorder and neuronal migration disorder due to ARFGEF2 mutation. Neurogenetics. 2009;10:333–336. doi: 10.1007/s10048-009-0192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci U S A. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferland RJ, Batiz LF, Neal J, Lian G, Bundock E, Lu J, Hsiao YC, Diamond R, Mei D, Banham AH, Brown PJ, Vanderburg CR, Joseph J, Hecht JL, Folkerth R, Guerrini R, Walsh CA, Rodriguez EM, Sheen VL. Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum Mol Genet. 2009;18:497–516. doi: 10.1093/hmg/ddn377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, Berkovic SF, Huttenlocher PR, Walsh CA. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/S0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- Gargiulo A, Auricchio R, Barone MV, Cotugno G, Reardon W, Milla PJ, Ballabio A, Ciccodicola A, Auricchio A. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet. 2007;80:751–758. doi: 10.1086/513321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Garre P, Seijo M, Gutiérrez-Delicado E, Castro del Río M, de la Torre C, Gómez-Abad C, Morales-Corraliza J, Puig M, Serratosa JM. Ehlers-Danlos syndrome and periventricular nodular heterotopia in a Spanish family with a single FLNA mutation. J Med Genet. 2006;43:232–237. doi: 10.1136/jmg.2004.029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AW, Morgan JE, Schneider J, West K, McKie L, Bhattacharya S, Jackson IJ, Cross SH. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum Mol Genet. 2006;15:2457–2467. doi: 10.1093/hmg/ddl168. [DOI] [PubMed] [Google Scholar]
- Hastie LE, Patton WF, Hechtman HB, Shepro D. H2O2-induced filamin redistribution in endothelial cells is modulated by the cyclic AMP-dependent protein kinase pathway. J Cell Physiol. 1997;172:373–381. doi: 10.1002/(SICI)1097-4652(199709)172:3<373::AID-JCP11>3.0.CO%3B2-7. [DOI] [PubMed] [Google Scholar]
- Jay D, García EJ, Lara JE, Medina MA, de la Luz Ibarra M. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Arch Biochem Biophys. 2000;377:80–84. doi: 10.1006/abbi.2000.1762. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Robertson SP, Hannibal MC, Finn LS, Morgan T, van Kogelenberg M, Loren DJ. Diffuse abnormal layering of small intestinal smooth muscle is present in patients with FLNA mutations and x-linked intestinal pseudo-obstruction. Am J Surg Pathol. 2010;34:1528–1543. doi: 10.1097/PAS.0b013e3181f0ae47. [DOI] [PubMed] [Google Scholar]
- Lian G, Lu J, Hu J, Zhang J, Cross SH, Ferland RJ, Sheen VL. Filamin a regulates neural progenitor proliferation and cortical size through Wee1-dependent Cdk1 phosphorylation. J Neurosci. 2012;32:7672–7684. doi: 10.1523/JNEUROSCI.0894-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell. 2005;16:1744–1755. doi: 10.1091/mbc.E04-10-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sheen V. Periventricular heterotopia. Epilepsy Behav. 2005;7:143–149. doi: 10.1016/j.yebeh.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Lu J, Tiao G, Folkerth R, Hecht J, Walsh C, Sheen V. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. J Comp Neurol. 2006;494:476–484. doi: 10.1002/cne.20806. [DOI] [PubMed] [Google Scholar]
- Masurel-Paulet A, Haan E, Thompson EM, Goizet C, Thauvin-Robinet C, Tai A, Kennedy D, Smith G, Khong TY, Solé G, Guerineau E, Coupry I, Huet F, Robertson S, Faivre L. Lung disease associated with periventricular nodular heterotopia and an FLNA mutation. Eur J Med Genet. 2011;54:25–28. doi: 10.1016/j.ejmg.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Nagano T, Yoneda T, Hatanaka Y, Kubota C, Murakami F, Sato M. Filamin A-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nat Cell Biol. 2002;4:495–501. doi: 10.1038/ncb808. [DOI] [PubMed] [Google Scholar]
- Nagano T, Morikubo S, Sato M. Filamin A and FILIP (filamin A-interacting protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Randazzo PA. Arf GAPs and membrane traffic. J Cell Sci. 2006;119:1203–1211. doi: 10.1242/jcs.02924. [DOI] [PubMed] [Google Scholar]
- Nishimura YV, Shinoda T, Inaguma Y, Ito H, Nagata K. Application of in utero electroporation and live imaging in the analyses of neuronal migration during mouse brain development. Med Mol Morphol. 2012;45:1–6. doi: 10.1007/s00795-011-0557-0. [DOI] [PubMed] [Google Scholar]
- Norman JC, Jones D, Barry ST, Holt MR, Cockcroft S, Critchley DR. ARF1 mediates paxillin recruitment to focal adhesions and potentiates Rho-stimulated stress fiber formation in intact and permeabilized Swiss 3T3 fibroblasts. J Cell Biol. 1998;143:1981–1995. doi: 10.1083/jcb.143.7.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci U S A. 1999;96:2122–2128. doi: 10.1073/pnas.96.5.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000;14:1618–1626. doi: 10.1210/me.14.10.1618. [DOI] [PubMed] [Google Scholar]
- Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P, Marini C, Brilstra EH, Dalla Bernardina B, Goodwin L, Bodell A, Jones MC, Nangeroni M, Palmeri S, Said E, Sander JW, Striano P, Takahashi Y, Van Maldergem L, Leonardi G, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129:1892–1906. doi: 10.1093/brain/awl125. [DOI] [PubMed] [Google Scholar]
- Phoenix TN, Temple S. Spred1, a negative regulator of Ras-MAPK-ERK, is enriched in CNS germinal zones, dampens NSC proliferation, and maintains ventricular zone structure. Genes Dev. 2010;24:45–56. doi: 10.1101/gad.1839510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayner SL, Balczon R, Frank DW, Cooper DM, Stevens T. Filamin A is a phosphorylation target of membrane but not cytosolic adenylyl cyclase activity. Am J Physiol Lung Cell Mol Physiol. 2011;301:L117–L124. doi: 10.1152/ajplung.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, Ganesh V, Underwood T, Wiley J, Leventer R, Vaid RR, Ruiz DE, Hutchins GM, Menasha J, Willner J, Geng Y, Gripp KW, Nicholson L, Berry-Kravis E, Bodell A, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, Guerrini R, Walsh CA. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- Smythe E, Ayscough KR. Actin regulation in endocytosis. J Cell Sci. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- Solé G, Coupry I, Rooryck C, Guérineau E, Martins F, Devés S, Hubert C, Souakri N, Boute O, Marchal C, Faivre L, Landré E, Debruxelles S, Dieux-Coeslier A, Boulay C, Chassagnon S, Michel V, Routon MC, Toutain A, Philip N, et al. Bilateral periventricular nodular heterotopia in France: frequency of mutations in FLNA, phenotypic heterogeneity and spectrum of mutations. J Neurol Neurosurg Psychiatry. 2009;80:1394–1398. doi: 10.1136/jnnp.2008.162263. [DOI] [PubMed] [Google Scholar]
- Soumiya H, Fukumitsu H, Furukawa S. Stem cell factor induces heterotopic accumulation of cells (heterotopia) in the mouse cerebral cortex. Biomed Res. 2009;30:121–128. doi: 10.2220/biomedres.30.121. [DOI] [PubMed] [Google Scholar]
- Sun Y, Almomani R, Aten E, Celli J, van der Heijden J, Venselaar H, Robertson SP, Baroncini A, Franco B, Basel-Vanagaite L, Horii E, Drut R, Ariyurek Y, den Dunnen JT, Breuning MH. Terminal osseous dysplasia is caused by a single recurrent mutation in the FLNA gene. Am J Hum Genet. 2010;87:146–153. doi: 10.1016/j.ajhg.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Sasaki K, Kamata R, Sakai R. The C-terminus of ephrin-B1 regulates metalloproteinase secretion and invasion of cancer cells. J Cell Sci. 2007;120:2179–2189. doi: 10.1242/jcs.008607. [DOI] [PubMed] [Google Scholar]
- Turner CE, West KA, Brown MC. Paxillin-ARF GAP signaling and the cytoskeleton. Curr Opin Cell Biol. 2001;13:593–599. doi: 10.1016/S0955-0674(00)00256-8. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z, Teng L, Ingber DE, Mammoto A, Kumar R, Alaoui-Jamali MA. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207:2421–2437. doi: 10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Neal J, Lian G, Shi B, Ferland RJ, Sheen V. Brefeldin A-inhibited guanine exchange factor 2 regulates filamin A phosphorylation and neuronal migration. J Neurosci. 2012;32:12619–12629. doi: 10.1523/JNEUROSCI.1063-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]