

Abstract
Impediments to DNA access due to assembly of the eukaryotic genome into chromatin are in part overcome by the activity of ATP-dependent chromatin-remodeling complexes. These complexes employ energy derived from ATP hydrolysis to destabilize histone—DNA interactions and alter nucleosome positions, thereby increasing the accessibility of DNA-binding factors to their targets. However, the mechanism by which theses complexes accomplish this task remains unresolved. We review aspects of nucleosome alteration by the SWI/SNF complex, the archetypal remodeling enzyme. We focus on experiments that provide insights into how SWI/SNF induces nucleosome movement along DNA. Numerous biochemical activities have been characterized for this complex, all likely providing clues as to the molecular mechanism of translocation.
Keywords: chromatin, DNA structure, gene expression, remodeling enzymes, SWI/SNF
Strategies to regulate chromatin accessibility
The chromosomes of eukaryotic cells have the remarkable ability to condense and organize their genetic material and control access to genetic information. Chromosomes are comprised of chromatin, a multifaceted and hierarchical nucleoprotein complex containing both histones and non-histone proteins. The primary structural unit of chromatin is the nucleosome, which consists of a nucleosome core and linker DNA. The nucleosome core is comprised of ~147 bp of DNA wrapped in ~1¾ left-handed superhelical turns around a octameric structure containing two molecules each of the core histones H2A, H2B, H3, and H4.[1] Cores are connected by 10–90 bp of linker DNA, which is not in tight association with the core histones.[2] Strings of nucleosomes constitute a primary arrangement that appears as an extended “beads-on-a-string” structure when viewed in low-salt (non-physiological) conditions by electron microscopy.[3,4] However, in solutions containing physiological ionic strengths, strings of nucleosomes spontaneously fold and condense into higher-order secondary chromatin structures such as the 30 nM diameter chromatin fiber[2, 5] and these further condense into tertiary structures such as the ~350 nM chromonema fibers[6] and perhaps even higher-order structures in the interphase nucleus. Chromatin fibers and other higher-order structures are considerably stabilized by the interaction of a single linker histone (typically H1 or H5) with the nucleosome core and linker DNA.[5, 7, 8]
The packaging of DNA by chromatin has a repressive effect on a variety of cellular processes such as replication, DNA repair, and gene transcription due to the reduced access of trans-acting factors to the DNA. In this regard, aspects of eukaryotic chromatin structure have been functionally integrated into basic regulatory processes. For example, nucleosomes are required to fully repress inducible genes in yeast cells in the absence of effectors.[9] Reduced DNA access in chromatin results both from the wrapping of DNA into nucleosomes and from the folding of nucleosome arrays into higher-order structures.[10] Not surprisingly, cells have devised several strategies to overcome the barriers of chromatin structure. These include directly (thermodynamically) competing with histone proteins for binding DNA, incorporation of histone post-translational modifications, and histone variants to allow facile disruption of chromatin and enzymatic disruption of chromatin, driven principally by dedicated “chromatin remodeling” complexes.
Far from being static structures, nucleosomes possess dynamic features in vivo and in vitro. For example, nucleosome DNA undergoes spontaneous unwrapping/rewrapping at rates and with probabilities that are likely relevant to in vivo processes.[11, 12] Thus biologically significant DNA binding by transacting factors can occur to nucleosome DNA and result in displacement of histone–DNA interactions (in the absence of extraneous activities) if concentrations of factors and binding free energies are sufficient to overcome the ~103–105 probability of DNA exposure within the nucleosome.[13] Moreover, multiple binding sites within the same nucleosome might lead to cooperative binding of otherwise unrelated factors.[14] In addition, the salt-dependent folding of nucleosome arrays can lead to significant reductions in the accessibility of binding sites within linker DNA but does not appear to significantly alter site exposure within the nucleosome core.[10] Thus, at its most basic level, chromatin primarily represents a thermodynamic barrier to DNA access that might be overcome without further modification to the structure.
The core histones are targets for numerous post-translational modifications (PTMs), including acetylation, methylation, phosphorylation, and ubiquitylation, many of which have been directly linked with specific chromatin-associated processes.[15] For example, increased levels of acetylation of specific lysine residues within the core histones, catalyzed by histone acetyl-transferases (HATs) and removed by histone deacetylases (HDACs), are found in transcriptionally active regions whereas histones are nominally hypoacetylated in regions of chromatin that are transcriptionally silent. Although the majority of PTMs, including acetylation, occur within the tail domains, these modifications—or indeed the presence of the tail domains themselves—appear to have only modest effects on the stability and dynamic properties of individual nucleosomes.[16–19] However, some histone PTMs have been found at strategic sites within the histone fold domains so as to directly alter the stability of DNA wrapping within the nucleosome.[20–22] Moreover, nucleosomes containing combinations of the H3 variant H3.3 and the H2A variant H2A.Z have been reported to exhibit increased salt sensitivity that might be important for maintenance of transcriptionally active loci in vivo.[23]
Acetylation appears to have a much greater impact on the stability of higher-order chromatin structures than on individual nucleosomes. Hansen and colleagues demonstrated that an increase in acetylation of only ~6 acetyl groups per nucleosome resulted in a loss of the ability of 12-mer nucleosome arrays to fold in Mg+2-containing buffers and a 15-fold increase in array transcription by RNA polymerase III.[24] Rather than simply weakening or eliminating histone-tail–DNA interactions, acetylation might alter both structure and interactions of the core histone tail domains that are important for higher-order structures.[25–27] For example, acetylation of lysine 16 in the H4 tail domain severely diminishes salt-dependent folding and condensation of nucleosome arrays, presumably due to disruption of an interaction between the H4 tail domain and a charged pocket on the H2A/H2B surface of a neighboring nucleosome.[28] Interestingly, acetylation within the H2B and H4 tail domains appears to most drastically alter the formation of higher-order tertiary chromatin structures as measured by the propensity of model nucleosome arrays to undergo self-association in vitro.[29]
In addition to direct effects on nucleosome and higher-order chromatin structure, distinct histone post-translational modifications can act sequentially or in combination to form a “his-tone code” that is recognized by effector proteins to indirectly regulate chromatin structures. For example, di- and tri-methylated lysine 9 of H3 is recognized and bound by the chromo-domain of the protein HP-1 or homologues and stimulates formation of repressive heterochromatin. On the other hand, methylation of H3 lysine 4 is generally associated with actively transcribed loci and recruits PHD-domain-containing proteins that function at such loci.[30] Often these effectors bring activities such as HATs to directly affect the structure or to recruit other factors via a variety of specific histone PTM recognition motifs.[31]
Eukaryotic cells also harbor chromatin-remodeling complexes that disrupt chromatin structure to increase access to the underlying DNA. These complexes are typically comprised of multiple subunits and use energy derived from ATP hydrolysis to distort nucleosome structure, mobilize nucleosomes, and possibly to alter higher-order structures. It is important to point out that whereas some remodelers alter chromatin structure to make specific genes more accessible for transcription machinery, others clearly play a role in transcriptional repression.[32, 33] Related complexes are also responsible for the deposition of histone variants whereas others function in other processes including DNA repair and DNA replication.[34–36] There are at least 4 classes of ATP-dependent chromatin remodelers that have been defined depending on the presence of conserved sequence motifs other than the ATPase domain belonging to the SNF2 subfamily of DNA helicases/ATPases: SWI/SNF, ISWI, NURD/Mi-2/CHD, INO80, and SWR1 (for an excellent review, see ref. [36]). This review focuses on the SWI/SNF family of complexes.
Basic properties of the SWI/SNF family of nucleosome-remodeling complexes
The SWI/SNF complex was the first biochemically characterized ATP-dependent chromatin-remodeling complex; it was originally purified from Saccharomyces cerevisiae[37] and human[38] cells. Genes encoding components within the complex were described as non-essential but required for the activation of genes involved in mating-type switching (SWI) and for sucrose fermentation (SNF: sucrose non-fermenting).[39] Yeast SWI/SNF is present at a relatively low abundance, about 100–200 molecules per cell[37] and a genome-wide analysis showed that about 5 % of yeast genes are regulated by SWI/SNF. A related SWI/SNF subfamily member in yeast that remodels the structure of chromatin (RSC) has also been characterized. Compared to ySWI/SNF, RSC is ~10 times more abundant and is essential for viability.[40] Whereas ySWI/SNF is a regulator of transcription and mitosis, RSC functions in transcriptional activation and chromosome segregation. Both complexes play distinct roles in DNA repair.[34, 41–43]
SWI/SNF purified from both yeast and human cells exhibits DNA-stimulated ATPase activity, but, despite homology to the family of DNA helicases, does not exhibit detectable helicase activity.[37, 38] The yeast SWI/SNF complex is 1.14 MDa and contains 12 subunits.[44] The Swi2p/Snf2p subunit is the primary catalytic component and harbors an ATPase domain, which contains seven ATPase/helicase subdomains. Structural studies indicate that these subunits are split into two parts: an N-terminal RecA-like DExx (I, Ia, II, and III) module, that plays role in ATP binding and hydrolysis, and a C-terminal HELICc (IV, V, and VI) module, that functions in DNA translocation (Figure 1).[36] These two motifs are separated by a short insertion. The cleft formed by these two motifs can bind ATP and drive translocation of the complex on DNA coupled to ATP hydrolysis (see below). Motif V is required to couple the energy of ATP hydrolysis to the remodeling activity of SWI/SNF as shown by a deletion of eight residues within this motif.[45] The related RSC complex in yeast contains the SNF2-orthologue Sth1, and has similar domain structure (Figure 1).
Figure 1.
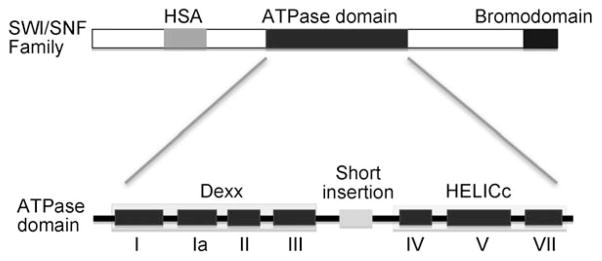
Schematic of subunit organization of SWI/SNF family. All SWI/SNF family members contain an ATPase domain, which is flanked by a HSA domain (light gray) at the N-terminal and a bromodomain (dark gray) at the C-terminal. The ATPase domain is comprised of seven ATPase/Helicase subdomains (I–VI) that are structurally split in two parts: Dexx and HELICc. Between motifs III and VI, there is a characteristic short insertion (light gray).
In mice and humans, there are two homologues of SWI/SNF, BAF (BRG1 associated factors) and PBAF (polybromo associated factors), which contain BRG1 and BRM (Brahma) as the ATPase domains, respectively.[36] In Drosophila two related SWI/SNF family members have been identified, BAP (Brahma Associated Proteins) and PBAP (polybromo associated proteins) that use the same ATPase subunit Brahma. What distinguishes SWI/SNF from other remodelers is that Swi2p/Snf2 also includes an HSA (helicase-SANT) domain and a post-HSA domain at the N-terminal flanking region, which contains a bromodomain within the C-terminal region.[36] Recent co-IP experiments indicate that the HSA domain is the platform for the binding of nuclear ARP–ARP (ySWI/SNF and RSC) or ARP-actin (human) modules. This ARP–HSA module interacts with post-HSA and protrusions 1 domain, which is a part of the insertion between motifs III and IV, to regulate the ATPase domain.[36, 46] The C-terminal bromodomain in the ATPase can recognize specific acetylated histone residues in histone tail-domains.[47–49] Acetylated H3Lys14 is specifically recognized by the bromodomain of RSC and recruits the complex for gene activation. Interestingly, RSC, PBAP and PBAF contain multiple bromodomains in contrast to a single domain in ySWI/SNF, which indicates the function for cooperatively recognizing several acetylated lysine residues during chromatin remodeling.[50] Direct interaction between SWI/SNF and gene-specific transcriptional factors can recruit SWI/SNF to target genes both in vitro and in vivo.[51]
Multiple biochemical activities have been described for the SWI/SNF complex, including disruption of bona fide nucleosome structure,[52, 53] DNA translocation and sliding of the his-tone octamer along the DNA in cis,[54–56] formation of novel disomic structures from monosomes,[57] ejecting the histone octamer to expose DNA,[58] displacement of H2A–H2B dimers,[59] and generating changes in DNA topology,[60] among others. The isolated Swi2p–Arp7–Arp9 complex shows equivalent ATPase and remodeling activity as intact SWI/SNF except that it no longer has H2A–H2B dimer-displacement activity.[61] Swi3p has a conserved SANT domain, which was first identified as a histone tail interaction domain.[62] SANT domains are found in several remodelers such as RSC and ISWI, as well as in histone-modifying enzymes such as Ada2p, Rsc8, and Gcn5p-containing HAT complexes. Recent data show that the N-terminal acidic domain of Swi3p is a H2A–H2B binding site and plays an essential role in histone dimer loss.[61] The SANT domain also has a crucial function in SWI/SNF assembly because a deletion within this domain results in SWI/SNF disassembly in vitro.[61, 62] Snf5 and Swi1p are partially functionally redundant and required for the recruitment of SWI/SNF to specific target genes by interaction with gene-specific activators.[63]
Overall structure of SWI/SNF
The three-dimensional structure of the ySWI/SNF complex was first characterized by the Peterson and Woodcock groups by electron microscopy (EM). The analysis showed that six protein lobes form a conical cavity on the surface of the complex, about 15 nm in width and 5 nm in depth, presumably creating an excellent binding pocket for a nucleosome.[44] Recently, a more detailed structure analysis by Bartholomew and colleagues by using cryo-EM (cryoelectron microscopy) revealed that ySWI/SNF contains a large asymmetric trough region on its surface with a contour appropriate for accommodating a mononucleosome; this is consistent with the results of DNA footprinting when SWI/SNF is targeted to the nucleosome through Gal4-VP16.[64] In this model, the nucleosome has an extensive interaction with SWI/SNF; it binds closely to the high wall of this trough, which results in one entire gyre of DNA interacting with the complex, providing protection from DNA cleavage, whereas the other half of the DNA faces toward the low wall of SWI/SNF, resulting in greater accessibility. Site-directed DNA crosslinking data show that the catalytic ATPase motor interacts with the nucleosome two helical turns from the dyad axis, which is supported by a series of experiments showing that the presence of DNA gaps at SHL2 prevents nucleosome movement catalyzed by remodelers (see below).[56, 65] Whereas this model provides some insights for the mechanism of nucleosome sliding, confirmation awaits a high-resolution structure of nucleosome-remodeler co-complex.
Interestingly, structural studies employing EM and OTR (orthogonal tilt reconstruction) methods reveal that the RSC complex, which shares general features with human PBAF, also contains a large and deep central cavity surrounded by two “long arms” and a lid that might embrace a single nucleosome. Due to conformational changes of the protein arms, “open” and “closed” conformers are resolved, which might reflect different states of either nucleosome interaction or remodeling. The contour of a nucleosome fits well into the cavity of either the open or closed conformation with the DNA entry/exit sites and nucleosome dyad facing out of the cavity. This RSC structure and conformation change is further demonstrated by recent cryo-EM studies of a RSC–nucleosome complex.[66] In this complex, the Sth1 subunit binds to the nucleosome at a location close to the dyad and destabilizes the structure, resulting in an overall rearrangement of histone–DNA interactions structure. This observation is consistent with the reduction of the histone–DNA interactions at this location detected by DNase I digestion, which indicates that DNA might form a small loop that will induce nucleosome sliding.
SWI/SNF–nucleosome interactions
A recent single-molecule DNA unzipping analysis provides a high-resolution view of histone–DNA interactions in the nucleosome.[67] Interestingly, this analysis detects contributions to histone binding from each of the two strands independently, and shows the strongest contacts around the nucleosome dyad, and two relatively weaker regions of contacts about 50 to 60 bp to each side of dyad. Conversely, some of the weakest histone–DNA interactions are detected at SHL2.5, about 25 bp to either side of the nucleosome dyad. Importantly, this site overlaps with a region centered about two turns from the dyad where several related ATP-dependent remodeling complexes make contacts with DNA that are critical for nucleosome mobilization.[56, 68] Moreover, the location of these interactions is consistent with hydroxyl radical footprinting of a SWI/SNF-nucleosome complex, which indicates that the complex contacts an entire turn of DNA and linker DNA, with protection of the DNA beginning 1–2 turns from the dyad, and extending well into the extra-nucleosomal DNA.[64] The interaction of SWI/SNF in this study was directed and oriented via recruitment by Gal4–VP16, thus allowing details of the structure to become apparent that would otherwise be obscured by random binding to either side of the nucleosome. Interestingly, SWI/SNF contacts sites on the side of the nucleosome distal from the Gal4 site; this suggests a bridging interaction with an extended portion of the complex. As mentioned above, these results are consistent with one half of the nucleosome sitting down into a pocket on the surface of the SWI/SNF complex.[64]
Mechanistic studies of nucleosome remodeling
Early studies show that nucleosome remodeling occurs without gross changes in either histone protein stoichiometry or configuration,[69–71] and indeed nucleosome substrates in which histones were cross-linked together appears to be remodeled in a facile manner.[71, 72] Although it is clear that histone–DNA interactions are altered during remodeling, the exact details remain unclear. A comparison of nucleosomes assembled with end-labeled versus body-labeled DNAs suggested that even though the register of specific DNA contacts to histones is altered, the DNA remains more or less in association with the histone surface.[73] By using a site-directed histone→DNA crosslinking approach, the Hayes group demonstrated that cross-linking does not significantly inhibit hSWI/SNF remodeling activity as monitored by DNase I footprinting.[74] However, a single crosslink between the DNA and the histone surface drastically reduced SWI/SNF-induced increases in the accessibility of the nucleosome DNA to restriction enzymes—even at sites distant from the crosslink—this indicates that hSWI/SNF remodeling requires transient global disruption of histone–DNA interactions.[74] This result is consistent with other in vitro studies that show that SWI/SNF alters nucleosome structure by at least transiently disrupting histone–DNA interactions.[52, 72]
A significant clue as to the mechanism of ATP-dependent remodeling was brought to light in studies by the Cairns, Peterson, and Owen-Hughes’ groups. Because the helicase-related domain of the Snf2 subunit did not exhibit classic helicase strand-displacement activity, these groups suspected that SWI/SNF and related complexes might exhibit a translocase activity, and that such an activity might result in an increase in super-helical torsion in the DNA.[75] Indeed, the Owen-Hughes group found that purified SWI/SNF complex (as well as several other remodeling complexes) induced detectable torsional stress in naked DNA fragments in an ATP-dependent manner, which was detected as extrusion of cruciform structures. These results imply that not only can SWI/SNF induce torsional stress in DNA, but also that the complex must contact DNA at a distal site from the ATPase domain to create a topologically isolated domain in which the stress can be transiently contained.[75] In a separate study, the Cairns group showed that the related RSC complex does indeed exhibit DNA translocase activity coupled to ATP hydrolysis, with a processivity limit of about 70 bp.[76] Interestingly, Peterson and co-workers demonstrated that nucleosomes within topologically constrained minicircles were refractory to remodeling as determined by restriction enzyme accessibility.[60] Moreover, they found that the disruption of his-tone–DNA interactions is nucleosome-specific; no such disruption was observed when non-specific histone–DNA complexes were confronted with the SWI/SNF complex.[60] These results imply that SWI/SNF and related complexes specifically bind to nucleosomes and employ torsional stress within the DNA of the nucleosome substrate to remodel nucleosome structure. A model emerges whereby the SWI/SNF complex contacts at least two sites in nucleosomes: a static anchoring interaction, and a site that translocates along the helix, pumping superhelical stress into the DNA to disrupt histone–DNA interactions.
Though the exact manner by which SWI/SNF-dependent torsional stress results in increased access to nucleosomal DNA remains unclear, data suggest that that remodeling results in the generation of a bona fide structurally altered nucleosome intermediate. Quantitative restriction enzyme analysis of remodeling by the BRG1 subunit suggests that short stretches of DNA sites within the nucleosome are transiently released from histone–DNA interactions and available for trans-acting factor binding.[52] The data indicate that multiple distinct remodeled species are generated by a mechanism that is not easily explained by simple uncoiling of DNA from the edges of the nucleosome or nucleosome sliding and that these species are in-terconverted by the enzyme. Although the stability and lifetime of these structurally altered states have been the subject of debate,[77, 78] current data suggest that such intermediates rapidly decay into a bona fide nucleosome that encompasses a different segment of DNA than the original nucleosome, whereas other remodelers appear to produce a “moved” nucleosome with even more fleeting intermediates.[55] Therefore, a primary outcome of ATP-dependent remodeling by SWI/SNF, RSC and other remodelers is the movement or mobilization of nucleosomes along the DNA.[54, 55] Such mobilization would stably expose sites previously blocked by association with the histone octamer. Thus, much of the work regarding ATP-dependent remodeling has focused on the mechanism of nucleosome mobilization.
Several crystal structures of nucleosome core particles suggest that nucleosome movement or mobilization could occur by a “twist-diffusion” mechanism.[79, 80] Within the nucleosome, strong sites of interactions occur approximately once every 10 bp of DNA, such that essentially each 10 bp stretch is, to an extent, topologically isolated. The twist diffusion mechanism posits that a DNA twist “defect” containing ±1 base pairs compared to the nominal helical twist can be accommodated within any 10 bp stretch of DNA. If the defect diffused through the entire length of the nucleosome, the histone octamer would be advanced along the DNA by one base pair. This model has the attribute that the majority of high-energy his-tone–DNA interactions would be preserved at any one moment in time, while the DNA screws along its long axis, along the ‘grooves’ in the surface of the histone octamer. However, the one-step nature of the advancement would require a complete rotation of the DNA on the nucleosome surface and thus requires passage through a strongly disfavored rotational orientation for nucleosome positioning sequences. Nevertheless, it is likely that the free energy derived from ATP hydrolysis would more than overcome this barrier.
To test whether a twist-diffusion mechanism is responsible for ATP-dependent remodeler-induced nucleosome mobilization, Aoyagi and co-workers placed branched DNA structures in the center of a nucleosome and measured the extent and rate of remodeling by the human SWI/SNF complex and the Xenopus Mi-2 complex.[81, 82] If a pure twist diffusion mechanism is used by the remodelers for nucleosome mobilization, it is likely that the steric bulk of the branched DNA structures would interfere or block movement of the histone octamer along the DNA. However, both enzymes were unaffected by either single or double-stranded branched structures in the center of the nucleosome, strongly indicating that this mechanism does not account for mobilization. A similar result was obtained by Längst and co-workers in which the attachment of streptavidin-bound beads to the center of the nucleosome DNA did not inhibit nucleosome mobilization by the ACF complex.[83] It is interesting to note that branched DNA structures placed outside of the nucleosome can hinder SWI/SNF-dependent mobilization in cis, perhaps due to steric interference with binding of the enzyme to the DNA.[54]
The dynamic process of spontaneous unwrapping of DNA from the edges of the nucleosome[12] also has been hypothesized to be related to the ATP-dependent remodeling and nucleosome-mobilization process. Upon uncoiling, rewrapping, or capturing the end of the nucleosome DNA at a position ~10n base pairs beyond the original point would lead to a loop of ~10n bp. This loop could then be propagated through the nucleosome without further net loss of histone–DNA contacts. Whereas a tenable and attractive model, only modest direct evidence for the existence of a DNA loop during nucleosome mobilization has been obtained. By using a single-molecule “molecular tweezers” approach, Bustamante, Peterson, and colleagues showed that RSC and SWI/SNF translocate DNA in a nucleosome-dependent fashion, likely by forming large internal loops within the nucleosomes.[84] Several labs have attempted to measure the “step size” — the size of one continuous movement of the nucleosome — that might be related to the size of the loop.[83, 85] Crosslinking results, mentioned above, indicating that remodeling as measured by restriction enzyme accessibility assays requires transient global disruption of his-tone–DNA interactions are also consistent with the loop-recapture mechanism.[74] In addition, the Längst group employed a novel assay exploiting the preferential intercalation of ethidium bromide to naked DNA to provide evidence for a region free of histone–DNA interactions within the nucleosome during ACF nucleosome remodeling.[83] Moreover, a recent cryo-electron microscopy and biochemical study of RSC-remodeled mononucleosomes provides evidence for remodeling intermediates containing internal loops.[53] In addition, these workers found that remodeling intermediates were not yet moved from their original position but contained more DNA than canonical nucleosomes. A similar result was recently obtained by Bartholomew and colleagues by precisely mapping histone–DNA interactions by using a DNA methylase protection assay known as MapIT, which provides single-molecule information[86] and revealed a subset of SWI/SNF remodeled products that exhibited protection of more than one nucleosome’s worth of DNA.[58] These intermediates might have been brought about by a loop-recapture mechanism, but other possibilities exist (see below).
A simple model for remodeling would thus be that a translocating enzyme encounters a dynamic nucleosome and peels off DNA as it proceeds around the structure, disrupting his-tone–DNA interactions and exposing sites to trans-acting factors. However, recent work provides evidence that the primary site of the translocation of SWI/SNF along DNA is actually located within the nucleosome, about two helical turns from the dyad axis. Two studies pinpointed the sites of interaction of SWI/SNF and RSC, respectively, by using gapped DNA substrates, which are known to block progress of translocases along DNA.[55, 56] These workers showed that translocation is blocked when the gaps reach the site ~two helical turns from the dyad; the effect is directional, such that translocation in the opposite direction is not affected.[55, 56] These results are consistent with footprinting results and for the smaller ISWI complex, which contacts nucleosome DNA at approximately the same site and also is affected similarly by gaps in the DNA.[56]
Thus a model emerges whereby DNA is drawn into the nucleosome by the translocase activity, which engages the nucleosome internally, to form some sort of distorted structure, perhaps with an internal bulge or loop induced by movement of the DNA-binding domain (Figure 2). The loop could form stochastically, being trapped by binding of the enzyme, or perhaps the translocase activity draws DNA in from the nucleosome edge by forcing movement of the DNA-binding domain (Figure 2, steps A and B). The loop is then free to translocate around the perimeter of the nucleosome to advance the DNA (steps C, D and F) or the loop DNA might be drawn further into the nucleosome by the translocase domain (steps E and F). Interestingly, very recent results from the Dimitrov group have documented an intermediate that arises during nucleosome remodeling by RSC, which they have dubbed the “Remo-some” (Figure 2, inset). In this intermediate the histone octa-mer is not yet moved along the DNA but AFM and cryo-EM work clearly show that the remosomes contain apparently randomly oriented internal loops and harbor more DNA than a typical nucleosome.[53] A basis for leverage against which the translocase activity works is provided by the ultrastructural results, which indicate that a significant portion of the remodeled nucleosomes is grasped by the remodeling complex. Interestingly, a requirement for DNA bulges in nucleosome remodeling or mobilization has not yet been established. Hopefully, in the future, probes capable of detecting internal DNA loops in nucleosomes or bulged regions will be devised, allowing the role of bulges to be investigated. It should be noted that exactly how the translocase activity imparts bulges or distortions in general remains unclear and likely awaits atomic-level structural analysis of the interaction between the ATPase domain and nucleosome.
Figure 2.

Model for the mechanism of SWI/SNF induced nucleosome mobilization. The enzyme interacts with the nucleosome DNA from about two turns from the dyad (horizontal line) to well into the linker DNA through the DNA-binding domain (DBD). A translocase domain makes critical interactions with the DNA about two turns from the dyad (star). Only the top turn of DNA in the nucleosome is shown (bold line) with associated linker DNA (dashed line). See text for details.
Recent results also have shed light on a potential mechanism for nucleosome displacement by SWI/SNF. Previous work had shown that SWI/SNF is required to evict nucleosomes upon activation of specific promoters in vivo, with histone chaperones such as Asf-1.[87–89] Yet, the mechanism of eviction is poorly understood. As mentioned above, in vitro studies show that the SWI/SNF complex or paralogues, such as the RSC complex, can displace H2A/H2B dimers as well as the complete histone octamer, resulting in naked DNA after remodeling activity.[90] However, the efficiency of histone displacement is relatively low with the mononucleosomes substrates typically employed in such studies and observable displacement requires the presence of large amounts of acceptor or competitor species.
In recent work, Bartholomew and colleagues discovered that remodeling of di- or trinucleosomes resulted in significant amounts of species with reduced histone content consistent with the loss of entire nucleosomes.[58] Remodeling of these small oligonucleosome substrates resulted in more rapidly migrating products, which previous workers might have assumed were due to nucleosome mobilization and altered nucleosome translational positions.[91] However, by using a quantitative double-label technique, Bartholomew and colleagues demonstrated that an entire core histone octamer was lost in the products of dinucleosome remodeling, whereas little or no loss is observed with mononucleosomes. Histone loss occurs in stages, beginning with the loss of one H2A/H2B dimer, whereas an entire histone octamer is lost in a second major product. Interestingly, histone displacement occurs in the absence of chaperones or naked DNA as acceptors. Thus, in contrast to the remodeling of mononucleosomes, the data indicate SWI/SNF remodeling of dinucleosomes results in efficient eviction of one of the two original nucleosomes. Precise mapping of nucleosome positions during remodeling indicated nucleosomes are primarily moved to one end of the DNA fragment and that one of the two nucleosomes is evicted during the process, which is consistent with the histone content data. Moreover, by recruiting SWI/SNF to one end of the dinucleosome template through the binding of the transcriptional activator Gal4–VP16, the mapping evidence suggests that the nearby (proximal) nucleosome is moved downstream and eventually runs into and displaces the distal nucleosome.
These results correlate well with a recent report from the Owen-Hughes laboratory that remodeling can result in one nucleosome invading the region of DNA occupied by a neighbor.[92] However, the Bartholomew group finds that no remodeling occurs if the linker DNA between the two nucleosomes is too short.[58] Moreover, mapping data indicate that SWI/SNF appears to contact DNA ahead of the advancing nucleosome and thereby encounters the evicted nucleosome itself. A model then emerges whereby one nucleosome occupies a large pocket on the surface of the SWI/SNF complex and stimulates its ATPase-driven DNA translocase activity. The nucleosome in the pocket retains all of its histones, although its structure might be drastically altered, whereas a neighboring nucleosome in the path of the mobilized nucleosome–SWI/SNF complex is evicted from the DNA.
Acknowledgments
This work was supported by a National Institutes of Health (NIH, USA) grant GM 52426 to J.J.H.
Biographies

Ning Liu received her B.S. in Biological Engineering from the Northeast Normal University (Jilin, China) in 2000 and an M.S. in Biochemistry in 2006 from the University of Rochester (Rochester, NY, USA). She is now working towards her Ph.D. in the Department of Biochemistry at the University of Rochester. Her current research interests include the application of protein chemistry to elucidate the mechanisms of ATP-dependent chromatin remodeling complexes.

Angela Balliano received her B.S. in Biochemistry from Nazareth College (Ro-chester, NY, USA) in 2007. She is currently working towards a Ph.D. in Biochemistry from the University of Rochester (Rochester, NY, USA). Her research interests include determining the mechanism of ATP-dependent chromatin remodeling complexes, as well as understanding the specific interactions between these complexes and the nucleosome.

Jeff Hayes received his Ph.D. in Chemistry from The Johns Hopkins University (Baltimore, MD, USA). He was a postdoctoral fellow with Prof. Alan P. Wolffe at the National Institutes of Health (Bethesda, MD, USA) and has been a faculty member at The University of Rochester (Rochester, NY, USA) since 1995. He is now a Professor of Biochemistry and Biophysics, and his research focuses on various aspects of chromatin structure and function.
References
- 1.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.van Holde KE. Chromatin. Springer; New York: 1989. [Google Scholar]
- 3.Olins AL, Olins DE. Science. 1974;183:330–332. doi: 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- 4.Woodcock CFL. J Cell Biol. 1973;59:368. [Google Scholar]
- 5.Thoma F, Koller T, Klug A. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belmont AS, Bruce K. J Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allan J, Hartman G, Crane-Robinson C, Aviles FX. Nature. 1980;288:675–679. doi: 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- 8.Carruthers LM, Bednar J, Woodcock CL, Hansen JC. Biochemistry. 1998;37:14776–14787. doi: 10.1021/bi981684e. [DOI] [PubMed] [Google Scholar]
- 9.Han M, Grunstein M. Cell. 1988;55:1137–1145. doi: 10.1016/0092-8674(88)90258-9. [DOI] [PubMed] [Google Scholar]
- 10.Poirier MG, Bussiek M, Langowski J, Widom J. J Mol Biol. 2008;379:772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li G, Levitus M, Bustamante C, Widom J. Nat Struct Mol Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 12.Polach KJ, Widom J. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 13.Li G, Widom J. Nat Struct Mol Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 14.Polach KJ, Widom J. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 15.Wolffe A, Hayes JJ. Nucleic Acids Res. 1999;27:711–720. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson JD, Lowary PT, Widom J. J Mol Biol. 2001;307:977–985. doi: 10.1006/jmbi.2001.4528. [DOI] [PubMed] [Google Scholar]
- 17.Ausio J, Dong F, van Holde KE. J Mol Biol. 1989;206:451–463. doi: 10.1016/0022-2836(89)90493-2. [DOI] [PubMed] [Google Scholar]
- 18.Polach KJ, Lowary PT, Widom J. J Mol Biol. 2000;298:211–233. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 19.Vitolo JM, Yang Z, Basavappa R, Hayes JJ. Mol Cell Biol. 2004;24:697–707. doi: 10.1128/MCB.24.2.697-707.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. J Biol Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira H, Somers J, Webster R, Flaus A, Owen-Hughes T. Mol Cell Biol. 2007;27:4037–4048. doi: 10.1128/MCB.02229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin C, Felsenfeld G. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tse C, Sera T, Wolffe AP, Hansen JC. Mol Cell Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Hayes JJ. J Biol Chem. 2007;282:32867–32876. doi: 10.1074/jbc.M706035200. [DOI] [PubMed] [Google Scholar]
- 26.Kan PY, Caterino TL, Hayes JJ. Mol Cell Biol. 2009;29:538–546. doi: 10.1128/MCB.01343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kan PY, Lu X, Hansen JC, Hayes JJ. Mol Cell Biol. 2007;27:2084–2091. doi: 10.1128/MCB.02181-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Hayes JJ. Mol Cell Biol. 2008;28:227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mellor J. Cell. 2006;126:22–24. doi: 10.1016/j.cell.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 31.Suganuma T, Workman JL. Cell. 2008;135:604–607. doi: 10.1016/j.cell.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 32.Whitehouse I, Tsukiyama T. Nat Struct Mol Biol. 2006;13:633–640. doi: 10.1038/nsmb1111. [DOI] [PubMed] [Google Scholar]
- 33.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Nat Genet. 1999;23:62–66. doi: 10.1038/12664. [DOI] [PubMed] [Google Scholar]
- 34.Peterson CL, Côté J. Genes Dev. 2004;18:602–616. doi: 10.1101/gad.1182704. [DOI] [PubMed] [Google Scholar]
- 35.Smith CL, Peterson CL. Curr Top Dev Biol. 2004;65:115–148. doi: 10.1016/S0070-2153(04)65004-6. [DOI] [PubMed] [Google Scholar]
- 36.Clapier CR, Cairns BR. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 37.Cote J, Quinn J, Workman JL, Peterson CL. Science. 1994;265:53–60. doi: 10.1126/science.8016655. [DOI] [PubMed] [Google Scholar]
- 38.Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nature. 1994;370:477–481. doi: 10.1038/370477a0. [DOI] [PubMed] [Google Scholar]
- 39.Peterson CL, Herskowitz I. Cell. 1992;68:573–583. doi: 10.1016/0092-8674(92)90192-f. [DOI] [PubMed] [Google Scholar]
- 40.Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD. Cell. 1996;87:1249–1260. doi: 10.1016/s0092-8674(00)81820-6. [DOI] [PubMed] [Google Scholar]
- 41.Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Cell Cycle. 2008;7:1067–1074. doi: 10.4161/cc.7.8.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang B, Qiu J, Ratnakumar K, Laurent BC. Curr Biol. 2007;17:1432–1437. doi: 10.1016/j.cub.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kent NA, Chambers AL, Downs JA. J Biol Chem. 2007;282:27693–27701. doi: 10.1074/jbc.M704707200. [DOI] [PubMed] [Google Scholar]
- 44.Smith CL, Horowitz-Scherer R, Flanagan JF, Woodcock CL, Peterson CL. Nat Struct Biol. 2003;10:141–145. doi: 10.1038/nsb888. [DOI] [PubMed] [Google Scholar]
- 45.Smith CL, Peterson CL. Mol Cell Biol. 2005;25:5880–5892. doi: 10.1128/MCB.25.14.5880-5892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szerlong H, Hinata K, Viswanathan R, Erdjument-Bromage H, Tempst P, Cairns BR. Nat Struct Mol Biol. 2008;15:469–476. doi: 10.1038/nsmb.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hassan AH, Neely KE, Workman JL. Cell. 2001;104:817–827. doi: 10.1016/s0092-8674(01)00279-3. [DOI] [PubMed] [Google Scholar]
- 48.Hassan AH, Awad S, Prochasson P. J Biol Chem. 2006;281:18126–18134. doi: 10.1074/jbc.M602851200. [DOI] [PubMed] [Google Scholar]
- 49.Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- 50.Kasten M, Szerlong H, Erdjument-Bromage H, Tempst P, Werner M, Cairns BR. Embo J. 2004;23:1348–1359. doi: 10.1038/sj.emboj.7600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peterson CL. FEBS Lett. 2000;476:68–72. doi: 10.1016/s0014-5793(00)01673-2. [DOI] [PubMed] [Google Scholar]
- 52.Narlikar GJ, Phelan ML, Kingston RE. Mol Cell. 2001;8:1219–1230. doi: 10.1016/s1097-2765(01)00412-9. [DOI] [PubMed] [Google Scholar]
- 53.Shukla MS, Syed SH, Montel F, Faivre-Moskalenko C, Bednar J, Travers A, Angelov D, Dimitrov S. Proc Natl Acad Sci USA. 2010;107:1936–1941. doi: 10.1073/pnas.0904497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitehouse I, Flaus A, Cairns BR, White MF, Workman JL, Owen-Hughes T. Nature. 1999;400:784–787. doi: 10.1038/23506. [DOI] [PubMed] [Google Scholar]
- 55.Saha A, Wittmeyer J, Cairns BR. Nat Rev Mol Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 56.Zofall M, Persinger J, Kassabov SR, Bartholomew B. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
- 57.Schnitzler G, Sif S, Kingston RE. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 58.Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, Bartholomew B. Mol Cell. 2010;38:590–602. doi: 10.1016/j.molcel.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruno M, Flaus A, Stockdale C, Rencurel C, Ferreira H, Owen-Hughes T. Mol Cell. 2003;12:1599–1606. doi: 10.1016/s1097-2765(03)00499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gavin I, Horn PJ, Peterson CL. Mol Cell. 2001;7:97–104. doi: 10.1016/s1097-2765(01)00158-7. [DOI] [PubMed] [Google Scholar]
- 61.Yang X, Zaurin R, Beato M, Peterson CL. Nat Struct Mol Biol. 2007;14:540–547. doi: 10.1038/nsmb1238. [DOI] [PubMed] [Google Scholar]
- 62.Boyer LA, Langer MR, Crowley KA, Tan S, Denu JM, Peterson CL. Mol Cell. 2002;10:935–942. doi: 10.1016/s1097-2765(02)00634-2. [DOI] [PubMed] [Google Scholar]
- 63.Prochasson P, Neely KE, Hassan AH, Li B, Workman JL. Mol Cell. 2003;12:983–990. doi: 10.1016/s1097-2765(03)00366-6. [DOI] [PubMed] [Google Scholar]
- 64.Dechassa ML, Zhang B, Horowitz-Scherer R, Persinger J, Woodcock CL, Peterson CL, Bartholomew B. Mol Cell Biol. 2008;28:6010–6021. doi: 10.1128/MCB.00693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwanbeck R, Xiao H, Wu C. J Biol Chem. 2004;279:39933–39941. doi: 10.1074/jbc.M406060200. [DOI] [PubMed] [Google Scholar]
- 66.Chaban Y, Ezeokonkwo C, Chung WH, Zhang F, Kornberg RD, Maier-Davis B, Lorch Y, Asturias FJ. Nat Struct Mol Biol. 2008;15:1272–1277. doi: 10.1038/nsmb.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. Nat Struct Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saha A, Wittmeyer J, Cairns BR. Nat Struct Mol Biol. 2005;12:747–755. doi: 10.1038/nsmb973. [DOI] [PubMed] [Google Scholar]
- 69.Boyer LA, Logie C, Bonte E, Becker PB, Wade PA, Wolffe AP, Wu C, Imbalzano AN, Peterson CL. J Biol Chem. 2000;275:18864–18870. doi: 10.1074/jbc.M002810200. [DOI] [PubMed] [Google Scholar]
- 70.Lorch Y, Zhang M, Kornberg RD. Mol Cell. 2001;7:89–95. doi: 10.1016/s1097-2765(01)00157-5. [DOI] [PubMed] [Google Scholar]
- 71.Boyer LA, Shao X, Ebright RH, Peterson CL. J Biol Chem. 2000;275:11545–11552. doi: 10.1074/jbc.275.16.11545. [DOI] [PubMed] [Google Scholar]
- 72.Bazett-Jones DP, Côté J, Landel CC, Peterson CL, Workman JL. Mol Cell Biol. 1999;19:1470–1478. doi: 10.1128/mcb.19.2.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cote J, Peterson CL, Workman JL. Proc Natl Acad Sci USA. 1998;95:4947–4952. doi: 10.1073/pnas.95.9.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aoyagi S, Narlikar G, Zheng C, Sif S, Kingston RE, Hayes JJ. Mol Cell Biol. 2002;22:3653–3662. doi: 10.1128/MCB.22.11.3653-3662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Havas K, Flaus A, Phelan M, Kingston R, Wade PA, Lilley DM, Owen-Hughes T. Cell. 2000;103:1133–1142. doi: 10.1016/s0092-8674(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 76.Saha A, Wittmeyer J, Cairns BR. Genes Dev. 2002;16:2120–2134. doi: 10.1101/gad.995002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guyon JR, Narlikar GJ, Sullivan EK, Kingston RE. Mol Cell Biol. 2001;21:1132–1144. doi: 10.1128/MCB.21.4.1132-1144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaskelioff M, Gavin IM, Peterson CL, Logie C. Mol Cell Biol. 2000;20:3058–3068. doi: 10.1128/mcb.20.9.3058-3068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richmond TJ, Davey CA. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 80.Ong MS, Richmond TJ, Davey CA. J Mol Biol. 2007;368:1067–1074. doi: 10.1016/j.jmb.2007.02.062. [DOI] [PubMed] [Google Scholar]
- 81.Aoyagi S, Wade PA, Hayes JJ. J Biol Chem. 2003;278:30562–30568. doi: 10.1074/jbc.M304148200. [DOI] [PubMed] [Google Scholar]
- 82.Aoyagi S, Hayes JJ. Mol Cell Biol. 2002;22:7484–7490. doi: 10.1128/MCB.22.21.7484-7490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strohner R, Wachsmuth M, Dachauer K, Mazurkiewicz J, Hochstatter J, Rippe K, Langst G. Nat Struct Mol Biol. 2005;12:683–690. doi: 10.1038/nsmb966. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Smith CL, Saha A, Grill SW, Mihardja S, Smith SB, Cairns BR, Peterson CL, Bustamante C. Mol Cell. 2006;24:559–568. doi: 10.1016/j.molcel.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flaus A, Owen-Hughes T. Curr Opin Genet Dev. 2004;14:165–173. doi: 10.1016/j.gde.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 86.Kilgore JA, Hoose SA, Gustafson TL, Porter W, Kladde MP. Methods. 2007;41:320–332. doi: 10.1016/j.ymeth.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adkins MW, Howar SR, Tyler JK. Mol Cell. 2004;14:657–666. doi: 10.1016/j.molcel.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 88.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Mol Cell. 2003;11:1587–1598. doi: 10.1016/s1097-2765(03)00231-4. [DOI] [PubMed] [Google Scholar]
- 89.Reinke H, Horz W. Mol Cell. 2003;11:1599–1607. doi: 10.1016/s1097-2765(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 90.Lorch Y, Maier-Davis B, Kornberg RD. Proc Natl Acad Sci USA. 2006;103:3090–3093. doi: 10.1073/pnas.0511050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu N, Hayes JJ. Mol Cell. 2010;38:484–486. doi: 10.1016/j.molcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Engeholm M, de Jager M, Flaus A, Brenk R, van Noort J, Owen-Hughes T. Nat Struct Mol Biol. 2009;16:151–158. doi: 10.1038/nsmb.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]