

Abstract
Shank3, which encodes a scaffolding protein at glutamatergic synapses, is a genetic risk factor for autism. In this study, we examined the impact of Shank3 deficiency on the NMDA-type glutamate receptor, a key player in cognition and mental illnesses. We found that knockdown of Shank3 with a small interfering RNA (siRNA) caused a significant reduction of NMDAR-mediated ionic or synaptic current, as well as the surface expression of NR1 subunits, in rat cortical cultures. The effect of Shank3 siRNA on NMDAR currents was blocked by an actin stabilizer, and was occluded by an actin destabilizer, suggesting the involvement of actin cytoskeleton. Since actin dynamics is regulated by the GTPase Rac1 and downstream effector p21-activated kinase (PAK), we further examined Shank3 regulation of NMDARs when Rac1 or PAK was manipulated. We found that the reducing effect of Shank3 siRNA on NMDAR currents was mimicked and occluded by specific inhibitors for Rac1 or PAK, and was blocked by constitutively active Rac1 or PAK. Immunocytochemical data showed a strong reduction of F-actin clusters after Shank3 knockdown, which was occluded by a PAK inhibitor. Inhibiting cofilin, the primary downstream target of PAK and a major actin depolymerizing factor, prevented Shank3 siRNA from reducing NMDAR currents and F-actin clusters. Together, these results suggest that Shank3 deficiency induces NMDAR hypofunction by interfering with the Rac1/PAK/cofilin/actin signaling, leading to the loss of NMDAR membrane delivery or stability. It provides a potential mechanism for the role of Shank3 in cognitive deficit in autism.
Introduction
Autism spectrum disorders (ASDs) are a group of neurodevelopmental diseases characterized by core symptoms including impairments in social interactions, deficient language skills, repetitive behaviors, decreased cognitive abilities, and attention deficits (Levy et al., 2010). Genetic studies have demonstrated that haploinsufficiency of Shank3 gene or de novo mutations in Shank3 gene are associated with ASD (Bonaglia et al., 2001; Durand et al., 2007; Moessner et al., 2007; Sebat et al., 2007). Animal models expressing mutant forms of Shank3 exhibit ASD-like symptoms, such as abnormal social behaviors, aberrant vocalizations, and deficits in learning and memory, along with synaptic dysfunction (Bozdagi et al., 2010; Peça et al., 2011; Wang et al., 2011).
Shank3 is an integral scaffolding protein located at the postsynaptic density of glutamatergic synapses (Naisbitt et al., 1999; Sheng and Kim, 2000). Previous work has demonstrated the ability for Shank proteins to interact with various proteins at the postsynaptic density, such as Homer, glutamate receptor complexes, and cytoskeleton proteins (Naisbitt et al., 1999; Böckers et al., 2001). While Shank3 has been linked to spine morphology and synapse formation (Boeckers et al., 2002; Roussignol et al., 2005; Durand et al., 2012), the mechanisms underlying its role in regulating synaptic proteins and synaptic transmission and plasticity remain unclear. Because effective neuronal communication is dependent upon the proper arrangement of glutamate receptor channels, we speculate that Shank3 may control brain functions by affecting the trafficking and anchoring of glutamate receptors at the synapse.
The NMDA-type glutamate receptor, which is primarily responsible for synaptic plasticity and memory, physically associates with Shank3 (Ehlers, 1999; Naisbitt et al., 1999). NMDAR dysfunction has been linked to mental disorders including schizophrenia and ASD (Carlson, 2012; Moghaddam and Javitt, 2012), exemplified by the fact that administration of NMDAR antagonists exacerbates schizophrenia-like symptoms (Luby et al., 1959; Javitt and Zukin, 1991; Krystal et al., 1994) and induces autistic-like deficits in mice (Wu et al., 2005; Zou et al., 2008). Furthermore, alterations in the expression of NMDAR-associated postsynaptic proteins have been found in postmortem brains of schizophrenic individuals (Clinton et al., 2003). Altered NMDAR function has been found in mice lacking Shank2 (Schmeisser et al., 2012; Won et al., 2012), and drugs acting at various sites on the NMDAR to enhance its function lead to the significant recovery of social deficits in Shank2-mutant mice (Won et al., 2012), suggesting their therapeutic potential for ASD treatment (Moskal et al., 2011).
The aforementioned results highly implicate NMDARs as a potential key target of Shank proteins in mental health and disorders; thus, we sought to uncover the mechanisms and critical molecules involved in Shank3 regulation of NMDAR function. We found that Shank3 deficiency leads to NMDAR hypofunction through a mechanism involving the actin cytoskeleton and several actin regulators. Our studies should help to reveal the pathophysiological basis of and new therapeutic targets for autism.
Materials and Methods
Reagents.
The Shank3 siRNA (Santa Cruz Biotechnology) is a mixture of three oligonucleotides, all of which target the N-terminal ankyrin repeat domain of rat Shank3: 5′-GAUACAAGCGGAGAGUUUAtt, CCUGAAGGUUCUUCGCAAUtt, and CUUGCCGAGGUAAUCAAGAtt. The N-terminal ankyrin domain-deleted Shank3 was used as the siRNA-resistant Shank3 (Shank3R). To test the knockdown effect, the full-length Shank3 (kindly provided by Dr. Carlo Sala in CNR Institute of Neuroscience, Milano, Italy) or Shank3R (kindly provided by Dr. Eunjoon Kim in Korea Advanced Institute of Science and Technology, Daejeon, South Korea) was transfected to HEK293 cells with Shank3 siRNA or control siRNA. Two days after transfection, the cells were harvested and subjected to Western blotting with anti-Shank3 (1:500, Neuromab). Tubulin (1:20,000, Sigma) was used as a loading control.
For mutant Rac1, T17N (dominant negative) and Q61L (constitutively active) were obtained from Addgene. TAT-PAK18 inhibitory peptide (RKKRRQRRvR-G-PPVIAPRPEHTKSVYTRS), TAT-p-cofilin peptide (RKKRRQRRR-MAS(p)GVAVSDGVIKVFN), and TAT control peptide (RKKRRQRRR) were synthesized by Keck Biotechnology Resource Laboratory (Yale University, New Haven, CT). Generation of PAK mutants, PAK-AID (autoinhibitory domain), DN-PAK1 (H83L, H86L, K299R), and CA-PAK1 (T423E), was performed with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All constructs were verified by DNA sequencing.
Primary neuronal culture and transfection.
Rat cortical cultures were prepared as described previously (Gu et al., 2005; Yuen et al., 2011). Briefly, frontal cortex was dissected from embryonic day 18 rat embryos of either sex, and cells were dissociated using trypsin and trituration through a Pasteur pipette. Neurons were plated on coverslips coated with poly-l-lysine in DMEM with 10% fetal calf serum at a density of 1 × 105 cells/cm2. When neurons were attached to the coverslip within 24 h, the medium was changed to Neurobasal with B27 supplement (Invitrogen). Cytosine arabinoside (AraC, 5 μm) was added at DIV 3 to stop glial proliferation. Cultured neurons were transfected with EGFP plus various constructs or siRNA (33.3 nm) using the Lipofectamine 2000 method. Drugs were added 1 d after transfection and electrophysiological recordings were performed on GFP-positive neurons at 2–3 d after transfection.
Electrophysiological recordings.
Whole-cell NMDA-elicited currents in cultured neurons were recorded using standard voltage-clamp techniques (Gu et al., 2005; Yuen et al., 2011, 2012). The internal solution contained the following (in mm): 180 N-methyl-d-glucamine, 4 MgCl2, 40 HEPES, 0.5 BAPTA, 12 phosphocreatine, 3 Na2ATP, and 0.5 Na2GTP, with pH 7.2–7.3 and 265–270 mOsm. The external solution consisted of the following (in mm): 127 NaCl, 20 CsCl, 1 CaCl2, 5 BaCl2, 10 HEPES, 12 glucose, 0.02 glycine, and 0.001 tetrodotoxin, pH 7.4, and 300 mOsm. Recordings were obtained using Axopatch200B patch-clamp amplifier (Molecular Devices), controlled with a computer running Clampex 10.2 with a DigiData 1440A series interface (Molecular Devices). A tight seal (>2 GΩ) was obtained using negative pressure, with membrane disruption after additional suction. The cell membrane was held at −60 mV, and NMDAR-mediated current was elicited by bath application of NMDA (100 μm) for 2 s every 30 s.
For synaptic current recordings, the coverslip containing cultured neurons was placed in the recording chamber and continuously perfused with 95% O2 and 5% CO2 artificial CSF (ACSF, in mm: 130 NaCl, 26 NaHCO3, 3 KCl, 5 MgCl2, 1.25 NaH2PO4, 1 CaCl2, 10 glucose, pH 7.4, and 300 mOsm). GABAA receptor antagonist bicuculline (10 μm) was added. The patch pipette was filled with an internal solution containing the following (in mm): 130 Cs methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 5 EGTA, 10 HEPES, 2 QX-314, 12 phosphocreatine, 5 MgATP, 0.5 Na2GTP, 0.2 leupeptin, pH 7.2–7.3, 265–270 mOsm. Recordings were obtained with a Multiclamp 700A amplifier and Digidata 1320A data acquisition system (Molecular Devices). Evoked EPSCs were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Grass Technologies). A concentric bipolar stimulating electrode (FHC) was placed ∼100 μm from the neuron under recording. Membrane potential was maintained at −70 mV for AMPAR-EPSC recordings. For NMDAR-EPSC, the cell (clamped at −70 mV) was depolarized to +40 mV for 3 s before stimulation to fully relieve the voltage-dependent Mg2+ block. Both AMPAR and NMDAR channels open at +40 mV, therefore NMDAR responses were calculated at 30 ms after stimulation when AMPAR-EPSCs had mostly (>90%) decayed. Data analysis used Clampfit (Molecular Devices) and Kaleidagraph (Albeck Software).
Immunocytochemical staining.
Cultured neurons were fixed in 4% paraformaldehyde in PBS for 20 min at room temperature and then washed three times with PBS. Neurons were then permeabilized with 0.1% Triton X-100 in PBS for 5 min followed by 1 h incubation with 5% bovine serum albumin (BSA) to block nonspecific staining. For the staining of surface NR1, neurons were fixed but not permeabilized. Neurons were incubated with the primary antibodies at 4°C overnight. Antibodies used include anti-Shank3 (1:500, Neuromab) (Durand et al., 2012), anti-NR1 (1:500, Neuromab) (Murata and Constantine-Paton, 2013), anti-MAP2 (1:200, Santa Cruz Biotechnology) (Yuen and Yan, 2011), anti-PSD95 (1:1000, Neuromab) (Verpelli et al., 2011), and anti-Thr423p-PAK1 (1:100, Cell Signaling Technology) (Zhan et al., 2003). After washing, neurons were incubated with Alexa Fluor 647- or 568-conjugated secondary antibody (Invitrogen, 1:200 and 1:500) for 2 h at room temperature. For the staining of F-actin, neurons were incubated with Alexa Fluor 568 -conjugated phalloidin (1 U/ml, Invitrogen) at room temperature for 20 min. After washing in PBS three times, coverslips were mounted on slides with VECTASHIELD mounting media (Vector Laboratories).
Fluorescent images were obtained using a 100× objective with a cooled charge-coupled device camera mounted on a Nikon microscope. All specimens were imaged under identical conditions and analyzed using identical parameters with the ImageJ software. The surface NR1 clusters or F-actin clusters were measured in GFP-positive neurons. To define dendritic clusters, threshold was chosen manually, so that clusters corresponded to puncta with the mean intensity equal to ∼2-fold of the diffuse fluorescent mean intensity on the dendritic shaft. On each coverslip, the cluster density of 3–6 neurons (2–3 dendritic segments of 30 μm length per neuron) was measured. The fluorescence intensity of p-PAK1 or MAP2 staining was assessed by measuring the mean intensity of pixels in proximal dendrites (background was subtracted using the threshold method). Three to four independent experiments for each of the treatments were performed.
Statistics.
All data are expressed as the mean ± SEM. Experiments with two groups were analyzed statistically using unpaired Student's t tests. Experiments with more than two groups were subjected to one-way ANOVA, followed by post hoc Tukey's tests.
Results
Knockdown of Shank3 selectively inhibits NMDA receptor function and surface expression
To investigate the consequence of Shank3 deficiency on NMDAR function, we used the RNA interference approach. As shown in Figure 1A, Shank3 siRNA caused a marked suppression of Shank3 protein expression in transfected HEK293 cells. Co-transfecting Shank3 siRNA with a siRNA-resistant Shank3 (Shank3R) construct prevented the Shank3 knockdown. In neuronal dendrites of cortical cultures (Fig. 1B), Shank3 siRNA transfection also significantly decreased Shank3 cluster density (# clusters/30 μm dendrite) (control siRNA: 12.1 ± 1.6, n = 20; Shank3 siRNA: 4.8 ± 0.5, n = 28, p < 0.001, ANOVA), Shank3 cluster size (μm2) (control siRNA: 0.24 ± 0.02, n = 20; Shank3 siRNA: 0.15 ± 0.01, n = 28, p < 0.001, ANOVA), and Shank3 cluster intensity (control siRNA: 160.9 ± 6.5, n = 20; Shank3 siRNA: 100.5 ± 5.2, n = 28, p < 0.001, ANOVA), confirming an effective knockdown of endogenous Shank3 in neurons. The expression of Shank3 clusters in neuronal dendrites was restored by co-transfecting Shank3 siRNA with Shank3R (cluster density: 11.1 ± 1.5, n = 18; cluster size: 0.23 ± 0.02, n = 18; cluster intensity: 145.6 ± 8.5, n = 18).
Figure 1.
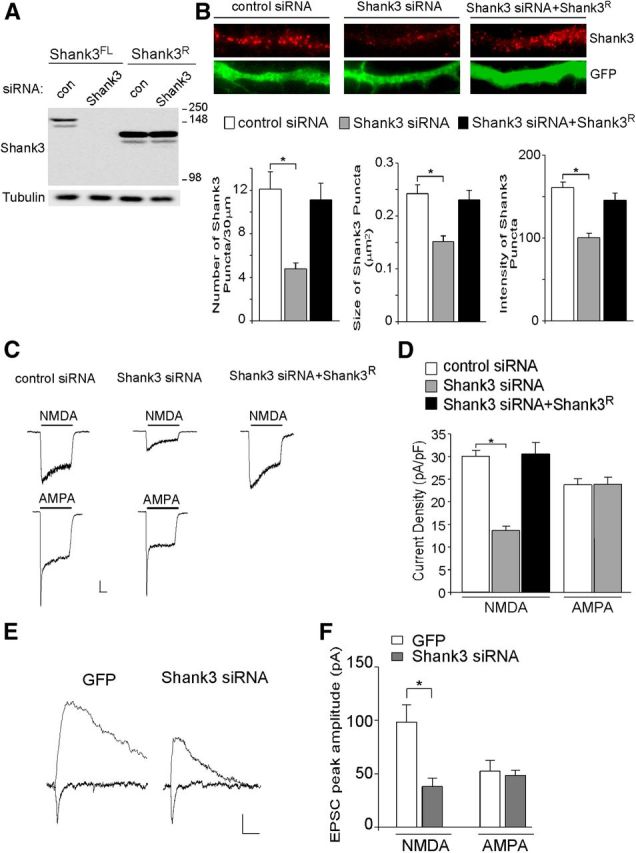
Cortical neurons with Shank3 knockdown show a selective reduction of NMDAR-mediated ionic and synaptic currents. A, Representative Western blots of Shank3 in HEK293 cells transfected with full-length rat Shank3 (Shank3FL) or the N-terminal deleted, siRNA-resistant Shank3 construct (Shank3R) in the presence of Shank3 siRNA or a scrambled control siRNA. B, Top, Immunostaining of Shank3 in cortical cultures (DIV19–22) transfected with control siRNA, Shank3 siRNA, or Shank3 siRNA plus Shank3R. Bottom, Bar graphs (mean ±SEM) showing the density, size, and intensity of Shank3 clusters (red) in neurons (GFP-positive) with different transfections. *p < 0.01, ANOVA. C, Representative traces of NMDAR- or AMPAR-mediated ionic currents in cultured cortical neurons (DIV 21) transfected with control siRNA, Shank3 siRNA, or Shank3 siRNA plus Shank3R. Calibration: 200 pA, 0.5 s. D, Bar graph summary of the NMDAR- or AMPAR-current density in cortical cultures with different transfections. *p < 0.01, ANOVA. E, Representative traces of evoked NMDAR-EPSC and AMPAR-EPSC in cortical neurons (DIV 14–18) transfected with Shank3 siRNA or GFP alone. Calibration: 20 pA, 50 ms. F, Bar graphs showing the peak amplitude of evoked NMDAR-EPSC and AMPAR-EPSC in cortical cultures with different transfections. *p < 0.01, t test.
In cortical cultures transfected with Shank3 siRNA, the NMDA-elicited current density (pA/pF) was significantly decreased (Fig. 1C,D, control siRNA: 30.1 ± 1.3, n = 15; Shank3 siRNA: 13.7 ± 0.9, n = 15, p < 0.01, ANOVA). The reducing effect of Shank3 knockdown on NMDAR current density was rescued by expressing the siRNA-resistant Shank3 (30.6 ± 2.5 pA/pF, n = 10), suggesting the specificity of the Shank3 siRNA. In contrast, AMPAR-mediated current density (pA/pF) was unchanged by Shank3 knockdown (control siRNA: 23.7 ± 1.3, n = 7; Shank3 siRNA: 23.8 ± 1.6, n = 8, p > 0.05, t test).
To examine the role of Shank3 in glutamatergic transmission, we examined the NMDAR- and AMPAR-mediated EPSCs evoked by stimulating neighboring neurons in Shank3 siRNA-transfected cells. As shown in Figure 1, E and F, knockdown of Shank3 significantly decreased the amplitude of NMDAR-EPSC (control siRNA: 98.1 ± 16.1 pA, n = 10, Shank3 siRNA: 37.9 ± 7.5 pA, n = 12, p < 0.01, t test) without affecting AMPAR-EPSC amplitude (control siRNA: 52.3 ± 10.1 pA, n = 10, Shank3 siRNA: 48.0 ± 5.1 pA, n = 12, p > 0.05, t test). These data suggest that Shank3 is required for proper NMDAR signaling.
Because NMDAR signaling could be affected by changes in channel gating or receptor localization (Wenthold et al., 2003), immunocytochemical experiments were performed to examine the impact of Shank3 knockdown on NMDAR surface expression. As shown in Figure 2, A and B, transfection of Shank3 siRNA significantly reduced surface NR1 cluster density (# clusters/30 μm dendrite) (control siRNA: 14.5 ± 0.6, n = 23; Shank3 siRNA: 7.5 ± 0.4, n = 29, p < 0.01, ANOVA), surface NR1 cluster size (μm2) (control siRNA: 0.45 ± 0.03, n = 23; Shank3 siRNA: 0.29 ± 0.03, n = 29, p < 0.01, ANOVA), and surface NR1 cluster intensity (control siRNA: 172.0 ± 3.3, n = 23; Shank3 siRNA: 146.9 ± 2.6, n = 29, p < 0.01, ANOVA). The loss of surface NR1 clusters was rescued when Shank3 siRNA was co-transfected with Shank3R (cluster density: 13.4 ± 0.9, n = 20; cluster size: 0.49 ± 0.1, n = 20; cluster intensity: 160.2 ± 4.5, n = 20).
Figure 2.

Cortical neurons with Shank3 knockdown show reduced surface NR1 clusters. A, Immunostaining of surface NR1 in cortical cultures (DIV 19–22) transfected with control siRNA, Shank3 siRNA, or Shank3 siRNA plus Shank3R. Scale bar, 10 μm. B, Bar graphs (mean ± SEM) showing the density, size, and intensity of surface NR1 clusters (red) in neurons (GFP-positive) with different transfections. *p < 0.01, ANOVA. C, Immunostaining of PSD-95 in cortical cultures transfected with control siRNA or Shank3 siRNA. D, Bar graphs (mean ± SEM) showing the density, size, and intensity of PSD-95 clusters (red) in control siRNA- or Shank3 siRNA-transfected neurons (GFP-positive).
To find out whether the loss of NMDAR clusters from Shank3 knockdown is due to an overall reduction in the number of synapses, we examined PSD-95, a synaptic marker. As shown in Figure 2C and D, Shank3 siRNA did not have a significant effect on PSD-95 puncta density (control siRNA: 11.4 ± 0.8, n = 20; Shank3 siRNA: 11.3 ± 0.7, n = 23, p > 0.05, t test), PSD-95 puncta size (control siRNA: 0.31 ± 0.03, n = 22; Shank3 siRNA: 0.30 ± 0.02, n = 25, p > 0.05, t test), or PSD-95 puncta intensity (control siRNA: 197.9 ± 2.1, n = 22; Shank3 siRNA: 198.1 ± 2.8, n = 25, p > 0.05, t test). It suggests that Shank3 knockdown induces the specific loss of NMDA receptors on the membrane of existing synapses, which may be responsible for the selectively decreased NMDAR currents in Shank3-deficient neurons. Because previous work using RNA interference of Shank3 has failed to find changes in total protein levels of major synaptic proteins (Verpelli et al., 2011), we aimed to uncover the mechanism by which Shank3 alters the transport or surface stability of the NMDA receptor.
Alterations in actin dynamics mediate Shank3 regulation of NMDAR currents
The NMDAR is closely tied to actin filaments through actin-binding proteins (Wyszynski et al., 1997). Mounting evidence suggests that the integrity of actin cytoskeleton is critically involved in the regulation of NMDA receptor trafficking and NMDAR-mediated synaptic transmission (Rosenmund and Westbrook, 1993; Allison et al., 1998; Lei et al., 2001; Gu et al., 2005, 2012). This led us to investigate the potential involvement of actin in Shank3 regulation of NMDAR signaling. As shown in Figure 3, A and B, in the presence of the actin-stabilizing drug phalloidin (1 μm, 12–16 h), the reducing effect of Shank3 knockdown on NMDAR current density (pA/pF) was abolished (phalloidin+control siRNA: 31.1 ± 2.1, n = 6; phalloidin+Shank3 siRNA: 31.7 ± 1.0, n = 7, p > 0.05, ANOVA). Furthermore, the application of latrunculin B (5 μm, 12–16 h), an actin destabilizer, induced a reduction of NMDAR current density (pA/pF) and occluded the effect of Shank3 knockdown (latrunculin+control siRNA: 17.2 ± 1.0, n = 10; latrunculin+Shank3 siRNA: 15.0 ± 0.5, n = 6, p > 0.05, ANOVA). Conversely, taxol (10 μm, 12–16 h), a microtubule stabilizer, failed to alter the effects of Shank3 siRNA (taxol+control siRNA: 27.2 ± 0.8, n = 9, taxol+Shank3 siRNA: 13.6 ± 0.7, n = 9, p < 0.01, ANOVA). Together, the data implicate actin dynamics for a mechanistic role in the NMDAR current reduction by Shank3 knockdown.
Figure 3.

The Shank3 regulation of NMDAR currents is influenced by agents that affect actin dynamics. A, Representative NMDAR currents in cultured cortical neurons transfected with control siRNA or Shank3 siRNA (DIV 19–22) in the presence of actin stabilizer phalloidin (1 μm), actin destabilizer latrunculin B (5 μm), or the microtubule stabilizer taxol (10 μm). Calibration: 200 pA, 0.5 s. B, Bar graph summary of NMDAR current density in control siRNA- or Shank3 siRNA-transfected neurons in the absence or presence of various cytoskeleton modifying agents. *p < 0.01, ANOVA.
The actin regulators Rac1 and PAK are involved in Shank3 regulation of NMDAR currents
The Rho family of small GTPases contains Rac1, a key molecule involved in actin cytoskeletal rearrangements and resulting morphological changes (Kaibuchi et al., 1999; Ridley, 2006). To examine the involvement of Rac1, we treated neurons with EHT1864, a specific inhibitor of Rac1 (Shutes et al., 2007). As shown in Figure 4, A and B, application of EHT (1 μm, 12–16 h) reduced NMDAR current density (pA/pF) and largely occluded the effect of Shank3 siRNA (EHT+control siRNA: 21.3 ± 1.4, n = 6; EHT+Shank3 siRNA: 18.8 ± 0.9, n = 7, p > 0.05, ANOVA), which was significantly different from nontreated cells (control siRNA: 29.5 ± 2.5, n = 5; Shank3 siRNA: 13.2 ± 1.1, n = 5, p < 0.01, ANOVA). To further test the role of Rac1, we transfected neurons with the constitutively active Rac1 (CA-Rac1, Q61L) or the dominant-negative Rac1 (DN-Rac1, T17N). As shown in Figure 4, A and B, CA-Rac1 blocked the reducing effect of Shank3 siRNA on NMDAR current density (CA-Rac1+control siRNA: 29.3 ± 1.0, n = 12; CA-Rac1+Shank3 siRNA: 29.1 ± 1.7, n = 10, p > 0.05, ANOVA), while DN-Rac1 occluded the effect of Shank3 siRNA (DN-Rac1+control siRNA: 15.5 ± 1.0, n = 11; DN-Rac1+Shank3 siRNA: 15.5 ± 0.9, n = 10, p > 0.05, ANOVA). These data suggest that Rac1 is required for Shank3 regulation of NMDA receptors.
Figure 4.

Rac1 is involved in Shank3 regulation of NMDAR currents. A, Representative NMDAR currents in cultured cortical neurons transfected with control siRNA or Shank3 siRNA (DIV 21) in the absence or presence of Rac1 inhibitor EHT (1 μm), or co-transfected with either CA-Rac1 (0.65 ng/μl, constitutively active form of Rac1) or DN-Rac1 (0.65 ng/μl, dominant-negative form of Rac1). Calibrations: 200 pA, 0.5 s. B, Bar graph summary of NMDAR current density in control siRNA- or Shank3 siRNA-transfected neurons in the absence of presence of various Rac1-altering agents. *p < 0.01, ANOVA.
Active Rac1 is able to regulate actin cytoskeleton assembly, mainly by targeting serine/threonine p21-activated kinase (PAK), a downstream molecule critical in cytoskeletal rearrangements (Manser et al., 1994; Sells et al., 1997). To test the involvement of PAK, we treated cortical cultures with IPA-3 (5 μm, 12–16 h), a pharmacological inhibitor of PAK (Deacon et al., 2008). As shown in Figure 5, A and B, the NMDAR current density (pA/pF) was significantly reduced by IPA and little further reduction occurred with Shank3 knockdown (IPA+control siRNA: 22.7 ± 0.6, n = 9; IPA+Shank3 siRNA: 19.2 ± 1.4, n = 7, p > 0.05, ANOVA).
Figure 5.
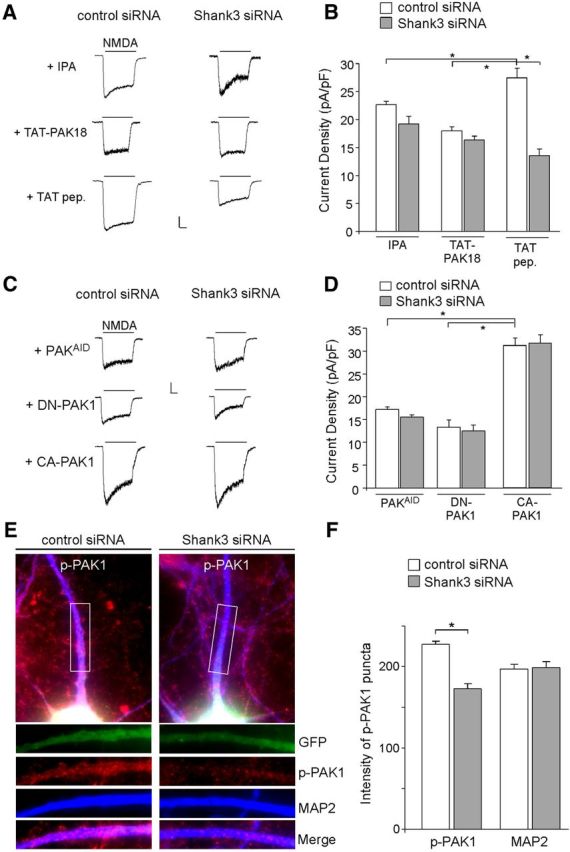
PAK activity is required for Shank3 regulation of NMDAR currents. A, Representative NMDAR currents in cultured cortical neurons transfected with control siRNA or Shank3 siRNA (DIV 21) in the presence of a pharmacological PAK inhibitor, IPA (5 μm), or a membrane-permeable PAK inhibitory peptide, TAT-PAK18 (10 μm). A peptide only containing the cell-permeable TAT sequence was used as a control. Calibrations: 200 pA, 0.5 s. B, Bar graph summary of NMDAR current density in control siRNA- or Shank3 siRNA-transfected neurons in the absence or presence of various PAK inhibiting agents. *p < 0.01, ANOVA. C, Representative NMDAR currents in cultured cortical neurons co-transfected with Shank3 siRNA and PAKAID (10 μm, dominant-negative form of PAK1–3), DN-PAK1 (0.95 ng/μl, dominant-negative form of PAK1), or CA-PAK1 (0.65 ng/μl, constitutively active form of PAK1). A control siRNA was also used. D, Bar graph summary of NMDAR current density in control siRNA- or Shank3 siRNA-transfected neurons in the presence of PAK mutants. *p < 0.01, ANOVA. E, Immunostaining of Thr423p-PAK1 (red) and MAP2 (blue) in cortical cultures transfected with control siRNA or Shank3 siRNA (green). F, Bar graph summary (mean ±SEM) of the intensity of p-PAK1 and MAP2 staining in control siRNA- or Shank3 siRNA-transfected neurons (GFP-positive). *p < 0.01, t test.
To further examine the involvement of PAK, we treated cultures with another PAK inhibitor, PAK18, a 18-mer peptide against the proline-rich domain of PAK that disrupts PAK interaction with the guanine nucleotide exchange factor PIX, reduces cellular PAK phosphorylation, and blocks PAK activation (Maruta et al., 2002; Zhao et al., 2006). The peptide was coupled to the protein transduction domain of the human immunodeficiency virus (HIV) TAT protein, which rendered it cell permeant (Schwarze et al., 1999). TAT-PAK18 (10 μm, 12–16 h) treatment led to decreased NMDAR current density (pA/pF) and occlusion of the effect of Shank3 knockdown (Fig. 5A,B, PAK18+control siRNA: 18.0 ± 0.8, n = 8, PAK18+Shank3 siRNA: 16.4 ± 0.7, n = 8, p > 0.05, ANOVA), which was significantly different from the results with TAT control peptide (TAT+control siRNA: 27.5 ± 1.7, n = 4; TAT+Shank3 siRNA: 13.6 ± 1.2, n = 4, p < 0.01, ANOVA).
To further test the role of PAK, we took advantage of a dominant-negative form of PAK, which contains the auto-inhibitory domain (AID, residues 70–150) of all the three PAKs, PAK1, 2, 3 (PAKAID). This domain inhibits PAK by blocking PAK auto-phosphorylation and therefore its catalytic activity (Hayashi et al., 2004; Kreis and Barnier, 2009). As shown in Figure 5, C and D, transfecting neurons with PAKAID (10 μm) also mimicked and occluded the effect of Shank3 siRNA on NMDAR current density (pA/pF) (PAKAID+control siRNA: 17.2 ± 0.6, n = 8; PAKAID+Shank3 siRNA: 15.6 ± 0.5, n = 9, p > 0.05, ANOVA).
To find out whether PAK1 is specifically involved, we transfected neurons with mutant forms of PAK1 (Sells et al., 1997). We used a dominant-negative PAK1 (DN-PAK1), which has the mutations of H83L, H86L (making it unable to bind Rac1/cdc42), and K299R (making it catalytically inactive), and a constitutively active (CA-PAK1), which has the mutation of T423E (making it catalytically active). As shown in Figure 5, C and D, transfecting neurons with DN-PAK1 reduced NMDAR current density (pA/pF) and occluded the effect of Shank3 siRNA (DN-PAK1+control siRNA: 13.3 ± 1.6, n = 11; DN-PAK1+Shank3 siRNA: 12.5 ± 1.3, n = 7, p > 0.05, ANOVA). Conversely, transfecting neurons with CA-PAK1 blocked the effect of Shank3 siRNA on NMDAR current density (CA-PAK1+control siRNA: 31.2 ± 1.7, n = 9; CA-PAK1+Shank3 siRNA: 31.7 ± 1.8, n = 8, p > 0.05, ANOVA). Together, these data suggest that Shank3 regulation of NMDARs depends on Rac1/PAK1-mediated control of actin dynamics.
Next, we examined the impact of Shank3 knockdown on PAK1 activity. It has been shown that Thr423 is a critical phosphorylation site in the PAK1 activation process (Yu et al., 1998; Gatti et al., 1999; Zenke et al., 1999). Thus, we performed immunocytochemical staining of Thr423-phosphorylated PAK1. As shown in Figure 5, E and F, the intensity of p-PAK1 was significantly lower on the dendrites of neurons transfected with Shank3 siRNA (control siRNA: 227.2 ± 4.0, n = 22; Shank3 siRNA: 173.0 ± 5.9, n = 25, p < 0.01, t test), while the intensity of MAP2, a dendritic marker, was unchanged (control siRNA: 197.1 ± 5.6, n = 15; Shank3 siRNA: 198.7 ± 7.4, n = 24, p > 0.05, t test). It suggests that Shank3 knockdown leads to the reduced PAK1 activity.
To directly examine the effect of Shank3 knockdown on actin dynamics, we performed immunocytochemical staining of F-actin. As shown in Figure 6, A and B, Shank3 siRNA-transfected neurons exhibited a significant reduction of F-actin cluster density (# clusters/30 μm dendrite) (control siRNA: 15.4 ± 0.7, n = 26; Shank3 siRNA: control: 11.0 ± 0.4, n = 25, p < 0.01, t test) and F-actin cluster size (μm2) (control siRNA: 0.72 ± 0.04, n = 26; Shank3 siRNA: 0.51 ± 0.03, n = 25, p < 0.01, t test). Treating these cultures with TAT-PAK18 peptide (10 μm, 12–16 h) reduced F-actin clusters and occluded the effect of Shank3 knockdown (Fig. 6C,D, PAK18+control siRNA: 11.2 ± 0.5 clusters/30 μm, 0.61 ± 0.03 μm2, n = 28; PAK18+Shank3 siRNA: 12.2 ± 0.5 clusters/30 μm, 0.60 ± 0.03 μm2, n = 34, p > 0.05, t test). It suggests that Shank3 knockdown reduces filamentous actin via a mechanism involving PAK.
Figure 6.
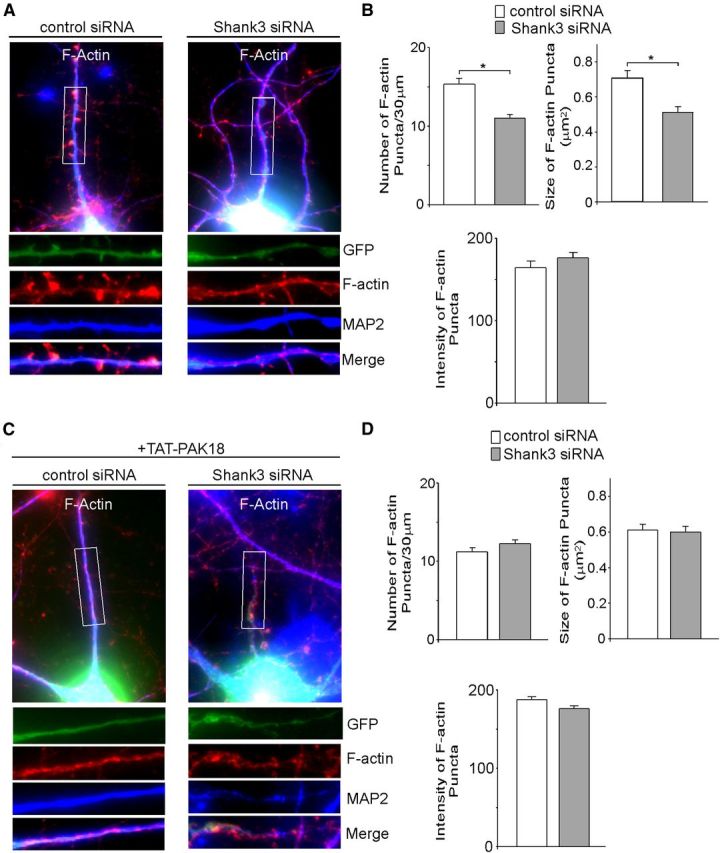
Shank3 knockdown reduces F-actin clusters, which is mimicked and occluded by PAK inhibitor. A, C, Immunostaining of F-actin (red) and MAP2 (blue) in cortical cultures (DIV 19–22) transfected with control siRNA or Shank3 siRNA (green) in the absence (A) or presence (C) of TAT-PAK18. B, D, Bar graph summary (mean ± SEM) showing the density, size, and intensity of F-actin puncta in control siRNA- or Shank3 siRNA-transfected neurons (GFP-positive) in the absence (B) or presence (D) of TAT-PAK18. *p < 0.01, t test.
Cofilin, the major actin depolymerizing factor, is required for Shank3 regulation of NMDAR currents.
Next, we examined the involvement of cofilin, an actin-binding protein whose activation depolymerizes actin filaments (dos Remedios et al., 2003). PAK, by phosphorylating cofilin via LIMK, inhibits its ability to depolymerize F-actin (Agnew et al., 1995). Since cofilin is inactivated by phosphorylation at Ser3 and reactivated by dephosphorylation (Morgan et al., 1993), a peptide consisting of 1–16 residues of cofilin with Ser3-phosphorylated (Aizawa et al., 2001; Zhou et al., 2004; Yuen and Yan, 2009) was used to serve as an inhibitor of endogenous cofilin. We treated cortical cultures with TAT-p-cofilin peptide, and examined the effect of Shank3 knockdown on NMDAR currents and F-actin clusters. As shown in Figure 7, A and B, TAT-p-cofilin peptide (10 μm, 12–16 h) blocked the ability of Shank3 siRNA to decrease NMDAR current density (pA/pF) (p-cofilin+control siRNA: 30.8 ± 1.1, n = 12; p-cofilin+Shank3 siRNA: 30.6 ± 1.0, n = 10, p > 0.05, ANOVA), which was in sharp contrast with the TAT control peptide (TAT+control siRNA: 26.8 ± 1.6, n = 4; TAT+Shank3 siRNA: 13.3 ± 1.4, n = 4, p < 0.01, ANOVA). Immunofluorescence data (Fig. 7C,D) also showed that treatment with the TAT-p-cofilin peptide prevented Shank3 siRNA-induced loss of F-actin clusters (p-cofilin+control siRNA: 16.5 ± 0.6 clusters/30 μm, 0.84 ± 0.04 μm2, n = 30; p-cofilin+Shank3 siRNA: 17.6 ± 0.7 clusters/30 μm, 0.78 ± 0.04 μm2, n = 29, p > 0.05, t test). Together, these results suggest that cofilin is an important component involved in Shank3 regulation of actin dynamics and NMDAR function.
Figure 7.
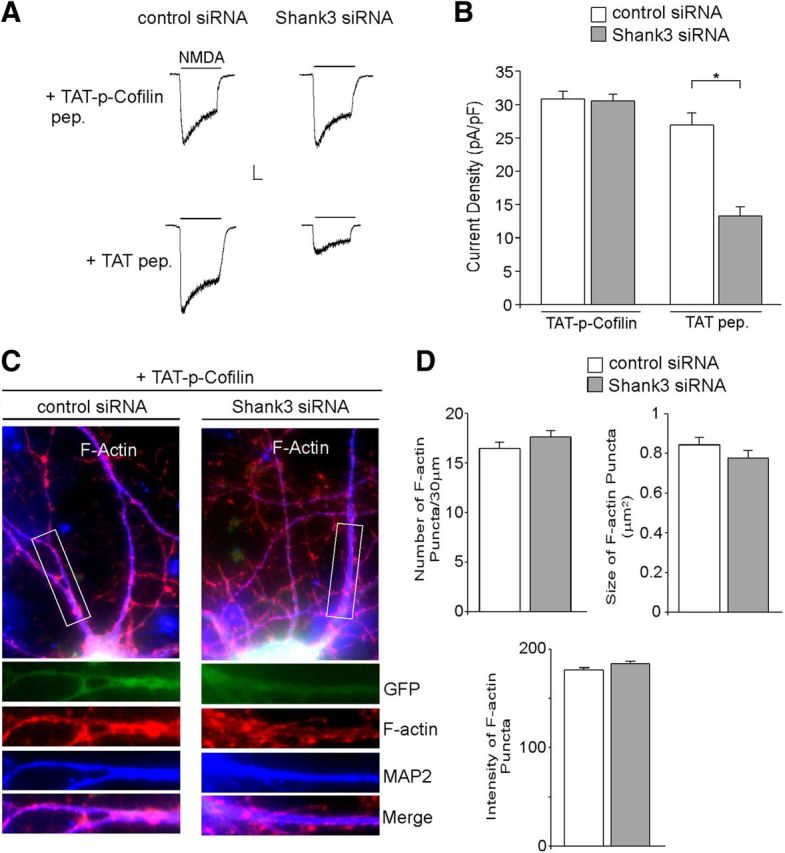
Inhibiting cofilin activity blocks the effects of Shank3 siRNA on NMDAR currents and F-actin. A, Representative NMDAR currents in cultured cortical neurons transfected with control siRNA or Shank3 siRNA (DIV 19–22) in the presence of a membrane-permeable cofilin inhibitory peptide, TAT-p-Cofilin (10 μm), or a TAT control peptide. B, Bar graph summary of NMDAR current density in control siRNA- or Shank3 siRNA-transfected neurons in the presence of TAT-p-Cofilin peptide or TAT control peptide. *p < 0.01, ANOVA. C, Immunostaining of F-actin (red) and MAP2 (blue) in cortical cultures transfected with control siRNA or Shank3 siRNA (green) treated with TAT-p-Cofilin peptide. D, Bar graph summary (mean ±SEM) showing the density, size, and intensity of F-actin puncta in control siRNA- or Shank3 siRNA-transfected neurons (GFP-positive) in the presence of TAT-p-Cofilin peptide.
Discussion
Alterations of glutamatergic synapses and excitatory transmission have been found in various ASD models expressing mutant or deleted Shank family proteins (Bozdagi et al., 2010; Wang et al., 2011; Schmeisser et al., 2012; Won et al., 2012). In this study, we have uncovered that Shank3 deficiency leads to the selective loss of NMDAR function and membrane trafficking through a mechanism involving the actin cytoskeleton and key regulators of actin dynamics. Understanding the mechanism through which Shank3 controls NMDARs will provide insights into the role of Shank3 in regulating NMDAR-dependent cognitive processes, which will help to identify key components that can restore NMDAR functions and mental capabilities in ASD conditions.
Actin is highly enriched in dendritic spines. Since the majority of excitatory synapses in the mammalian CNS are formed on dendritic spines, the remodeling and stability of actin is crucial to the formation and maintenance of glutamatergic synapses during development and at mature stages (Harris, 1999; Matus, 2000; Rao and Craig, 2000; Smart and Halpain, 2000; Hotulainen and Hoogenraad, 2010). The integrity of actin cytoskeleton is also critical for NMDAR channel activity and membrane delivery (Rosenmund and Westbrook, 1993; Allison et al., 1998), as well as the plasticity of NMDAR-mediated synaptic responses (Morishita et al., 2005). Shank proteins have been suggested to cross-link NMDAR/PSD-95 complexes to regulators of the actin cytoskeleton (Naisbitt et al., 1999). A recent study shows that the mutations of Shank3 identified in ASD affect the actin accumulation in synapses, therefore altering the dendritic spine development and growth cone motility (Durand et al., 2012). Consistently, our data suggest that Shank3 knockdown leads to the disruption of actin polymerization, resulting in the loss of NMDAR surface expression and channel functions.
In addition to the notion that the reduced NMDAR currents in Shank3-deficient neurons are mediated by a reduction in the number of surface NMDA receptors because of an actin-dependent change in their membrane trafficking, another possibility is that NMDAR function could be directly influenced by cytoskeletal interactions. It has been found that α-actinin (an actin-associating protein) binding to NMDARs decreases single NMDAR channel shut time, resulting in an increased open probability (Rycroft and Gibb, 2004). Expression of the mutant α-actinin reduces NMDAR currents and accelerates inactivation, which gives the idea that α-actinin supports NMDAR activity via tethering it to plasma membrane phospholipid (Michailidis et al., 2007). Thus, altering actin state could influence NMDAR function via multiple mechanisms.
A key player in the regulation of actin dynamics is Rac1 (Threadgill et al., 1997), a member of the family of Rho GTPases. Rho proteins, with RhoA, Rac1, and Cdc42 as the most studied members, act as molecular switches to orchestrate coordinated changes in the actin cytoskeleton essential for neurite outgrowth, spine development, and network formation (Nakayama et al., 2000; Negishi and Katoh, 2002; Ahnert-Hilger et al., 2004). Rac1 stimulates spine formation, dendrite initiation, elongation, and branching complexity (Kozma et al., 1997; Threadgill et al., 1997; Li et al., 2000; Wong et al., 2000). Emerging evidence indicates that aberrant Rac1 signaling results in abnormal neuronal connectivity and deficient cognitive functioning in humans (Ramakers, 2002; Newey et al., 2005). NMDA receptor stimulation is able to regulate Rac1-dependent actin remodeling, which is important for the development and structural remodeling of dendritic arbors and spines (Tolias et al., 2005; Hayashi-Takagi et al., 2010). Here, we show that NMDA receptor trafficking and function is also subject to the regulation by Rac1-controlled actin dynamics. These interactions not only link synaptic proteins to the actin cytoskeleton, but can also explain alterations in both synaptic plasticity and Rac signaling found in mental illness (Chen et al., 2010).
PAK, p21-activated kinase, is the key downstream effector of Rac1, which stimulates spine synapse formation and neurite outgrowth by facilitating actin filament assembly (Manser et al., 1994; Bokoch, 2003). Different mutations in the PAK genes have been identified in mental retardation cases (Allen et al., 1998; Bienvenu et al., 2000). Mice expressing a forebrain-specific dominant-negative form of PAK show fewer dendritic spines, altered spine morphology, and changes in synaptic strength (Hayashi et al., 2004). Shank proteins have been shown to form a complex with PAK and overexpression of Shank in cultured neurons promotes synaptic accumulation of PAK (Park et al., 2003). Consistently, we have found that Shank3 knockdown leads to reduced PAK1 activity. Moreover, inhibiting PAK1 decreases the basal NMDAR current, and the reducing effect of Shank3 knockdown on NMDARs and F-actin is occluded by PAK1 inhibitors and blocked by constitutively active PAK1. These data suggest that Rac1/PAK1-mediated actin dynamics is important for NMDAR membrane delivery/maintenance and its regulation by Shank3.
A major downstream effector of PAK is cofilin, an actin-binding protein essential for controlling the equilibrium between filamentous and monomeric actin (dos Remedios et al., 2003). Cofilin is inactivated by LIM kinase (a substrate of PAK)-mediated phosphorylation at Ser3, and is reactivated by Slingshot-mediated dephosphorylation (Agnew et al., 1995; Huang et al., 2006), providing a phosphoregulatory mechanism for actin reorganization (Bamburg, 1999; Huang et al., 2006). The dephosphorylated cofilin binds to F-actin, leading to actin severing and depolymerization (Morgan et al., 1993). Our previous studies have shown that cofilin-regulated actin dynamics is critically involved in the regulation of NMDAR trafficking and function (Mandal and Yan, 2009; Gu et al., 2012). Since the Ser3 residue of cofilin acts as a switch for actin assembly (F-actin stabilization) and disassembly (F-actin severing), we used a Ser3-phosphorylated cofilin peptide (Aizawa et al., 2001; Zhou et al., 2004) to inhibit the activation of endogenous cofilin. We have found that Shank3 siRNA-induced reduction of NMDAR currents and F-actin clusters is blocked by the cofilin inhibitor, suggesting that increased cofilin activity by Shank3 knockdown is responsible for the increased actin depolymerization and loss of NMDARs at the cell surface.
Based on our data, we suggest a model for the loss of NMDARs in Shank3-deficient conditions. Normally, Shank3 connects NMDARs to the actin cytoskeleton via intermediary elements (Naisbitt et al., 1999). Shank3 is located at the tip of actin filaments and enhances its polymerization (Durand et al., 2012). In neurons with the loss of Shank3, the reduced Rac1 and PAK activity results in the increased cofilin activity (due to reduced cofilin phosphorylation). Consequently, actin depolymerization is increased, leading to disrupted NMDAR membrane delivery through the actin cytoskeleton. Given the significance of NMDARs on cognition, our results provide a potential molecular basis for the behavioral deficits in Shank3 models of ASD (Bozdagi et al., 2010; Wang et al., 2011).
Footnotes
This work was supported by NIH Grants (R01-MH085774 and R21-MH101690) to Z.Y. We thank Xiaoqing Chen for her excellent technical support. We thank Drs. Carlo Sala and Eunjoon Kim for providing Shank3 plasmids.
The authors declare no competing financial interests.
References
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- Ahnert-Hilger G, Höltje M, Grosse G, Pickert G, Mucke C, Nixdorf-Bergweiler B, Boquet P, Hofmann F, Just I. Differential effects of Rho GTPases on axonal and dendritic development in hippocampal neurones. J Neurochem. 2004;90:9–18. doi: 10.1111/j.1471-4159.2004.02475.x. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I. Phosphorylation of cofilin by LIM-kinase is necessary for semaphoring 3A-induced growth cone collapse. Nat Neurosci. 2001;4:367–373. doi: 10.1038/86011. [DOI] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, McDonell N, Carrié A, Zemni R, Couvert P, Ropers HH, Moraine C, van Bokhoven H, Fryns JP, Allen K, Walsh CA, Boué J, Kahn A, Chelly J, Beldjord C. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am J Med Genet. 2000;93:294–298. doi: 10.1002/1096-8628(20000814)93:4<294::AID-AJMG8>3.0.CO%3B2-F. [DOI] [PubMed] [Google Scholar]
- Böckers TM, Mameza MG, Kreutz MR, Bockmann J, Weise C, Buck F, Richter D, Gundelfinger ED, Kreienkamp HJ. Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J Biol Chem. 2001;276:40104–40112. doi: 10.1074/jbc.M102454200. [DOI] [PubMed] [Google Scholar]
- Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins—a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem. 2002;81:903–910. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15. doi: 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GC. Glutamate receptor dysfunction and drug targets across models of autism spectrum disorders. Pharmacol Biochem Behav. 2012;100:850–854. doi: 10.1016/j.pbb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci. 2010;30:10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton SM, Haroutunian V, Davis KL, Meador-Woodruff JH. Altered transcript expression of NMDA receptor-associated postsynaptic proteins in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2003;160:1100–1109. doi: 10.1176/appi.ajp.160.6.1100. [DOI] [PubMed] [Google Scholar]
- Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, Peterson JR. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, Nosworthy NJ. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol Rev. 2003;83:433–473. doi: 10.1152/physrev.00026.2002. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17:71–84. doi: 10.1038/mp.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Synapse structure: glutamate receptors connected by the shanks. Curr Biol. 1999;9:R848–R850. doi: 10.1016/S0960-9822(00)80043-3. [DOI] [PubMed] [Google Scholar]
- Gatti A, Huang Z, Tuazon PT, Traugh JA. Multisite autophosphorylation of p21-activated protein kinase gamma-PAK as a function of activation. J Biol Chem. 1999;274:8022–8028. doi: 10.1074/jbc.274.12.8022. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Fu AK, Ip NY, Yan Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci. 2005;25:4974–4984. doi: 10.1523/JNEUROSCI.1086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Liu W, Wei J, Yan Z. Regulation of NMDA receptors by metabotropic glutamate receptor 7. J Biol Chem. 2012;287:10265–10275. doi: 10.1074/jbc.M111.325175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/S0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- Kozma R, Sarner S, Ahmed S, Lim L. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol. 1997;17:1201–1211. doi: 10.1128/mcb.17.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P, Barnier JV. PAK signalling in neuronal physiology. Cell Signal. 2009;21:384–393. doi: 10.1016/j.cellsig.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lei S, Czerwinska E, Czerwinski W, Walsh MP, MacDonald JF. Regulation of NMDA receptor activity by F-actin and myosin light chain kinase. J Neurosci. 2001;21:8464–8472. doi: 10.1523/JNEUROSCI.21-21-08464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SE, Giarelli E, Lee LC, Schieve LA, Kirby RS, Cunniff C, Nicholas J, Reaven J, Rice CE. Autism spectrum disorder and co-occurring developmental, psychiatric, and medical conditions among children in multiple populations of the United States. J Dev Behav Pediatr. 2010;31:267–275. doi: 10.1097/DBP.0b013e3181d5d03b. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3:217–225. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenic drug; sernyl. AMA Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- Mandal M, Yan Z. Phosphatidylinositol (4,5)-bisphosphate regulation of N-methyl-d-aspartate receptor channels in cortical neurons. Mol Pharmacol. 2009;76:1349–1359. doi: 10.1124/mol.109.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Maruta H, He H, Nheu T. Interfering with Ras signaling usingmembrane-permeable peptides or drugs. Methods Mol Biol. 2002;189:75–85. doi: 10.1385/1-59259-281-3:075. [DOI] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Michailidis IE, Helton TD, Petrou VI, Mirshahi T, Ehlers MD, Logothetis DE. Phosphatidylinositol-4,5-bisphosphate regulates NMDA receptor activity through alpha-actinin. J Neurosci. 2007;27:5523–5532. doi: 10.1523/JNEUROSCI.4378-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, Scherer SW. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Moskal JR, Burgdorf J, Kroes RA, Brudzynski SM, Panksepp J. A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci Biobehav Rev. 2011;35:1982–1988. doi: 10.1016/j.neubiorev.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Murata Y, Constantine-Paton M. Postsynaptic density scaffold SAP102 regulates cortical synapse development through EphB and PAK signaling pathway. J Neurosci. 2013;33:5040–5052. doi: 10.1523/JNEUROSCI.2896-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/S0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi M, Katoh H. Rho family GTPases as key regulators for neuronal network formation. J Biochem. 2002;132:157–166. doi: 10.1093/oxfordjournals.jbchem.a003205. [DOI] [PubMed] [Google Scholar]
- Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–19229. doi: 10.1074/jbc.M301052200. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–199. doi: 10.1016/S0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Signaling between the actin cytoskeleton and the postsynaptic density of dendritic spines. Hippocampus. 2000;10:527–541. doi: 10.1002/1098-1063(2000)10:5<527::AID-HIPO3>3.0.CO%3B2-B. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-Y. [DOI] [PubMed] [Google Scholar]
- Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci. 2005;25:3560–3570. doi: 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycroft BK, Gibb AJ. Regulation of single NMDA receptor channel activity by alpha-actinin and calmodulin in rat hippocampal granule cells. J Physiol. 2004;557:795–808. doi: 10.1113/jphysiol.2003.059212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, Balschun D, Skryabin BV, Dieck S, Smalla KH, Montag D, Leblond CS, Faure P, Torquet N, Le Sourd AM, Toro R, Grabrucker AM, Shoichet SA, Schmitz D, Kreutz MR, Bourgeron T, Gundelfinger ED, Boeckers TM. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/S0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113:1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–554. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO%3B2-7. [DOI] [PubMed] [Google Scholar]
- Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron. 1997;19:625–634. doi: 10.1016/S0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Verpelli C, Dvoretskova E, Vicidomini C, Rossi F, Chiappalone M, Schoen M, Di Stefano B, Mantegazza R, Broccoli V, Böckers TM, Dityatev A, Sala C. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J Biol Chem. 2011;286:34839–34850. doi: 10.1074/jbc.M111.258384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, Lorenzo I, Wu G, Weinberg RJ, Ehlers MD, Philpot BD, Beaudet AL, Wetsel WC, Jiang YH. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011;20:3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- Wong WT, Faulkner-Jones BE, Sanes JR, Wong RO. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. J Neurosci. 2000;20:5024–5036. doi: 10.1523/JNEUROSCI.20-13-05024.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zou H, Strong JA, Yu J, Zhou X, Xie Q, Zhao G, Jin M, Yu L. Bimodal effects of MK-801 on locomotion and stereotypy in C57BL/6 mice. Psychopharmacology. 2005;177:256–263. doi: 10.1007/s00213-004-1944-1. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Yu JS, Chen WJ, Ni MH, Chan WH, Yang SD. Identification of the regulatory autophosphorylation site of autophosphorylation-dependent protein kinase (auto-kinase). Evidence that auto-kinase belongs to a member of the p21-activated kinase family. Biochem J. 1998;334:121–131. doi: 10.1042/bj3340121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Yan Z. Dopamine D4 receptors regulate AMPAR trafficking and glutamatergic transmission in GABAergic interneurons of prefrontal cortex. J Neurosci. 2009;29:550–562. doi: 10.1523/JNEUROSCI.5050-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Yan Z. Cellular mechanisms for dopamine D4 receptor-induced homeostatic regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J Biol Chem. 2011;286:24957–24965. doi: 10.1074/jbc.M111.221416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psy. 2011;16:156–170. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–977. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–32573. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- Zhan Q, Ge Q, Ohira T, Van Dyke T, Badwey JA. p21-activated kinase 2 in neutrophils can be regulated by phosphorylation at multiple sites and by a variety of protein phosphatases. J Immunol. 2003;171:3785–3793. doi: 10.4049/jimmunol.171.7.3785. [DOI] [PubMed] [Google Scholar]
- Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with longterm depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zou H, Zhang C, Xie Q, Zhang M, Shi J, Jin M, Yu L. Low dose MK-801 reduces social investigation in mice. Pharmacol Biochem Behav. 2008;90:753–757. doi: 10.1016/j.pbb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]