

Abstract
Linker histones are multifunctional proteins that are involved in a myriad of processes ranging from stabilizing the folding and condensation of chromatin to playing a direct role in regulating gene expression. However, how this class of enigmatic proteins binds in chromatin and accomplishes these functions remains unclear. Here we review data regarding the H1 structure and function in chromatin, with special emphasis on the C-terminal domain (CTD), which typically encompasses approximately half of the mass of the linker histone and includes a large excess of positively charged residues. Owing to its amino acid composition, the CTD was previously proposed to function in chromatin as an unstructured polycation. However, structural studies have shown that the CTD adopts detectable secondary structure when interacting with DNA and macromolecular crowding agents. We describe classic and recent experiments defining the function of this domain in chromatin folding and emerging data indicating that the function of this protein may be linked to intrinsic disorder.
Keywords: core histones, nucleosome, tail domains, DNA, linker histone
Introduction
The genome of eukaryotic cells is packaged into chromatin, a complex structure in which DNA is associated with both histones and a multitude of nonhistone proteins. The basic unit of chromatin, the nucleosome, consists of a nucleosome core (composed of 2 copies each of the four core histones and 147 bp of DNA), linker DNA of variable length, and, in higher eukaryotes, typically one linker histone. The tight association of DNA within the nucleosome core is evidenced by the protection of approximately 147 bp of DNA from cleavage by the exo/endonuclease micrococcal nuclease (MNase), while the linker DNA between nucleosome cores is not in tight association with the core histones and is preferentially digested by such nucleases (Noll 1974; van Holde 1989). MNase digestion of chromatin containing linker histones also produces an additional intermediate product, termed the chromatosome, which contains 168 bp of DNA, the four core histones, and one molecule of H1 (Simpson 1978). The extra 20 bp of DNA protected from MNase digestion in chromatosomes appears to be symmetrically distributed on either side of the nucleosome core region in bulk chromatin (Simpson 1978). However, in reconstituted nucleosomes containing unique DNA sequences, asymmetric distributions are often observed (Hayes and Wolffe 1993; Sera and Wolffe 1998). Protection of the linker may result from H1-mediated formation of an apposed linker DNA stem-loop motif instead of wrapping of the additional base pairs of DNA around the nucleosome (Bednar et al. 1998). Interestingly, the linker DNA varies considerably in length in a cell- and species-specific fashion, perhaps in a quantized manner, whereby only integral numbers of helical twists can be accommodated within higher order chromatin structures (Widom 1992; Wang et al. 2008).
Linker histones are less evolutionarily conserved than the core histones. Multiple nonallelic variants are expressed among different eukaryotes and can vary between different tissues within the same organism (Lennox and Cohen 1983; Parseghian and Hamkalo 2001; Tanaka et al. 2001). Eleven different linker histone subtypes have been identified in mammals (Ausio 2006; Clausell et al. 2009; Happel and Doenecke 2009). Of the 11 subtypes, 7 are somatic (H1.1–H1.5, H1.0, and H1x), 3 are found in sperm (H1t, H1T2, and HILS1), and 1 is oocyte specific (H1oo) (Bucci et al. 1982; Lennox and Cohen 1984; Tanaka et al. 2001; Happel et al. 2005; Clausell et al. 2009). While it is known that these isoforms exist, very little is known about their specific actions at a molecular level. Knockouts of linker histones show that they are nonessential in many organisms such as yeast (Patterton et al. 1998), Tetrahymena thermophila (Shen et al. 1995), Arabidopsis thaliana (Wierzbicki and Jerzmanowski 2005), Caenorhabditis elegans (Jedrusik and Schulze 2001), and Ascobolus immersus (Barra et al. 2000), but defects in development and life span are observed. However, recent work indicates that linker histones are essential for life in higher organisms such as mice (Fan et al. 2005) and Drosophila (Lu et al. 2009b).
Likewise, much data suggest a critical role for H1 in regulating chromatin and chromosome structure. For example, deletion of the macronuclear (somatic) H1 gene in Tetrahymena thermophila caused a 1.5-fold increase in nuclear volume (Shen et al. 1995), and a similar increase in nuclear volume was observed in embryonic stem cells derived from H1 knockout mice (Fan et al. 2005). Moreover, despite earlier reports indicating that linker histones are not required for metaphase chromosome structure during pronuclei formation in cytostatic factor-arrested Xenopus laevis egg extracts (Ohsumi et al. 1993; Dasso et al. 1994), recent work with unarrested extracts showed that H1 is indeed required for normal morphology of replicated mitotic chromosomes (Maresca et al. 2005). Interestingly, chromosomes accumulated 4–10-fold more H1 after replication and immunodepletion of the embryonic H1 (B4) from these extracts resulted in abnormal mitotic chromosomes that did not segregate properly. Thus, H1 appears to be an essential element of higher order chromatin and chromosome structure.
Linker histones were traditionally considered to be general repressors of transcription and are depleted in chromatin regions containing transcriptionally active genes (Bresnick and Felsenfeld 1993). However, rather than producing global effects on transcription, knockouts of linker histones and even specific H1 subtypes indicate that these proteins are involved in the regulation of specific genes and in specific processes (Sancho et al. 2008). For example, knockout of Tetrahymena thermophila somatic H1 results in the up- and down-regulation of a specific subset of genes, but not global transcriptional deregulation (Shen and Gorovsky 1996). Interestingly, in some cases regulation of these specific genes by H1 appears to involve dephosphorylation-dependent targeting of Tetrahymena H1 to specific promoter elements (Song and Gorovsky 2007). Moreover, the variant H1.5 interacts with the transcription factor MSX1 to inhibit specific target genes during myogenesis (Lee et al. 2004) and H1.4 appears to be involved in heterochromatin formation via interaction with HP1 (Vaquero et al. 2004). Taking a more comprehensive approach, Sancho et al. (2008) used inducible short hairpin RNA mediated knockdown of each of the H1 variants in a breast cancer cell line and examined the effect on genome-wide gene expression using microarrays. A different subset of genes was found to be altered in each H1 knockdown, and depletion of each variant had different effects on cell survival. For example, loss of H1.2 and H1.4 resulted in arrest of cell proliferation, while depletion of H1.0, H1.3, or H1.5 resulted in normal cell growth. In addition, the first complete H1 knockout in chicken DT40 cells showed that H1 is not essential for mitotic chromatin compaction and global transcriptional repression in vertebrates. However, linker histone is important in interphase nuclear compaction and nucleosome spacing (Hashimoto et al. 2010). Together these studies demonstrate that linker histones and variants play both general and specific roles in regulating chromatin structure, gene transcription, and development in many eukaryotic organisms.
Linker histone structure
Linker histones bind to the surface of nucleosomes near where the DNA enters and exits the core and also interact with the linker DNA (Allan et al. 1980). Because of this exterior association, this class of histones exhibits much greater mobility about the nucleus than the core histones (Catez et al. 2004; Phair et al. 2004). While several crystal structures for nucleosome cores have been solved to high resolution (Luger et al. 1997) and the interactions of the core histones with the DNA are well understood, much less is known about the structure of linker histones and their interactions within chromatin. In higher eukaryotes, these small, basic proteins are composed of 3 distinct domains: a well-conserved globular domain, a long ~100 amino acid C-terminal tail, and a short N-terminal tail. This review will focus primarily on the C-terminal tail domain.
The globular domain
The structures of the globular domains of linker histones H5 and H1 have been characterized by single-crystal X-ray crystallography (Ramakrishnan et al. 1993) and NMR (Cerf et al. 1993), respectively. In general, these domains consist of a 3-helix “winged helix” fold containing a classical helix-turn-helix motif. The globular domain is fairly well conserved across species, is necessary for generating the 168 bp chromatosome particles observed during micrococcal nuclease digestion of native chromatin, and is sufficient for specific binding to the nucleosome in vitro (Allan et al. 1980). While the exact location of this binding site is unknown, several different models have been proposed and the reader is referred to the review of Crane-Robinson (1997) for further details; see also Brown et al. (2006). However, despite the controversy surrounding its location, it is generally accepted that the globular domain contacts at least 2 strands of DNA near the nucleosome dyad where DNA exits and enters the histone core and thus stabilizes DNA wrapping (Staynov and Crane-Robinson 1988; Goytisolo et al. 1996; Zhou et al. 1998; Brown et al. 2006). Consistent with this idea, in vivo FRAP (fluorescence recovery after photo-bleaching) studies of mutations in the H1° globular domain indicate 2 DNA-binding surfaces and have been interpreted as suggesting an off-axis binding site, about 10 bp from the nucleosome dyad, which includes one linker DNA arm wrapped in a continual superhelical fashion (Brown et al. 2006). However, recent molecular modeling suggests that the globular domain binds within the minor groove of DNA at the nucleosome dyad and orients linker DNA to form a stem structure (Fan and Roberts 2006), consistent with early work employing thymidine dimer formation as a probe of DNA structure in H1-containing chromatin (Pehrson 1989). However, it is worth noting that the homology of this domain to the winged-helix family of transcription factors suggests interaction with the major groove of DNA (Ramakrishnan et al. 1993). Perhaps the much-widened minor groove at the nucleosome dyad axis of symmetry sufficiently mimics the major groove in this regard. Indeed, very recent hydroxyl radical footprinting data strongly support a model whereby the globular domain contacts the minor groove at the dyad and the first helical turn of linker DNA on either side of the nucleosome core (Syed et al. 2010).
The N-terminal tail
The N-terminal tail of linker histone H1 is approximately 20–35 amino acid residues in length. When examining the amino acid composition of the tail, 2 distinct subregions are of note (Böhm and Mitchell 1985). The first half of the N-terminal tail is devoid of basic amino acid residues and is enriched in alanine, proline, and highly hydrophobic residues, and thus is not expected to interact strongly with DNA. In contrast, the region proximal to the globular domain is highly basic and contains 1 arginine and 5 lysine residues and resembles histone H3 in sequence and some posttranslational modifications (Kuzmichev et al. 2004). The proximity of this region to the globular domain and the high density of positively charged residues suggest that it may contribute to the binding stability of H1 in chromatin (Allan et al. 1986; Vila et al. 2001). Using circular dichroism, high-resolution NMR, and IR spectroscopy, Vila et al. (2001, 2002) investigated the structure of the N-terminal tail domain in aqueous and helix-stabilizing solutions. They found that this domain possessed little structure in water, where the circular dichroism spectra were dominated by contributions from random coil conformations. However, in trifluoroethanol the peptide acquired a high degree of helical structure. Thus, the N-terminal tail domain may acquire a distinct fold upon interaction with chromatin.
The C-terminal domain
The C-terminal tail domain (CTD) comprises approximately 100 amino acids and shows significant variability in primary sequence between individual linker histone subtypes and between species. This domain has diverged considerably throughout evolution and accounts for the bulk of sequence heterogeneity between H1 variants (Ponte et al. 2003). The CTD sequence is loosely based on a repeat of the sequence S/TPXK and has sequence content resembling that of the intrinsically disordered family of proteins (see below) (Hansen et al. 2006; Lu et al. 2009a). Interestingly, this motif is well conserved in the H1 and H1-related proteins of many protists, which typically lack a canonical globular domain, including the H1 of Tetrahymena thermophila (Kasinsky et al. 2001). Linker histone C-termini, on average, have 30–50 net positive charges (Subirana 1990), owing to an abundance of lysine residues distributed evenly across the domain (Kasinsky et al. 2001). With the exception of H5, most linker histone CTDs contain very few or no arginines. Interestingly, a general correlation exists between increased charge and the number of arginine residues in the CTDs of H1s as cells differentiate, with embryonic H1s (e.g., human H1oo, Xenopus laevis B4) containing the least overall charge, somatic H1 having intermediate charge, and H1s within quiescent cells (H1°, H5) containing the highest levels of overall positive charge and typically 1–2 arginine residues within their CTDs (Subirana 1990; Khochbin and Wolffe 1994).
The C-terminal domain stabilizes folding of nucleosome arrays into chromatin fibers (Allan et al. 1986) and is required for high-affinity binding to chromatin in vivo (Hendzel et al. 2004). However, data regarding the nucleosome binding and condensation properties of individual H1 variants are somewhat contradictory and do not paint a clear picture of their function. On one hand, researchers have found distinct differences in the ability of specific subtypes to induce chromatin condensation by different assays. Early work by Liao and Cole (1981) showed that individual sub-fractions of linker histones differed in their ability to bind and induce condensation of H1-stripped dinucleosomes and naked DNA. In contrast, Talasz et al. (1998) found that the affinities of H1 subtypes purified from mouse liver and testis for binding reconstituted mononucleosomes were virtually identical (Kd = 1–3 nmol/L) with the exception of H1b (H1.5) (Kd = 8–16 nmol/L). Moreover, only H1b and H1t exhibited a reduced affinity for binding naked DNA compared with the other subtypes, and higher amounts of these proteins were needed to aggregate a reconstituted polynucleosome template (Talasz et al. 1998). Differences in the results of these workers may be due to the method of H1 preparation. Khadake and Rao (1995) provided evidence that acid-extracted linker histones may have altered folding properties for both the globular domain and the CTD compared with salt-extracted proteins. Moreover, using salt-extracted H1 subtypes, they showed that a fraction containing H1b, H1d, H1e, and H1c had higher DNA condensing properties than a fraction containing H1a (H1.1) and H1t (Khadake and Rao 1995). Consistent with this data, a recent report employing chromatin assembled in Drosophila embryo extracts indicated that exogenously added human H1 isoforms have different affinities for chromatin, with the amount of subtype required to generate a nucleosome repeat length of 200 bp — [H1.5, H1.4] > [H1.3, H1.2, H1.0] > H1.1 > H1x — on minichromosomes reflecting the affinities of the H1 subtypes for chromatin (Clausell et al. 2009). These results were in agreement with FRAP data from Th’ng et al. (2005) which showed that H1.4 and H1.5 are most tightly bound in chromatin, while H1.3 and H1.0 have intermediate affinity and H1.1 and H1.2 are weakly bound. It is worth noting that these works examined the ability of linker histone subtypes to promote salt-induced aggregation of the chromatin in vitro, a phenomenon thought to be related to the formation of higher order tertiary chromatin structures. However, using analytical ultracentrifugation and model nucleosome arrays, Lu et al. (2009a) were able to analyze the ability of H1 isoforms to promote folding of secondary structures. They found no difference in the ability of mouse H1 isoforms H1°, H1S-1, H1S-2, H1S-3, and H1S-4 to induce folding of model nucleosome arrays (Lu et al. 2009a). This data shows that while linker histones stabilize higher order folding and chromatin condensation, the different isoforms may vary in their nucleosome binding affinity and their ability to induce aggregation of polynucleosomes, a variability likely attributable to differences in the CTDs (Clausell et al. 2009). However, the specific sites or residues within the H1 CTD required for chromatin binding and its structural arrangement within compacted chromatin remain undefined. These issues will be explored in the following sections.
The role of the H1 CTD in charge neutralization
Native chromatin undergoes several salt-dependent transitions in vitro, including a transition from extended nucleosome arrays to partially folded “zig-zag” structures, to fully condensed “30-nm” chromatin fibers, and condensation (self-association) of chromatin fibers to large, higher order assemblies (Thoma et al. 1979; Widom 1986; Hansen 2002). These transitions are thought to recapitulate the behavior and structures of native chromatin in vivo. As mentioned above, the CTD of linker histone H1 is required for stabilizing higher order chromatin structures and for high-affinity binding of H1 to chromatin in vivo (Allan et al. 1980, 1986; Lever et al. 2000; Misteli et al. 2000; Hendzel et al. 2004). Importantly, H1 stimulates formation of fully folded and self-associated chromatin but apparently does not alter the intrinsic nucleosomal array folding pathway (Carruthers et al. 1998; Hansen 2002). The large excess of positively charged residues in H1s is believed to effect neutralization of negatively charged DNA to assist in this condensation. Indeed, early experiments demonstrated that H1-containing chromatin undergoes salt-dependent folding and condensation at much lower concentrations of counter-ions or monovalent salts (60 mmol/L NaCl or 0.3 mmol/L MgCl2), while H1-depleted chromatin requires divalent cations (2–5 mmol/L MgCl2) for condensation (Thoma et al. 1979). Thus, the H1 CTD appears to regulate chromatin condensation and higher order structural transitions through neutralization of negatively charged linker DNA.
As previously mentioned, the CTDs of most linker histones contain 30–50 net positive charges that are evenly distributed throughout the entire domain. Analyses of the electrostatic properties of the C-terminal tail of linker histone H1 found that assuming physiological conditions, all basic residues are fully charged (Subirana 1990), and nearly all positively charged residues in the CTD are involved in neutralization of the DNA backbone (Clark and Kimura 1990). A detailed comparison of the extent of DNA charge neutralization effected by H1 as predicted by Manning poly-electrolyte theory with the observed extent of chromatin condensation in various conditions of mono- and multivalent salts indicated that the extent of chromatin folding or condensation is directly related to the extent of charge neutralization by salt and (or) H1 (Clark and Kimura 1990). These results indicate that H1 behaves as a “directed” polycation and functions primarily to neutralize the charge of linker DNA in folded chromatin. Thus, a model emerges whereby the CTD is positioned with respect to the nucleosome by the globular domain to interact with linker DNA. Since the charged residues are distributed throughout the CTD and the C-terminal tail appears to be unstructured in solution, it is believed that the entire CTD may participate in chromatin condensation by effecting general charge neutralization of DNA in the interior of the chromatin fiber (Allan et al. 1986; Subirana 1990).
The CTD: basic blob or structured element?
The exact mechanism by which the C-terminal tail stabilizes chromatin by shielding negative charges on DNA is still unclear. Despite the view that this domain is unstructured and functions as a general and nonspecific cation within chromatin, physical evidence indicates that the C-terminal tail domain acquires a substantial amount of α-helical structure in the presence of secondary structure stabilizers such as trifluoroethanol and NaClO4 (Verdaguer et al. 1993; Clark et al. 1988), presumably through the promotion of hydrogen bonding (Clark et al. 1988). Moreover, IR spectroscopy showed that the CTD, while unstructured in aqueous solution, acquires substantial structure in the presence of DNA and physiological ionic strengths (Roque et al. 2005). In a recent study, the effects of charge neutralization on the secondary structure of the CTD independent of DNA interaction were studied using Fourier transformed infrared spectroscopy. By performing the experiments at alkaline pH in which the ε-amino groups of lysine are deprotonated, Roque et al. (2009) found folding of the CTD similar to that seen when complexed with DNA. These results indicate that CTD binding to linker DNA in chromatin could induce folding within this domain and stabilize specific structures that would provide scaffolding for appropriate higher order chromatin structures.
As mentioned above, neutralization of negative charges along the DNA backbone by the CTD likely stabilizes the close apposition of neighboring nucleosomes in condensed chromatin. A simple model of CTD function then predicts that incremental deletions of the CTD would result in incremental losses in the ability of the linker histone to stabilize folding of nucleosome arrays into chromatin fibers. To test this hypothesis, Lu and Hansen (2004) used model nucleosome arrays and examined the effect of systematic deletions within the CTD of H1 on the salt-dependent formation of secondary and tertiary chromatin structures. They created 4 truncation mutants in which approximately 24 amino acids were successively deleted from the C-terminal end. Results from this study indicated that amino acid residues primarily responsible for alteration of linker DNA structure and chromatin fiber condensation are confined to 2 discrete subdomains (regions 1 and 3, see Fig. 1) (Lu and Hansen 2004). Binding of the CTD to linker DNA may induce specific conformational changes within the CTD that are responsible for mediating linker DNA stem motif formation, stabilization of locally folded chromatin structures, and nucleosomal array self-association. However, as the CTD was progressively deleted, the concentration needed to achieve stoichiometric binding increased, suggesting that the CTD contributes to the overall binding free energy of H1 (Lu and Hansen 2004). Clearly, these data are not consistent with a simple charge neutralization model for CTD function and indicate that specific subdomains within the CTD act through alternative mechanisms to regulate chromatin structure.
Fig. 1.
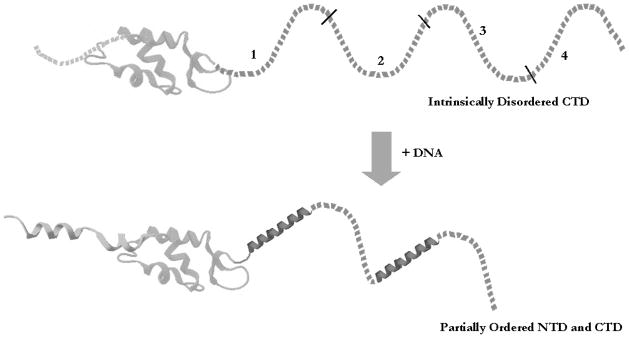
The C-terminal domains (CTDs) of linker histones are intrinsically disordered. The H1 CTD is disordered in the absence of interacting partners. Upon interaction with DNA or other targets in chromatin, the domain adopts a complementary structure. Interaction with other targets may induce alternative structures. Numbers indicate regions deleted in the work of Lu and Hansen (2004). Globular domain adapted from Vila et al. (2002).
H1 phosphorylation and CTD function
A conserved feature of all linker histone variants is the serine/threonine kinase phosphorylation sites distributed throughout these molecules, including the CTD. Linker histone phosphorylation levels are lowest in G1 and rise during both S phase and mitosis in response to activation by cdc2-family kinases (Roth and Allis 1992). Inhibition of H1 kinase activity and dephosphorylation of H1 can cause partial chromosome decondensation (Roth and Allis 1992). Molecular modeling reveals that the CTD may adopt a HMGB box-like structure and that the CTD S/TPXK motifs are sites of DNA binding and function in the compaction of DNA (Bharath et al. 2003). Hendzel et al. (2004) quantitatively defined the contributions of 2 S/TPXK phosphorylation sites, T152 and S183 in H1.1, to the stability of H1 binding in chromatin using FRAP. T152 aligns with a conserved TPKK site in which both lysines are predicted to bind extra-nucleosomal DNA (Bharath et al. 2003). The conserved SPAK site at S183 is 4 amino acids away from the last two lysines predicted to contact DNA (Bharath et al. 2003). Thus, phosphorylation of either or both of these sites within the CTD may have an impact on DNA binding in vivo.
Using glutamic acid substitutions to mimic phosphorylation, Hendzel et al. (2004) found that the substitution T152E increased H1 mobility in the nucleus by at least 5-fold with respect to the wild-type protein, an effect similar to that observed when the entire C-terminus was truncated up to this residue. The large effect of addition of a single negative charge suggests that phosphorylation of T152 disrupts the DNA binding activity of a larger element or domain within the CTD. Similar results were observed with glutamic acid substitution and truncation at S183. Moreover, substituting lysines for both T152 and S183 stabilized binding of H1.1 to DNA in vivo. These results indicate that phosphorylation of H1 within 2 separate S/TPXK phosphorylation sites, T152 and S183, significantly disrupts linker histone binding to chromatin, perhaps by altering CTD conformation(s) required for interaction with DNA.
CTD and the charged patch model
As mentioned above, the linker histones of unicellular organisms are diverse and often lack the typical tripartite structure of the linker histones of higher eukaryotes. Such is the case in Tetrahymena thermophila H1, which lacks a conserved globular domain. Nevertheless, Tetrahymena macro-nuclear H1 has a primary structure closely resembling the CTD of higher eukaryotes and is rapidly phosphorylated in somatic chromatin (Allis and Gorovsky 1981). Since phosphorylation is largely confined to both the N- and C-terminal tails in both higher and lower eukaryotes, Tetrahymena H1 has been used as a model to investigate the function of phosphorylation within these domains (Roth et al. 1991). Five in vivo phosphorylation sites in Tetrahymena H1 have been identified and are clustered in a region encompassing 20 residues towards the N-terminal region of the protein (Roth et al. 1988; Mizzen et al. 1999). Of these 5 sites, 3 are canonical S/TPXK p34(cdc2) kinase sites and the other 2 are atypical non-proline directed sites. Deletion or mutation of these sites demonstrated that macronuclear phosphorylation is not essential for viability in Tetrahymena (Dou et al. 1999; Dou and Gorovsky 2000). However, chromatin in macronuclear H1 knockout strains is less condensed than that in wild-type cells (Shen et al. 1995). Mutagenesis of these phosphorylation sites to residues that mimicked either the dephosphorylated (alanine) or the phosphorylated (glutamic acid) state showed that phosphorylation of H1 in Tetrahymena phenocopies the H1-null phenotype in its positive and negative effects on transcription (Dou et al. 1999). For example, deletion of H1 was previously shown to result in down-regulation of the CyP1 gene in starved Tetrahymena cells. When the phosphorylated region was mutagenized to mimic the charge of fully phosphorylated H1, CyP1 expression was similarly inhibited. Interestingly, simply altering the total charge within the phosphorylated region by substitution of non-phosphorylated residues, as long as the combination of acidic and basic residues had a net charge similar to the dephosphorylated protein, yielded a similar phenotype to cells that contain a constitutively de-phosphorylated mutant of H1 (Dou and Gorovsky 2000). These effects were shown to be independent of the hydrophobicity of the region and did not require residues that structurally resembled phosphorylated serines. Thus, the change in net charge within this region as a result of phosphorylation appears to be the critical event for H1-regulated gene transcription (Dou and Gorovsky 2000). Further studies by Dou and Gorovsky (2002) showed that charge-altering mutations placed in the phosphorylated region or throughout the molecule had the same effect on CyP1 transcription, provided that they were clustered in a small region. These studies demonstrated that phosphorylation of H1 in Tetrahymena regulates the expression of specific genes by changing the overall charge of a small domain termed a “charge patch”. This domain can be located anywhere in the H1 protein, with the critical feature for transcriptional regulation being the phosphorylation-dependent creation of a localized patch of negative charge. The similarity of the H1 knockout and phosphorylation mimic phenotypes suggests that the charge patch produced by phosphorylation may act to facilitate the dissociation of H1 from chromatin (Dou and Gorovsky 2002).
Intrinsic disorder: a model for H1 CTD mode of action
Linker histones are multifunctional proteins that are involved in a myriad of processes within chromatin, including roles in nucleosome spacing, chromatin condensation, and gene expression. In addition, linker histones are the binding targets for other nuclear regulatory proteins such as DFF40/CAD (Widlak et al. 2000; Widlak et al. 2005) and BAF (Montes de Oca et al. 2005). However, how these small, relatively simple proteins are able to specifically recognize and interact with so many different types of macromolecules and structures is not known.
The amino acid composition of the linker histone CTD may give clues as to its mode of action. The CTD sequences of linker histones are not well conserved, yet these domains have remarkably similar amino acid compositions, predominantly containing lysines, alanines, and prolines. Serines, glycines, threonines, and valines are also found, but to a lesser extent. The C-terminus of H1 is almost completely devoid of acidic and highly hydrophobic amino acids (Kasinsky et al. 2001; Hansen et al. 2006). Protein regions containing this characteristic amino acid composition are believed to be intrinsically disordered (Hansen et al. 2006). Intrinsically disordered proteins are not completely unstructured in solution, but rather are envisioned to have stretches of polypeptides exhibiting molten globule-like properties in their native state that adopt defined structures upon interaction with macromolecular partners.
Intrinsic disorder may explain the observed behavior of the linker histone CTD. Lu and Hansen (2004) demonstrated that the ability to alter linker DNA structure and stabilize folding and oligomerization of nucleosome arrays is localized to 2 discontinuous regions of ~24 amino acid residues (subdomains 1 and 3; see above) of the H1 CTD. They hypothesized that it is the overall amino acid residue content and not the exact primary sequence of the protein that is important. This hypothesis is supported by data indicating that chromatin-condensing functions of mouse linker histone isoforms are similar, despite their differences in CTD primary sequences (Lu et al. 2009a). To further test this idea, Lu et al. (2009a) examined the intrinsic function of subdomains 1 and 3 versus regions 2 and 4 in the H1 CTD (see Fig. 1) (Lu and Hansen 2004). As mentioned above, deletion through region 1 was found to be required for the formation of the stem-loop linker DNA motif and stabilization of fiber folding and oligomerization, while region 3 was found to stabilize folded chromatin fibers (Lu et al. 2009a). Recombinant technology was then used to scramble the primary sequence without altering amino acid composition, and to switch the position of the functional and nonfunctional CTD regions of recombinant H1 Intrinsic disorder may explain the observed behavior of the linker histone CTD. Lu and Hansen (2004) demonstrated that the ability to alter linker DNA structure and stabilize folding and oligomerization of nucleosome arrays is localized to 2 discontinuous regions of. Surprisingly, when the primary sequence of region 1 was scrambled, the ability of the CTD to bind linker DNA and stabilize condensed chromatin was not altered. Also, regions 2, 3, and 4 in the CTD could all replace functional region 1 if they were placed in the proper position along the polypeptide chain. Owing to the interchangeability of the four CTD subdomains, it is hypothesized that the binding of the globular domain helps to position regions 1 and 3 of the CTD (Lu et al. 2009a).
Analysis of H1 isoforms showed that approximately 40% of the residues are lysine and 20%–30% are alanine, with serine, threonine, valine, glycine, and proline being significantly represented (Hansen et al. 2006). Replacement of 3 valine residues and 1 threonine residue in CTD region 1 with asparagine and proline, residues not normally found in the CTD, led to an altered H1 function in chromatin folding (Lu et al. 2009a). The specific amino acid composition of these regions may allow the formation of short α-helices in these two regions in response to DNA binding that mediates CTD function in chromatin (Clark et al. 1988; Roque et al. 2009). Since only two 24 amino acid regions are required to stabilize higher order chromatin structure, and the linker histone CTD is approximately 100 amino acids long, the basis for linker histones all having CTDs of this length, and with a similar amino acid composition, may be to allow the CTD to interact with a multitude of ancillary factors.
Thus, a model emerges whereby regions of the CTD become structured upon binding to DNA and nucleosome surfaces in chromatin to mediate alteration of linker DNA structure and chromatin condensation (Fig. 1). An evolutionary advantage of intrinsically disordered proteins is the ability to mediate several different functions by participating in different macromolecular interactions and to allow high specificity in molecular interactions with appropriately attenuated affinities. However, confirmation of this model awaits determination of the exact structures formed within the CTD upon intertactions with different macromolecular partners and further structural analysis of H1-bound nucleosomes and arrays.
Acknowledgments
This work was supported by NIH grant GM52426.
Footnotes
This paper is one of a selection of papers published in a Special Issue entitled 31st Annual International Asilomar Chromatin and Chromosomes Conference, and has undergone the Journal’s usual peer review process.
References
- Allan J, Hartman PG, Crane-Robinson C, Aviles FX. The structure of histone H1 and its location in chromatin. Nature. 1980;288(5792):675–679. doi: 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- Allan J, Mitchell T, Harborne N, Bohm L, Crane-Robinson C. Roles of H1 domains in determining higher order chromatin structure and H1 location. J Mol Biol. 1986;187(4):591–601. doi: 10.1016/0022-2836(86)90337-2. [DOI] [PubMed] [Google Scholar]
- Allis CD, Gorovsky MA. Histone phosphorylation in macro- and micronuclei of Tetrahymena thermophila. Biochemistry. 1981;20(13):3828–3833. doi: 10.1021/bi00516a025. [DOI] [PubMed] [Google Scholar]
- Ausio J. Histone variants—the structure behind the function. Brief Funct Genomics Proteomics. 2006;5(3):228–243. doi: 10.1093/bfgp/ell020. [DOI] [PubMed] [Google Scholar]
- Barra JL, Rhounim L, Rossignol JL, Faugeron G. Histone H1 is dispensable for methylation-associated gene silencing in Ascobolus immersus and essential for long life span. Mol Cell Biol. 2000;20(1):61–69. doi: 10.1128/mcb.20.1.61-69.2000. 10.1128/MCB.20.1.61-69. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci USA. 1998;95(24):14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharath MM, Chandra NR, Rao MR. Molecular modeling of the chromatosome particle. Nucleic Acids Res. 2003;31 (14):4264–4274. doi: 10.1093/nar/gkg481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm L, Mitchell TC. Sequence conservation in the N-terminal domain of histone H1. FEBS Lett. 1985;193(1):1–4. doi: 10.1016/0014-5793(85)80067-3. [DOI] [PubMed] [Google Scholar]
- Bresnick EH, Felsenfeld G. Evidence that the transcription factor USF is a component of the human β-globin locus control region heteromeric protein complex. J Biol Chem. 1993;268(25):18824–18834. [PubMed] [Google Scholar]
- Brown DT, Izard T, Misteli T. Mapping the interaction surface of linker histone H1° with the nucleosome of native chromatin in vivo. Nat Struct Mol Biol. 2006;13(3):250–255. doi: 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci LR, Brock WA, Meistrich ML. Distribution and synthesis of histone 1 subfractions during spermatogenesis in the rat. Exp Cell Res. 1982;140(1):111–118. doi: 10.1016/0014-4827(82)90162-8. [DOI] [PubMed] [Google Scholar]
- Carruthers LM, Bednar J, Woodcock CL, Hansen JC. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry. 1998;37(42):14776–14787. doi: 10.1021/bi981684e. [DOI] [PubMed] [Google Scholar]
- Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol Cell Biol. 2004;24(10):4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. 10.1128/MCB.24.10.4321-4328. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerf C, Lippens G, Muyldermans S, Segers A, Ramakrishnan V, Wodak SJ, et al. Homo- and heteronuclear two-dimensional NMR studies of the globular domain of histone H1: sequential assignment and secondary structure. Biochemistry. 1993;32(42):11345–11351. doi: 10.1021/bi00093a011. [DOI] [PubMed] [Google Scholar]
- Clark DJ, Kimura T. Electrostatic mechanism of chromatin folding. J Mol Biol. 1990;211(4):883–896. doi: 10.1016/0022-2836(90)90081-V. [DOI] [PubMed] [Google Scholar]
- Clark DJ, Hill CS, Martin SR, Thomas JO. α-Helix in the carboxy-terminal domains of histones H1 and H5. EMBO J. 1988;7(1):69–75. doi: 10.1002/j.1460-2075.1988.tb02784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausell J, Happel N, Hale TK, Doenecke D, Beato M, Aramayo R. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS One. 2009;4(10):e0007243. doi: 10.1371/journal.pone.0007243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane-Robinson C. Where is the globular domain of linker histone located on the nucleosome? Trends Biochem Sci. 1997;22(3):75–77. doi: 10.1016/S0968-0004(97)01013-X. [DOI] [PubMed] [Google Scholar]
- Dasso M, Dimitrov S, Wolffe AP. Nuclear assembly is independent of linker histones. Proc Natl Acad Sci USA. 1994;91(26):12477–12481. doi: 10.1073/pnas.91.26.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Gorovsky MA. Phosphorylation of linker histone H1 regulates gene expression in vivo by creating a charge patch. Mol Cell. 2000;6(2):225–231. doi: 10.1016/s1097-2765(00)00024-1. 10.1016/S1097-2765(00) 00024-1. [DOI] [PubMed] [Google Scholar]
- Dou Y, Gorovsky MA. Regulation of transcription by H1 phosphorylation in Tetrahymena is position independent and requires clustered sites. Proc Natl Acad Sci USA. 2002;99(9):6142–6146. doi: 10.1073/pnas.092029599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Mizzen CA, Abrams M, Allis CD, Gorovsky MA. Phosphorylation of linker histone H1 regulates gene expression in vivo by mimicking H1 removal. Mol Cell. 1999;4(4):641–647. doi: 10.1016/S1097-2765(00)80215-4. [DOI] [PubMed] [Google Scholar]
- Fan L, Roberts VA. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. Proc Natl Acad Sci USA. 2006;103(22):8384–8389. doi: 10.1073/pnas.0508951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell. 2005;123(7):1199–1212. doi: 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Goytisolo FA, Gerchman SE, Yu X, Rees C, Graziano V, Ramakrishnan V, Thomas JO. Identification of two DNA-binding sites on the globular domain of histone H5. EMBO J. 1996;15(13):3421–3429. [PMC free article] [PubMed] [Google Scholar]
- Hansen JC. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31(1):361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- Hansen JC, Lu X, Ross ED, Woody RW. Intrinsic protein disorder, amino acid composition, and histone terminal domains. J Biol Chem. 2006;281(4):1853–1856. doi: 10.1074/jbc.R500022200. 10.1074/jbc. R500022200. [DOI] [PubMed] [Google Scholar]
- Happel N, Doenecke D. Histone H1 and its isoforms: contribution to chromatin structure and function. Gene. 2009;431(1–2):1–12. doi: 10.1016/j.gene.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Happel N, Schulze E, Doenecke D. Characterisation of human histone H1x. Biol Chem. 2005;386(6):541–551. doi: 10.1515/BC.2005.064. 10. 1515/BC.2005.064. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Takami Y, Sonada E, Iwasaki T, Iwano H, Tachibana M, et al. Histone H1 null vertebrate cells exhibit altered nucleosome architecture. Nucl Acids Res. 2010;38(11):3533–3545. doi: 10.1093/nar/gkq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JJ, Wolffe AP. Preferential and asymmetric interaction of linker histones with 5S DNA in the nucleosome. Proc Natl Acad Sci USA. 1993;90(14):6415–6419. doi: 10.1073/pnas.90.14.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel MJ, Lever MA, Crawford E, Th’ng JP. The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J Biol Chem. 2004;279(19):20028–20034. doi: 10.1074/jbc.M400070200. [DOI] [PubMed] [Google Scholar]
- Jedrusik MA, Schulze E. A single histone H1 isoform (H1.1) is essential for chromatin silencing and germline development in Caenorhabditis elegans. Development. 2001;128(7):1069–1080. doi: 10.1242/dev.128.7.1069. [DOI] [PubMed] [Google Scholar]
- Kasinsky HE, Lewis JD, Dacks JB, Ausió J. Origin of H1 linker histones. FASEB J. 2001;15(1):34–42. doi: 10.1096/fj.00-0237rev. [DOI] [PubMed] [Google Scholar]
- Khadake JR, Rao MR. DNA- and chromatin-condensing properties of rat testes H1a and H1t compared to those of rat liver H1bdec; H1t is a poor condenser of chromatin. Biochemistry. 1995;34(48):15792–15801. doi: 10.1021/bi00048a025. [DOI] [PubMed] [Google Scholar]
- Khochbin S, Wolffe AP. Developmentally regulated expression of linker-histone variants in vertebrates. Eur J Biochem. 1994;225(2):501–510. doi: 10.1111/j.1432-1033.1994.00501.x. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different Ezh2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14(2):183–193. doi: 10.1016/S1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- Lee H, Habas R, Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science. 2004;304(5677):1675–1678. doi: 10.1126/science.1098096. [DOI] [PubMed] [Google Scholar]
- Lennox RW, Cohen LH. The histone H1 complements of dividing and nondividing cells of the mouse. J Biol Chem. 1983;258(1):262–268. [PubMed] [Google Scholar]
- Lennox RW, Cohen LH. The alterations in H1 histone complement during mouse spermatogenesis and their significance for H1 subtype function. Dev Biol. 1984;103(1):80–84. doi: 10.1016/0012-1606(84)90009-5. [DOI] [PubMed] [Google Scholar]
- Lever MA, Th’ng JPH, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408(6814):873–876. doi: 10.1038/35048603. [DOI] [PubMed] [Google Scholar]
- Liao LW, Cole RD. Condensation of dinucleosomes by individual subfractions of H1 histone. J Biol Chem. 1981;256(19):10124–10128. [PubMed] [Google Scholar]
- Lu X, Hansen JC. Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J Biol Chem. 2004;279(10):8701–8707. doi: 10.1074/jbc.M311348200. [DOI] [PubMed] [Google Scholar]
- Lu X, Hamkalo B, Parseghian MH, Hansen JC. Chromatin condensing functions of the linker histone C-terminal domain are mediated by specific amino acid composition and intrinsic protein disorder. Biochemistry. 2009a;48(1):164–172. doi: 10.1021/bi801636y. 10. 1021/bi801636y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wontakal SN, Emelyanov AV, Morcillo P, Konev AY, Fyodorov DV, Skoultchi AI. Linker histone H1 is essential for Drosophila development, the establishment of pericentric heterochromatin, and a normal polytene chromosome structure. Genes Dev. 2009b;23(4):452–465. doi: 10.1101/gad.1749309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. 10. 1038/38444. [DOI] [PubMed] [Google Scholar]
- Maresca TJ, Freedman BS, Heald R. Histone H1 is essential for mitotic chromosome architecture and segregation in Xenopus laevis egg extracts. J Cell Biol. 2005;169(6):859–869. doi: 10.1083/jcb.200503031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408(6814):877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- Mizzen CA, Dou Y, Liu Y, Cook RG, Gorovsky MA, Allis CD. Identification and mutation of phosphorylation sites in a linker histone. Phosphorylation of macronuclear H1 is not essential for viability in Tetrahymena. J Biol Chem. 1999;274(21):14533–14536. doi: 10.1074/jbc.274.21.14533. [DOI] [PubMed] [Google Scholar]
- Montes de Oca R, Lee KK, Wilson KL. Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J Biol Chem. 2005;280(51):42252–42262. doi: 10.1074/jbc.M509917200. [DOI] [PubMed] [Google Scholar]
- Noll M. Subunit structure of chromatin. Nature. 1974;251(5472):249–251. doi: 10.1038/251249a0. [DOI] [PubMed] [Google Scholar]
- Ohsumi K, Katagiri C, Kishimoto T. Chromosome condensation in Xenopus mitotic extracts without histone H1. Science. 1993;262(5142):2033–2035. doi: 10.1126/science.8266099. [DOI] [PubMed] [Google Scholar]
- Parseghian MH, Hamkalo BA. A compendium of the histone H1 family of somatic subtypes: an elusive cast of characters and their characteristics. Biochem Cell Biol. 2001;79(3):289–304. doi: 10.1139/bcb-79-3-289. [DOI] [PubMed] [Google Scholar]
- Patterton HG, Landel CC, Landsman D, Peterson CL, Simpson RT. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J Biol Chem. 1998;273(13):7268–7276. doi: 10.1074/jbc.273.13.7268. 10. 1074/jbc.273.13.7268. [DOI] [PubMed] [Google Scholar]
- Pehrson JR. Thymine dimer formation as a probe of the path of DNA in and between nucleosomes in intact chromatin. Proc Natl Acad Sci USA. 1989;86(23):9149–9153. doi: 10.1073/pnas.86.23.9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair RD, Scaffidi P, Elbi C, Vecerová J, Dey A, Ozato K, et al. Global nature of dynamic protein–chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol Cell Biol. 2004;24 (14):6393–6402. doi: 10.1128/MCB.24.14.6393-6402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte I, Vila R, Suau P. Sequence complexity of histone H1 subtypes. Mol Biol Evol. 2003;20(3):371–380. doi: 10.1093/molbev/msg041. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM. Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature. 1993;362(6417):219–223. doi: 10.1038/362219a0. [DOI] [PubMed] [Google Scholar]
- Roque A, Iloro I, Ponte I, Arrondo JL, Suau P. DNA-induced secondary structure of the carboxyl-terminal domain of histone H1. J Biol Chem. 2005;280(37):32141–32147. doi: 10.1074/jbc.M505636200. [DOI] [PubMed] [Google Scholar]
- Roque A, Ponte I, Suau P. Role of charge neutralization in the folding of the carboxy-terminal domain of histone H1. J Phys Chem B. 2009;113(35):12061–12066. doi: 10.1021/jp9022579. [DOI] [PubMed] [Google Scholar]
- Roth SY, Allis CD. Chromatin condensation: does histone H1 dephosphorylation play a role? Trends Biochem Sci. 1992;17(3):93–98. doi: 10.1016/0968-0004(92)90243-3. [DOI] [PubMed] [Google Scholar]
- Roth SY, Schulman IG, Richman R, Cook RG, Allis CD. Characterization of phosphorylation sites in histone H1 in the amitotic macronucleus of Tetrahymena during different physiological states. J Cell Biol. 1988;107(6 Pt 2):2473–2482. doi: 10.1083/jcb.107.6.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Collini MP, Draetta G, Beach D, Allis CD. A cdc2-like kinase phosphorylates histone H1 in the amitotic macronucleus of Tetrahymena. EMBO J. 1991;10(8):2069–2075. doi: 10.1002/j.1460-2075.1991.tb07738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M, Diani E, Beato M, Jordan A, Malik HS. Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 2008;4(10):e1000227. doi: 10.1371/journal.pgen.1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sera T, Wolffe AP. Role of histone H1 as an architectural determinant of chromatin structure and as a specific repressor of transcription on Xenopus oocyte 5S rRNA genes. Mol Cell Biol. 1998;18(7):3668–3680. doi: 10.1128/mcb.18.7.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Gorovsky MA. Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell. 1996;86(3):475–483. doi: 10.1016/S0092-8674(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Shen X, Yu L, Weir JW, Gorovsky MA. Linker histones are not essential and affect chromatin condensation in vivo. Cell. 1995;82(1):47–56. doi: 10.1016/0092-8674(95)90051-9. [DOI] [PubMed] [Google Scholar]
- Simpson RT. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry. 1978;17(25):5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- Song X, Gorovsky MA. Unphosphorylated H1 is enriched in a specific region of the promoter when CDC2 is down-regulated during starvation. Mol Cell Biol. 2007;27(5):1925–1933. doi: 10.1128/MCB.01619-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staynov DZ, Crane-Robinson C. Footprinting of linker histones H5 and H1 on the nucleosome. EMBO J. 1988;7(12):3685–3691. doi: 10.1002/j.1460-2075.1988.tb03250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subirana JA. Analysis of the charge distribution in the C-terminal region of histone H1 as related to its interaction with DNA. Biopolymers. 1990;29(10–11):1351–1357. doi: 10.1002/bip.360291003. 10.1002/bip. 360291003. [DOI] [PubMed] [Google Scholar]
- Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, et al. Single-base resolution mapping of H1–nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci USA. 2010;108(21):9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasz H, Sapojnikova N, Helliger W, Lindner H, Puschendorf B. In vitro binding of H1 histone subtypes to nucleosomal organized mouse mammary tumor virus long terminal repeat promotor. J Biol Chem. 1998;273(48):32236–32243. doi: 10.1074/jbc.273.48.32236. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Hennebold JD, Macfarlane J, Adashi EY. A mammalian oocyte-specific linker histone gene H1oo: homology with the genes for the oocyte-specific cleavage stage histone (cs-H1) of sea urchin and the B4/H1M histone of the frog. Development. 2001;128(5):655–664. doi: 10.1242/dev.128.5.655. [DOI] [PubMed] [Google Scholar]
- Th’ng JP, Sung R, Ye M, Hendzel MJ. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J Biol Chem. 2005;280(30):27809–27814. doi: 10.1074/jbc.M501627200. [DOI] [PubMed] [Google Scholar]
- Thoma F, Koller T, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol. 1979;83(2):403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde KE. Chromatin. Springer-Verlag; New York: 1989. [Google Scholar]
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16(1):93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Verdaguer N, Perelló M, Palau J, Subirana JA. Helical structure of basic proteins from spermatozoa. Comparison with model peptides. Eur J Biochem. 1993;214(3):879–887. doi: 10.1111/j.1432-1033.1993.tb17991.x. 10. 1111/j.1432-1033.1993.tb17991.x. [DOI] [PubMed] [Google Scholar]
- Vila R, Ponte I, Collado M, Arrondo JL, Jiménez MA, Rico M, Suau P. DNA-induced α-helical structure in the NH2-terminal domain of histone H1. J Biol Chem. 2001;276(49):46429–46435. doi: 10.1074/jbc.M106952200. [DOI] [PubMed] [Google Scholar]
- Vila R, Ponte I, Jiménez MA, Rico M, Suau P. An inducible helix-Gly-Gly-helix motif in the N-terminal domain of histone H1e: a CD and NMR study. Protein Sci. 2002;11(2):214–220. doi: 10.1110/ps.29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JP, Fondufe-Mittendorf Y, Xi L, Tsai GF, Segal E, Widom J, Stormo G. Preferentially quantized linker DNA lengths in Saccharomyces cerevisiae. PLOS Comput Biol. 2008;4(9):e1000175. doi: 10.1371/journal.pcbi.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlak P, Li P, Wang X, Garrard WT. Cleavage preferences of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease) on naked DNA and chromatin substrates. J Biol Chem. 2000;275(11):8226–8232. doi: 10.1074/jbc.275.11.8226. 10.1074/jbc. 275.11.8226. [DOI] [PubMed] [Google Scholar]
- Widlak P, Kalinowska M, Parseghian MH, Lu X, Hansen JC, Garrard WT. The histone H1 C-terminal domain binds to the apoptotic nuclease, DNA fragmentation factor (DFF40/CAD) and stimulates DNA cleavage. Biochemistry. 2005;44(21):7871–7878. doi: 10.1021/bi050100n. [DOI] [PubMed] [Google Scholar]
- Widom J. Physicochemical studies of the folding of the 100 Å nucleosome filament into the 300 Å filament. Cation dependence. J Mol Biol. 1986;190(3):411–424. doi: 10.1016/0022-2836(86)90012-4. [DOI] [PubMed] [Google Scholar]
- Widom J. A relationship between the helical twist of DNA and the ordered positioning of nucleosomes in all eukaryotic cells. Proc Natl Acad Sci USA. 1992;89(3):1095–1099. doi: 10.1073/pnas.89.3.1095. 10. 1073/pnas.89.3.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki AT, Jerzmanowski A. Suppression of histone H1 genes in Arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics. 2005;169(2):997–1008. doi: 10.1534/genetics.104.031997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YB, Gerchman SE, Ramakrishnan V, Travers A, Muyldermans S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998;395(6700):402–405. doi: 10.1038/26521. [DOI] [PubMed] [Google Scholar]